
Abstract
Background:
Although periodontitis is reportedly associated with increased cognitive decline in Alzheimer’s disease, the mechanisms underlying this process remain unknown. Porphyromonas gingivalis lipopolysaccharide (P.g-LPS) is an endotoxin associated with periodontal disease.
Objective:
We investigated the effect of periodontitis on learning capacity and memory of amyloid-β protein precursor (AβPP)/presenilin (PS1) transgenic mice along with the mechanisms underlying these effects.
Methods:
Mice were randomly assigned to three groups, namely AβPP/PS1 (control), P.g-LPS Injection, and P.g-LPS Injection + Ligation. Mice from the P.g-LPS Injection group were injected with P.g-LPS in the periodontal tissue three times per week for 8 weeks, while mice from the P.g-LPS Injection + Ligation group were injected with P.g-LPS and subjected to ligation of the gingival sulcus of the maxillary second molar.
Results:
Expression of gingival proinflammatory cytokines as well as alveolar bone resorption in P.g-LPS-injected and ligatured mice was increased compared to that in control mice. Mice in the P.g-LPS Injection + Ligation group exhibited cognitive impairment and a significant reduction in the number of neurons. Glial cell activation in the experimental groups with significantly increased amyloid-β (Aβ) levels was more pronounced relative to the control group. Induction of periodontitis was concurrent with an increase in cyclooxygenase-2, inducible nitric oxide synthase, AβPP, and beta-secretase 1 expression and a decrease in A disintegrin and metalloproteinase domain-containing protein 10 expression.
Conclusion:
These findings indicated that periodontitis exacerbated learning and memory impairment in AβPP/PS1 mice and augmented Aβ and neuroinflammatory responses. Our study provides a theoretical basis for risk prediction and early intervention of Alzheimer’s disease and periodontitis.
INTRODUCTION
Periodontitis, an inflammatory disease of the gums and surrounding tissues, is primarily caused by gram-negative anaerobic bacteria [1, 2]. During the development of periodontitis, the effects of bacteria and their toxic products, along with resultant inflammatory responses, are not only localized, but also become systemic throughout the periodontal pocket lining, resulting in bacteremia or endotoxemia [3, 4]. Therefore, a localized periodontal infection may spread throughout the body, facilitating the occurrence and development of systemic diseases, such as cardiovascular and cerebrovascular diseases, cancer, and diabetes, in addition to acting as a risk factor for premature birth and low birth weight babies [5]. Furthermore, studies have demonstrated that periodontitis is associated with cognitive decline in Alzheimer’s disease (AD) [6, 7].
AD causes progressive mental, behavioral, and cognitive decline, and may lead to reduced learning abilities. It is a progressive degenerative disease of the central nervous system and a major form of dementia, showing characteristic pathological changes including amyloid plaque formation caused by aggregation of extracellular amyloid proteins, formation of neurofibrillary tangles due to abnormal aggregation of intracellular tau proteins, and neuron loss [8, 9]. An imbalance in the production and clearance of amyloid-β (Aβ) is believed to play a key role in AD progression [10]. Evidently overproduction and excessive accumulation of Aβ, caused by cleavage of amyloid-β protein precursor (AβPP) in the brain, are principal contributors to the pathogenesis of AD [11]. Studies have also shown that Aβ is generated during AβPP metabolism, which occurs via two pathways, namely non-amyloid and amyloid pathways. In the non-amyloid pathway, AβPP is catalyzed by α-secretase (a member of a large metallopeptidase family), also known as A disintegrin and metalloprotease (ADAM), and subsequently by γ-secretase under normal physiological conditions, thus preventing Aβ generation. In the amyloid pathway, AβPP is activated by β-site APP cleavage 1 (BACE1), a rate-limiting secretase, leading to the production of toxic Aβ fragments that contribute to AD pathology, following which it is acted upon by γ-secretase, leading to the production of toxic Aβ fragments that contribute to AD pathology [12–14]. Studies have shown that Aβ deposition is associated with overactivation of microglia, which promotes neuroinflammation and neuronal dysfunction [15].
Porphyromonas gingivalis (P.g) is one of the most common pathogens associated with periodontitis, and P.g lipopolysaccharide (P.g-LPS) plays a major pathogenic role in P.g infection [16]. Poole et al. found P.g-LPS in the brains of autopsied AD victims, wherein P.g-LPS levels in brain tissues were significantly higher than those in non-AD subjects [17]. In periodontitis, the balance between bacterial levels and host responses is disrupted, leading to increased inflammatory cell infiltration and production of proinflammatory cytokines, such as interleukin (IL) 1 beta (IL-1β), IL-6, and tumor necrosis factor-alpha (TNF-α) [18, 19]. Recent studies have indicated that systemic inflammation plays a central role in AD progression if the acute injury is sufficiently intense (e.g., severe sepsis) or has a long duration (e.g., repeated stimulation by large doses of proinflammatory agents such as LPS) [20]. Sheng et al. observed that intraperitoneal injection of LPS induced neuroinflammation with increasing AβPP expression and Aβ deposition in mice, suggesting that peripheral inflammation might lead to central inflammation and further aggravation of AD [21]. During this process, high concentrations of inflammatory mediators dilate the blood vessels and increase endothelial permeability of the blood-brain barrier, thereby allowing entry of pathogenic factors into the central nervous system, which in turn, activates microglial cells that subsequently release a considerable number of inflammatory factors, thus damaging the functional central nervous system and causing brain inflammation [22, 23]. However, whether periodontitis contributes to cognitive impairment is unclear. In this study, we evaluated the pathogenic manifestations induced by P.g-LPS via the Aβ generation pathway. Notably, we used AβPP/PS1 transgenic mice to examine whether periodontitis induced by P.g-LPS and ligation exacerbated spatial learning and memory impairment. We also examined the biological mechanisms underlying behavioral changes.
MATERIALS AND METHODS
Animals
Thirty 6-month-old AβPP/PS1 double-transgenic male mice were purchased from Nanjing Biomedical Research Institute of Nanjing University-Nanjing Institute of Biomedical Sciences: License Number; SCXK (JiangSun, China) 2015-0001. All mice weighed 35–45 g and were provided ad libitum access to water and fed with standard rodent food in a 22±2°C environment under a 12-h light/12-h dark cycle for 24 h. Next, AβPP/PS1 mice were randomly divided into three groups based on the following treatments: AβPP/PS1 (control group); P.g-LPS (ultrapure, InvivoGen, Inc., San Diego, CA, USA) only; and P.g-LPS combined with ligation. To induce periodontitis, a 4-0 silk ligature was inserted into the gingival sulcus between the second maxillary molar (M2) and first maxillary molar (M1) of the maxilla in both sides of each mouse for 8 weeks, followed by interpapillary injection of 2μL of 1.0 mg/mL P.g-LPS (thrice a week) at the same position in a manner similar to that performed on mice of the P.g-LPS and P.g-LPS + Ligation groups. The injection was administered slowly, and the needle (Gaoge, Shanghai, China) was kept in place for a few seconds to ensure that there was no wastage or loss of P.g-LPS during needle extraction. Age-matched control animals were not injected. Animals were sacrificed 9 weeks after the first injection. Maxillae were dissected and immersed in 10%buffered formalin for 48 h. All efforts were made to minimize animal use as well as animal suffering. All animal experiments were undertaken in accordance with the relevant guidelines and regulations of the State Committee of Science and Technology of the People’s Republic of China, Order No. 2 on November 14, 1988 (revised 2011), and approved by the Animal Experimentation Ethics Committee of the Zunyi Medical University, which also meet the NIH guidelines for the care and use of laboratory animals.
Behavioral experiments
Morris water maze test
After 8 weeks of P.g-LPS injection, the Morris water maze test was performed to assess spatial learning and memory. The device consisted of a white plastic round pool (radius = 60 cm, height = 50 cm) filled with water (23±2°C) to a depth of 30 cm. The pool consisted of four equal quadrants; a movable platform made of Perspex was positioned under the third quadrant, submerged a centimeter from the water surface. The task consisted of two steps. The first step was direction sailing training. During the first four consecutive days, the mouse was released into the water at one of the four quadrants and allowed 60 s to reach the hidden platform, where it remained for 20 s, and escape latency (i.e., time taken to swim toward the hidden platform, climb, and remain on it for more than 3 s) was recorded. If a mouse failed to reach the platform within 60 s, it was manually guided to the platform and allowed to remain on it for 20 s, and the escape latency was recorded as 60 s [24]. The spatial probe test designed to measure final spatial memory consolidation was scheduled to take place on the 5th day; during this time, the hidden platform was withdrawn. These activities were automatically measured and analyzed using a behavior analyzing system (TopScan Version 3.00, CleverSys, Reston, VA, USA).
Novel object recognition task
The novel object recognition task involved the use of two reaction chambers of the same size: 40×40×40 cm. Two days of training sessions were conducted to allow the mice to be familiar with to the apparatus. Each session involved placing a mouse in the apparatus containing two similar objects and allowing it to explore for 5 min. The objects were positioned identically in two nearby corners and were similar in texture, color, size, and shape. On the 3rd day (test session), one of the objects was replaced with a new object of the same size but a different shape. The discrimination ratio for each mouse was expressed as (TN–TF)/(TN+ TF) ratio; where TF= time spent exploring familiar object; TN= time spent exploring the novel object. The objects were cleaned with 10%ethanol solution between trials [25, 26].
Y-maze test
The Y-maze, which is used to observe the spontaneous alternation rates displayed by mice, allows changes in short-term spatial learning and memory to be determined, according to previously published procedures [27, 28]. The Y-maze consisted of three arms with an angle of 120° between any two arms, which were 40 cm long and 8 cm wide with 15 cm high walls. The setup included a ceiling-mounted infrared camera tracking system attached to a computer with supporting software. The mice were initially placed at the end of one fixed arm and allowed to explore the Y-maze freely for 5 min. Entries into all arms were noted (presence of all four paws inside an arm was considered a valid entry), where consecutive entries into three different arms by an animal was considered as a spontaneous alternation. Next, the mouse was removed from the Y-maze to its house, and the Y-maze was cleaned until dry using 10%ethanol to remove odor [29, 30]. An infrared camera was used to record all trials on a video cassette recorder. The video recordings were later analyzed to determine the number of entries and time spent in each arm. These activities were automatically measured and analyzed using a behavior analysis system (TopScan Version 3.00).
Tissue preparation
Following the behavioral tests, the mice (n = 4–5 per group) were anesthetized with sodium pentobarbital and transcardially perfused with chilled 0.1 M phosphate-buffered saline (PBS). The brains were divided into left and right hemispheres and placed in pre-cooled 4%buffered paraformaldehyde at 4°C overnight, and subsequently embedded in paraffin. The hippocampi and cerebral cortices of few animals were isolated and immediately stored in Eppendorf micro test tubes at –80°C. Gingival tissues and alveolar bones were stored in 4%buffered paraformaldehyde at –80°C and at 4°C, respectively.
Methylene blue staining
The maxillary molar regions were fixed in 4%neutralized formalin for 24 h. De-fleshed molar samples were stained with 1%aqueous methylene blue (Dalian Meilun Biotechnology, Dalian, China) for 5 min [31]. Digital photographs were obtained using a 500-μm macro lens digital camera (Canon, Tokyo, Japan).
Micro-computed tomography (Micro-CT)
Maxilla specimens were collected and placed in 4%formaldehyde in PBS and subjected to micro-CT analysis using a μ-CT50 scanning system (Scanco Medical vivaCT 40, Brüttisellen, Switzerland) at a resolution of 10.5μm. The volume of interest was determined using the volume of alveolar bone of root bifurcation, within which trabecular plate thickness, bone volume, and bone volume per total volume were calculated. The volume of interest was drawn with a slice-based method, starting from the bottom of the pulp chamber of the second molar. Images were analyzed using the software μct V6.1 (Scanco Medical vivaCT 40).
Nissl staining
Nissl staining was performed to determine mor-phological changes occurring in hippocampal neurons as well as the number of surviving neurons, as per previously described methods [32]. Right brain hemispheres (n = 4) were serially cut into 35μm-thick sections in the coronal plane and stained with toluidine blue (Solarbio, Beijing, China). Nissl bo-dies in the CA1 and DG regions of the hippocampus and cerebral cortex were stained blue-purple. Nissl-positive neurons were counted using a light microscope with 400× magnification (KS300, Zeiss-Kontron, Oberkochen, Germany). Four different vis-ual fields were selected from each section, and the number of surviving neurons in the same area of the hippocampal CA1 and DG regions and cerebral cortex was counted as per previously described protocols [32]. The mean value for each mouse was obtained as the number of neurons in the mouse.
Western blotting
The hippocampal and cerebral cortical tissues (n = 4 per group) were homogenized using a protein extraction solution containing a complete protease inhibitor mixture. The homogenized samples were centrifuged at 12,000 rpm for 15 min, and the supernatant was extracted, aliquoted, and finally stored at –80°C. Protein concentrations of each sample were determined using the BCA protein assay kit (Biocolor Biotechnology, Antrim, UK). Equal amounts of proteins (25–30μg) were heated at 100°C for 5 min, separated by electrophoresis using a 10%SDS or 15%polyacrylamide gel, and transferred onto polyvinylidene difluoride membranes (0.45μm in thickness). Membranes were blocked for 2–3 h with 5%defatted milk in 1× Tris-buffered saline Tween-20 and probed with the corresponding primary antibodies against AβPP (1:5000, ab32136, Abcam, Cambridge, UK), BACE1 (1:2000, D220305, BBI Life Sciences, Shanghai, China), ADAM10 (1:2000, 25900-1-AP, Proteintech, Rosemont, IL, USA), inducible nitric ox-ide synthase (iNOS, 1:1000, bs-0162R, Bioss, Wob-urn, MA, USA), cyclooxygenase-2 (COX-2, 1:1000, 12375-1-AP, Proteintech), IL-6 (1:500, 21865-1-AP, Proteintech), TNF-α (1:2000, 60291-1-lg, Proteintech), glial fibrillary acidic protein (GFAP; 1:1000, #3670, Cell Signaling Technology, Danvers, MA, USA), ionized calcium-binding adaptor molecule 1 (Iba1; 1:1000, ab178847, Abcam), Aβ40 (1:2000, MAB2675, Abnova, Taipei, Taiwan), and Aβ42 (1:2000, #14974, Cell Signaling Technology) at 4°C overnight. The blot was then incubated with the corresponding anti-mouse or rabbit immunoglobulin horseradish peroxidase-conjugated antibodies for 1–2 h at 37°C. Next, ECL Reagent (Millipore Corpor-ation, Billerica, MA, USA) and Image Lab (Bio-Rad, Hercules, CA, USA) were used to analyze the protein bands [25].
Immunohistochemistry and immunofluorescence
Brain samples were processed for immunohistochemistry and immunofluorescence according to previously reported procedures [33]. Briefly, brain samples were immersed in xylene for 10 min twice and rehydrated by serially diluting in a descending ethanol gradient (absolute alcohol to 95%, to 80%, to 70%) for 5 min and washed thrice in distilled water for 5 min. Antigen retrieval was performed in citrate buffer (10 mM, pH 6.0) in a microwave for 6 min at a high temperature [34]. To quench endogenous peroxidase activity, sections were incubated with 3%H2O2 in PBS for 30 min. After rinsing thrice with PBS, the sections were blocked with 10%normal goat serum in PBS for 30 min and incubated with primary antibodies against GFAP and Iba1 overnight at 4°C. After washing with PBS thrice, the sections were probed with goat anti-rabbit lgG/hor-seradish peroxidase polymer or goat anti-mouse lgG/horseradish peroxidase polymer (1:500, Beyotime, Shanghai, China) at 37°C for 1 h. Sections were washed with PBS and visualized using the DAB chromogen solution (Dako, Glostrup, Denmark) for a few seconds. After counterstaining with hematoxylin, the sections were dehydrated with alcohol and cleared in xylene before being mounted under a glass coverslip using neutral balsam mounting medium [34]. The immunostained sections were then imaged using a microscope (Olympus, Tokyo, Japan). Images of the hippocampal and cortical regions were recorded, and the results are expressed as the average intensity of positive immunoreactive cells of GFAP and Iba1 immunohistochemistry. Immunofluorescence assays were performed to detect Aβ location [25]. Briefly, the sections were washed thrice in PBS and incubated with 10%normal goat serum in PBS for 1 h at 37°C. The sections were then incubated with the primary rabbit Polyclonal Aβ antibody (Cell Signaling Technology, 15126S) overnight at 4°C. After washing in PBS, the sections were incubated with the corresponding secondary antibodies for 2 h at 37°C and then incubated with DAPI for 5 min at 37°C.
Quantitative real-time PCR (qRT-PCR)
The gene expression of IL-6 and TNF-α genes was detected via qRT-PCR. Total mRNA from the gingiva was extracted using RNAiso Plus (Takara, Shiga, Japan) according to the manufacturer’s instructions. The quality and quantity of RNA were determined using Nano Drop (ND-2000, Thermo Fisher Scientific, Waltham, MA, USA), with a 260/280 ratio of 1.8/2.1. A total of 50 ng of extracted mRNA was reverse-transcribed into cDNA using the High Capacity Reverse Transcriptase Kit (Takara). Thereafter, a PCR sample of 15μL was prepared with 3μL cDNA (10 ng/μL), 7.5μL TB Green Premix EX Taq II (Takara), 0.5μL primer mix (10μM each), and 4μL ddH2O. The PCR conditions were as follows: initial denaturation at 95°C for 5 min; 50 cycles of denaturation at 95°C for 5 min; annealing at 60°C for 10 s; elongation at 60°C for 30 s [35]. Primer sequences (Table 1) of the selected genes were designed using the Primer 3 software. For data normalization purposes, mRNA expression of actin was determined, and the relative units were calculated using the comparative 2-ΔΔCT method [25].
Primer sequences used for qRT-PCR analysis
Statistical analysis
All statistical analyses were conducted out using the SPSS software, version 19.0 (SPSS, Inc., Chica-go, IL, USA). Normality of the data was checked using the Kolmogorov-Smirnov test. Normally distr-ibuted continuous variables are expressed as mean± SEM. One-way analysis of variance (ANOVA), fol-lowed by Dunnett’s test, was used for multiple comparisons. Statistical significance was set at p < 0.05.
RESULTS
Effects of periodontitis on alveolar bone loss and expression of proinflammatory cytokines in the gingiva
Methylene blue staining results showed that alv-eolar bone resorption in the P.g-LPS and P.g-LPS + Ligation groups was enhanced compared with that in the AβPP/PS1 mice (Fig. 1a). Micro-CT images showed severe alveolar bone loss had occurred in the P.g-LPS and P.g-LPS + Ligation groups compared to that in the control group (Fig. 1b). Furthermore, the trabecular plate thickness, bone volume, and bone volume per total volume values in the ligation group were significantly different compared to those in the control and P.g-LPS groups (p < 0.01; Fig. 1c–e). We also assessed levels of proinflammatory cytokines by evaluating mRNA expression of IL-6 and TNF-α in the gingiva tissues via qRT-PCR. The levels of both proinflammatory cytokines were notably enhanced in the P.g-LPS + Ligation group compared to those in the AβPP/PS1 mice group (p < 0.05 or p < 0.01; Figs. 1f, g).
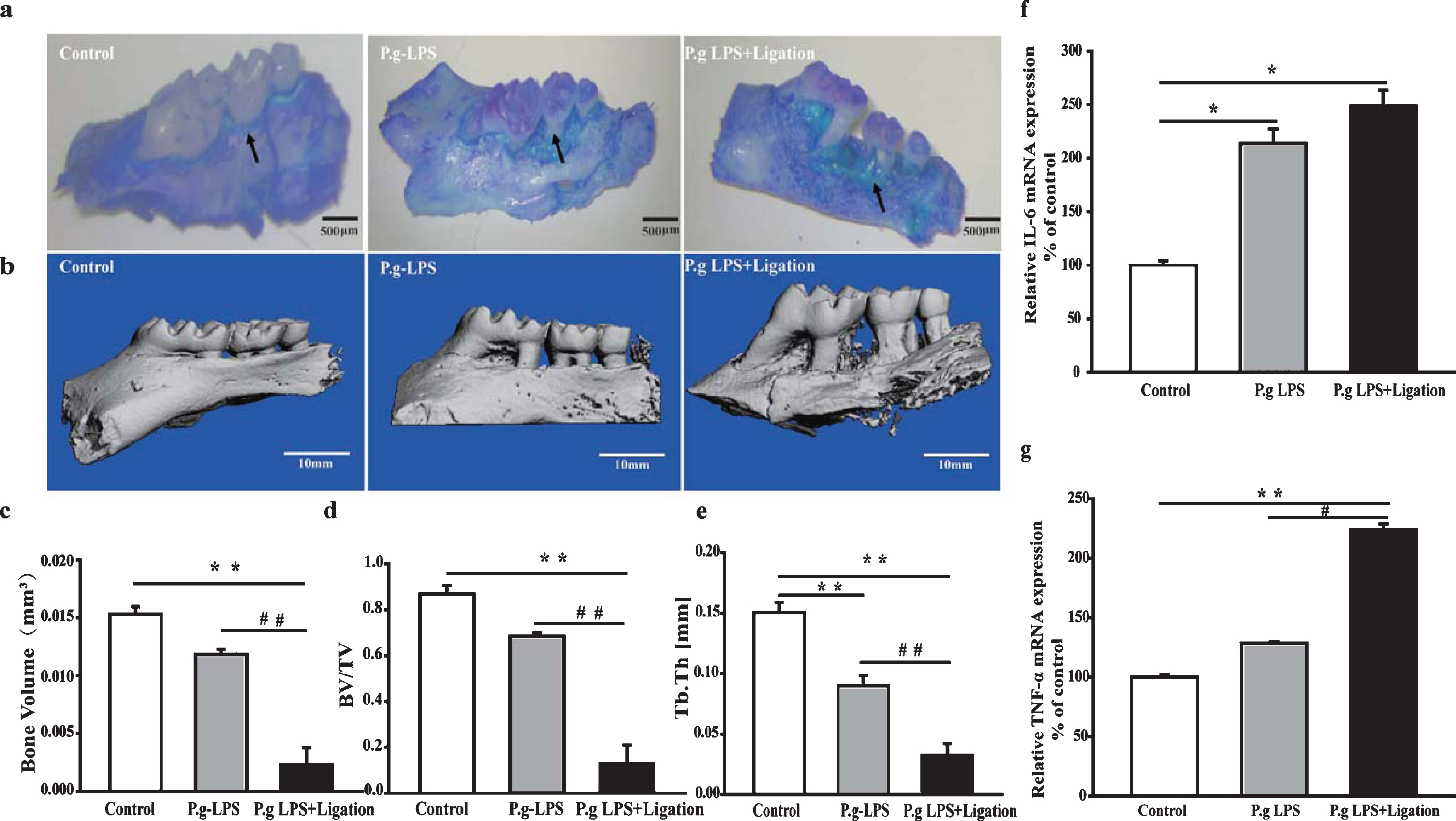
Effects of periodontitis on alveolar bone loss and proinflammatory cytokines in the gingiva. (a) Methylene blue staining, (b) Micro-CT, (c) bone volume, (d) bone volume per total volume, and (e) trabecular thickness in the region of interest in mouse alveolar bone. The mRNA levels of (f) IL-6 and (g) TNF- α in the gingiva of AβPP/PS1 mice. Values are expressed as mean±SEM (n = 4), *p < 0.05, **p < 0.01 versus control, #p < 0.05, # #p < 0.01 P.g-LPS versus P.g-LPS + Ligation.
Effects of periodontitis on spatial learning and memory impairment in AβPP/PS1 transgenic mice
To study the effects of periodontitis on learning and memory, as well as spatial learning and memory abilities of mice, the animals were subjected to behavioral tests 8 weeks after surgery. In the navigation test, escape latency of all mice was gradually shortened, while their capability to locate the platform was enhanced after training (Fig. 2a). A decrease in the mean escape latencies of AβPP/PS1 transgenic mice was observed during days 2–4. No distinct differences were observed between the mean escape latencies of P.g-LPS + Ligation and AβPP/PS1 groups on days 1 and 2 (p = 0.76 and 0.113). However, marked differences were observed on days 3 and 4 (p = 0.001 and 0.002). The percentage of time spent in the target quadrant by animals of the P.g-LPS + Ligation group while searching for the platform was lower than that spent by animals of the AβPP/PS1 control group, suggesting cognitive impairment in the former (Fig. 2b).
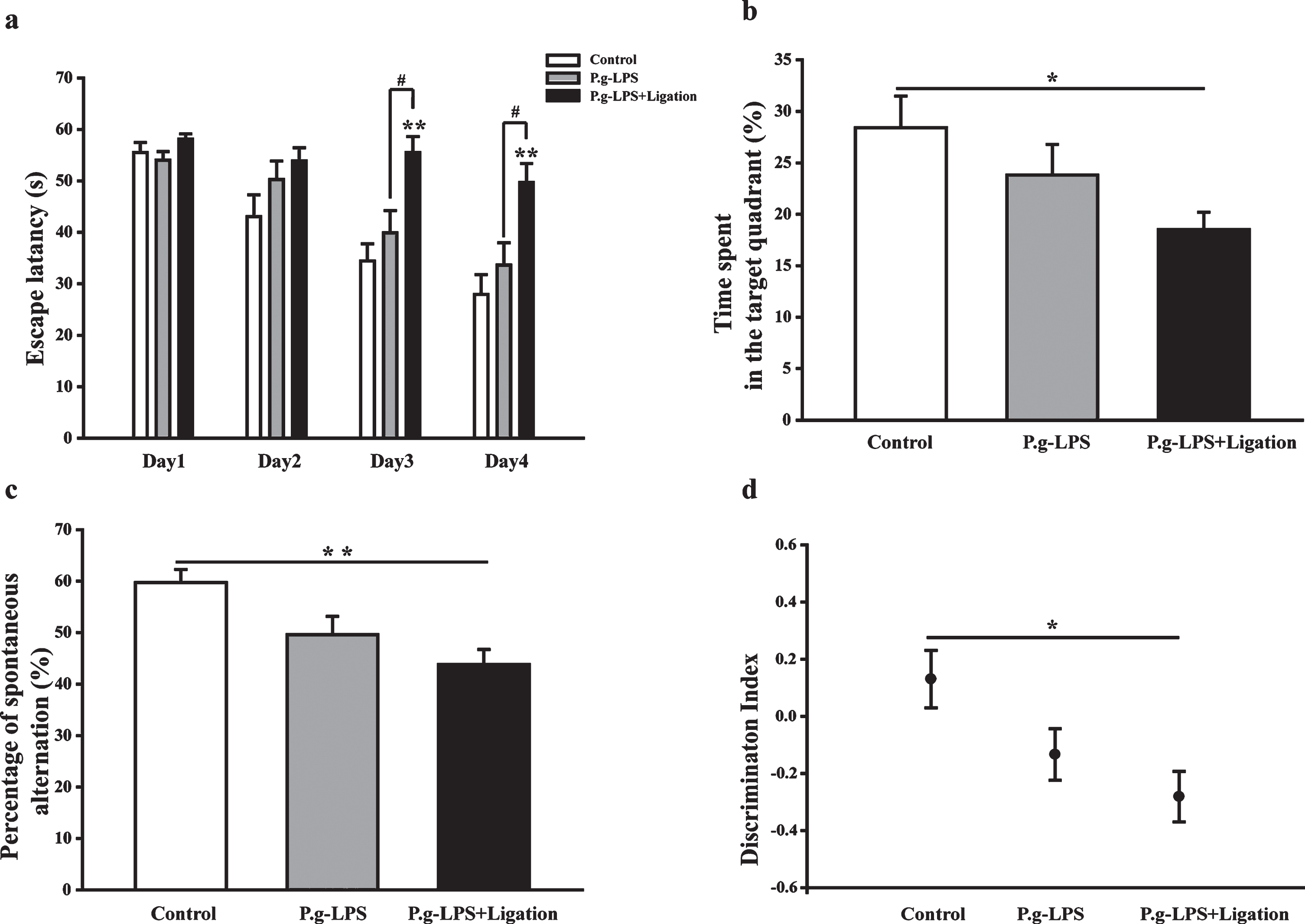
Effects of periodontitis on learning and memory functions of AβPP/PS1 mice. (a) Escape latency of mice to reach the hidden platform from day 1 to 4. (b) Percentage of time utilized by the mice for swimming in the target quadrant. (c) Spontaneous alternation rate of mice in the Y-maze assay. (d) New object discrimination index. Values are expressed as the mean±SEM (n = 10), *p < 0.05, **p < 0.01 versus control. #p < 0.05 P.g-LPS versus P.g-LPS + Ligation.
Spontaneous alternation shown by the P.g-LPS + Ligation group during the Y-maze test was notably decreased compared to that shown by the AβPP/PS1 group (p < 0.01, Fig. 2c). A similar result was obt-ained for the novel object recognition assay. Generally, the mice spent more time with the novel object compared to the time spent with the familiar object. Relative to behavior at consecutive training sessions, a notable decrease was observed in the new object discrimination index during the experimental session for the P.g-LPS + Ligation group compared to that observed for the control group (p < 0.05; Fig. 2d).
Effects of P.g-LPS and ligation-induced periodontitis on neuronal loss
The number of neuronal cells in the CA1 and DG regions of the hippocampus and cerebral cortex was determined to study the effects of P.g-LPS- and ligation-induced periodontitis on neuronal morphology and boundary. Structurally intact and highly dense pyramidal layer neurons were observed in the control group (Fig. 3). By contrast, the number of atrophied and pyknotic neurons in the P.g-LPS and P.g-LPS + Ligation groups was reduced.
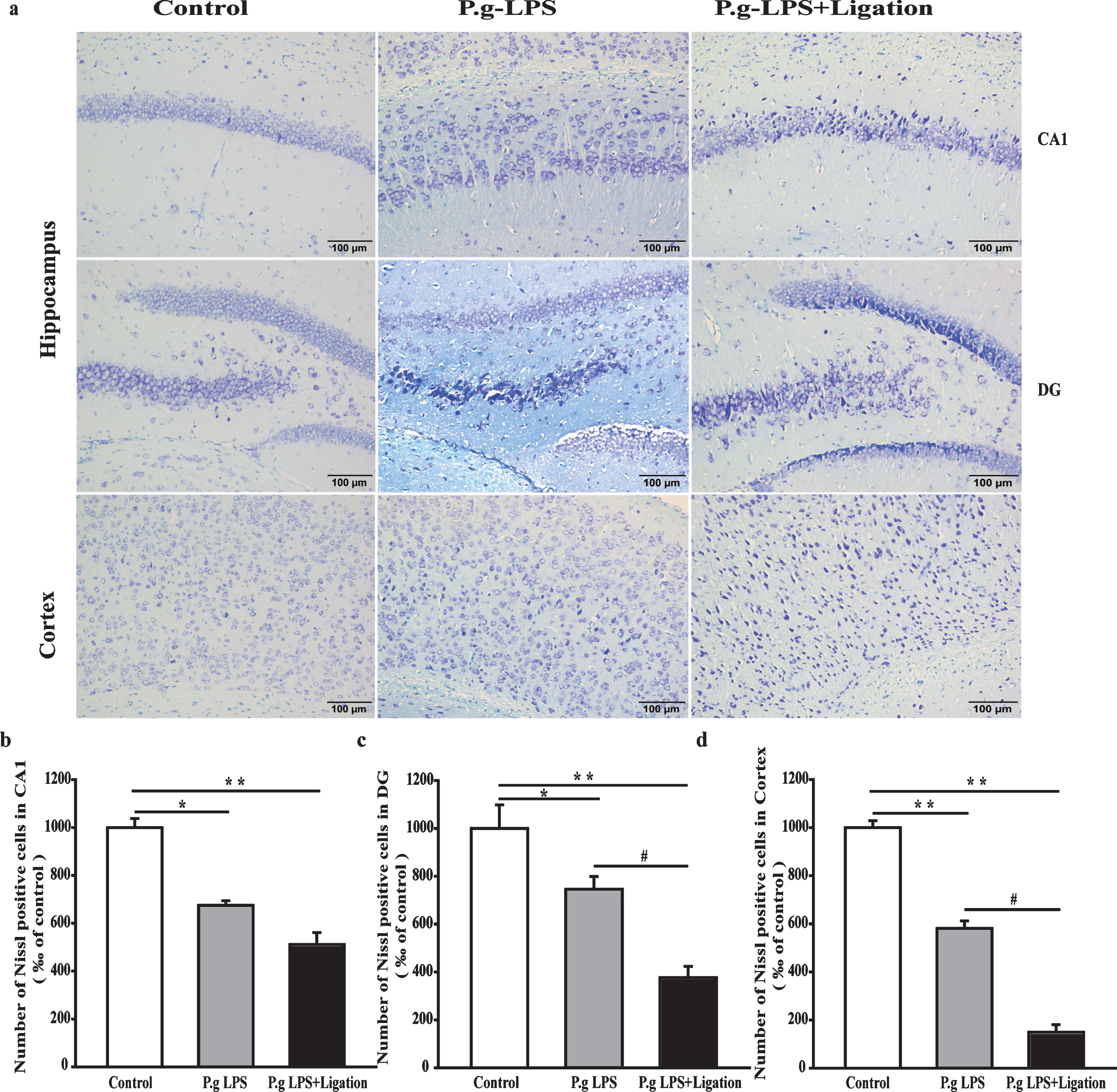
Effects of periodontitis on neurons in the hippocampus and cerebral cortex of mice. (a) Representative photomicrographs of Nissl staining. (b-d) Quantitative analysis of the number of neurons. Values are expressed as mean±SEM (n = 4), *p < 0.05 versus control, **p < 0.01 versus control, #p < 0.01 P.g-LPS versus P.g-LPS + Ligation.
Immunofluorescence staining with anti-Aβ antibody was used to assess general Aβ levels in the hippocampi and cerebral cortices of mouse brains (Fig. 4a–c). Aβ deposition in the hippocampal regions and cerebral cortices of mice from the P.g-LPS and P.g-LPS + Ligation groups was significantly increased relative to that in mice form the AβPP/PS1 control group.
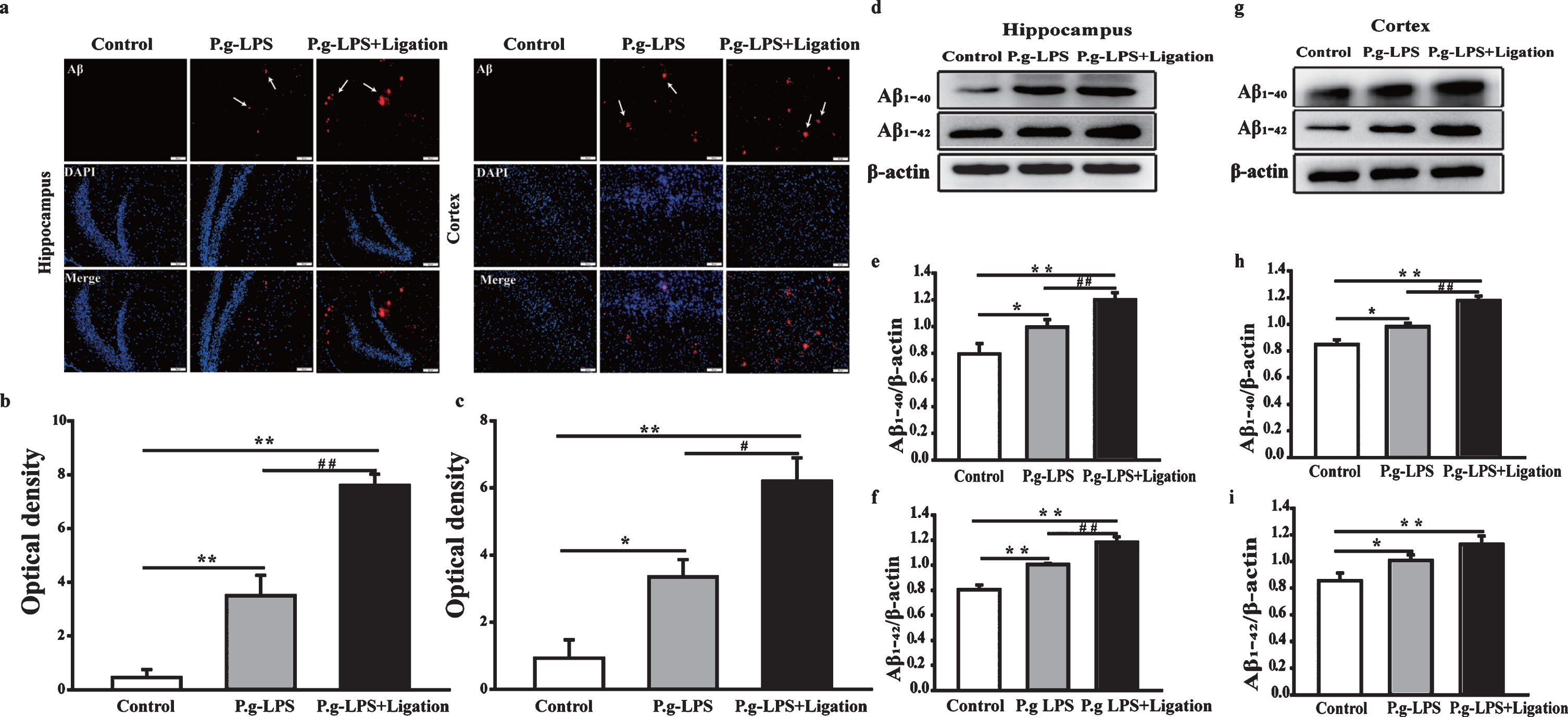
Effects of periodontitis on the levels of Aβ40 and Aβ42 in the hippocampus and cerebral cortex of mice. (a) Images of immunofluorescence in the hippocampus and cerebral cortex of AβPP/PS1 mice (magnification 200×). (b, c) Optical density statistics for Aβ deposition in the hippocampus and cerebral cortex. (d, g) Representative bands of Aβ40 and Aβ42 in the hippocampus and cerebral cortex of mice. (e, i) Quantitative analysis based on normalized with β-actin expression levels. Values are expressed as mean±SEM (n = 4), *p < 0.05, **p < 0.01 versus control, #p < 0.05, # #p < 0.01 P.g-LPS versus P.g-LPS + Ligation.
Effects of periodontitis on the levels of Aβ42 and Aβ40 in the hippocampus and cerebral cortex
Western blotting yielded similar results (Fig. 4d–i). Compared with the control group, a dramatic increase in the Aβ level was observed in the P.g-LPS and P.g-LPS + Ligation groups, including the level of Aβ40/42 (p < 0.05 or p < 0.01). Interestingly, significant differences were also observed between the P.g-LPS and P.g-LPS + Ligation groups (p < 0.05 or p < 0.01).
Effects of periodontitis on Aβ production pathway in the hippocampus and cerebral cortex
Abnormally increased Aβ production correlates closely with the activity of secretases of the AβPP cleavage process. Therefore, we evaluated the chan-ges in AβPP that might contribute to increased Aβ production (Fig. 5). AβPP protein levels were significantly elevated in the brains of mice of the P.g-LPS + Ligation group compared to those in mice of the control group (p < 0.01). Notably, there was a significant difference between P.g-LPS and P.g-LPS + Ligation groups (p < 0.05), indicating that ligation increased inflammation, leading to increased expression of AβPP. We further analyzed the roles of two crucial enzymes, ADAM10 and BACE1, in the cleavage of AβPP [28]. BACE1 and ADAM10 expression increased and decreased significantly, respectively, in the P.g-LPS + Ligation group compared with that in the AβPP/PS1 group (p < 0.05 or p < 0.01) in both cerebral cortices and hippocampi.
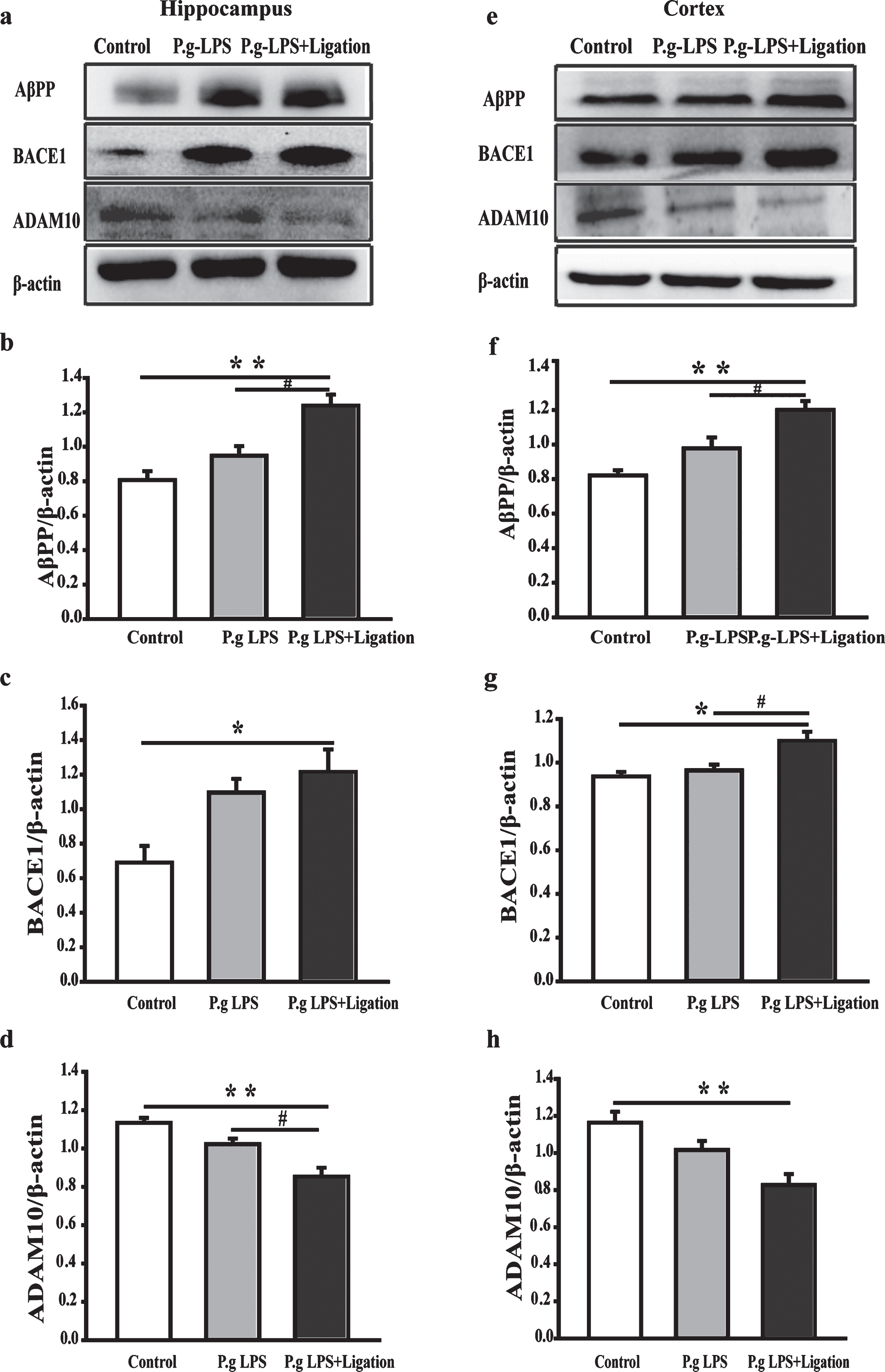
Effect of periodontitis on the process of amyloid-β production in the hippocampus and cerebral cortex of mice. (a, e) Antibody-reactive bands of AβPP, BACE1, and ADAM10 in the hippocampus and cerebral cortex of mice. (b–h) Quantitative analysis based on normalized with β-actin expression levels. Values are expressed as mean±SEM (n = 4), *p < 0.05, **p < 0.01 versus control. #p < 0.05 P.g-LPS versus P.g-LPS + Ligation.
Periodontitis promotes astrocyte and microglial activation
Intraperitoneal injection of LPS may induce neuroinflammation [21, 36]. Therefore, we determined whether P.g-LPS also caused neuroinflammation, by evaluating microglial and astrocytic responses via western blotting and immunohistochemistry staining of Iba1 and GFAP, respectively (Fig. 6). Iba1 immunostaining intensity in the P.g-LPS and P.g-LPS + Ligation groups increased compared to that in the AβPP/PS1 control group. Furthermore, the number of Iba1-immunoreactive microglia in these groups was increased compared with that in the control group. Additionally, GFAP-positive staining intensity in the hippocampal regions and the cerebral cortices increased significantly in the P.g-LPS and P.g-LPS + Ligation groups compared with that in the control group. Notably, the mean intensity of positive immunoreactive cells as well as the number of astrocytes in the hippocampal and cortical regions of mice with periodontitis increased. Similar results were observed via western blotting (Fig. 6g–l). Iba1 and GFAP expression in the hippocampi of the P.g-LPS + Ligation group was significantly increased compared to that in the control (p < 0.01 and p < 0.05, respectively) and cortex (p < 0.01) groups. Interestingly, Iba1 expression in the cerebral cortex of mice from the P.g-LPS group differed significantly from that of mice from the P.g-LPS + Ligation group (p < 0.05).
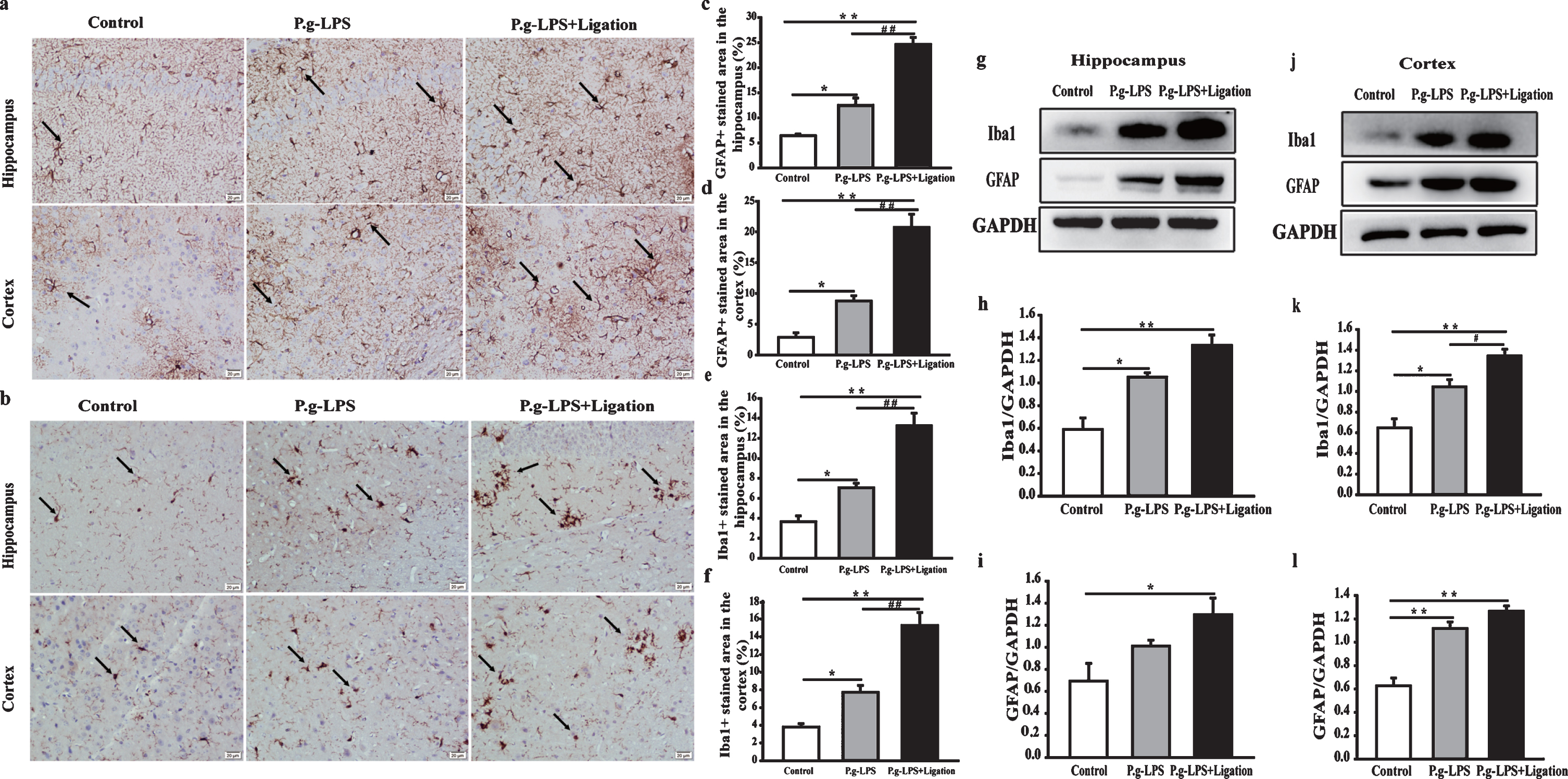
Periodontitis promotes astrocytes and microglial activation. (a, b) Micrographs of GFAP and Iba1 immunoreactivity in the hippocampus and cerebral cortex (magnification 200×, scale bar 200μm). (c, e) Quantitative analysis. (g, j) Antibody-reactive bands of Iba1 and GFAP in the hippocampus and cerebral cortex of mice, respectively. Quantitative analysis of (h, k) Iba1 and (i, l) GFAP expression in the hippocampus and cerebral cortex. The relative optical density was normalized to GAPDH values. Values are expressed as the mean±SEM (n = 4), *p < 0.05, **p < 0.01 versus control, #p < 0.05, # #p < 0.01 P.g-LPS versus P.g-LPS + Ligation.
Periodontitis increased the expression of proinflammatory cytokines in the hippocampus and cerebral cortex
Next, we explored whether P.g-LPS and ligation-induced periodontitis affected the proinflammatory response of glia by assessing the protein expression of iNOS, COX-2, interleukin 1 beta and TNF-α via western blotting. As shown in Fig. 7, the expression of inflammatory proteins in the hippocampal and cortical tissues of both P.g-LPS and P.g-LPS + Ligation groups was increased compared to that in the control group. Interestingly, a notable difference was observed between the cerebral cortices of mice from the P.g-LPS and P.g-LPS + Ligation groups.
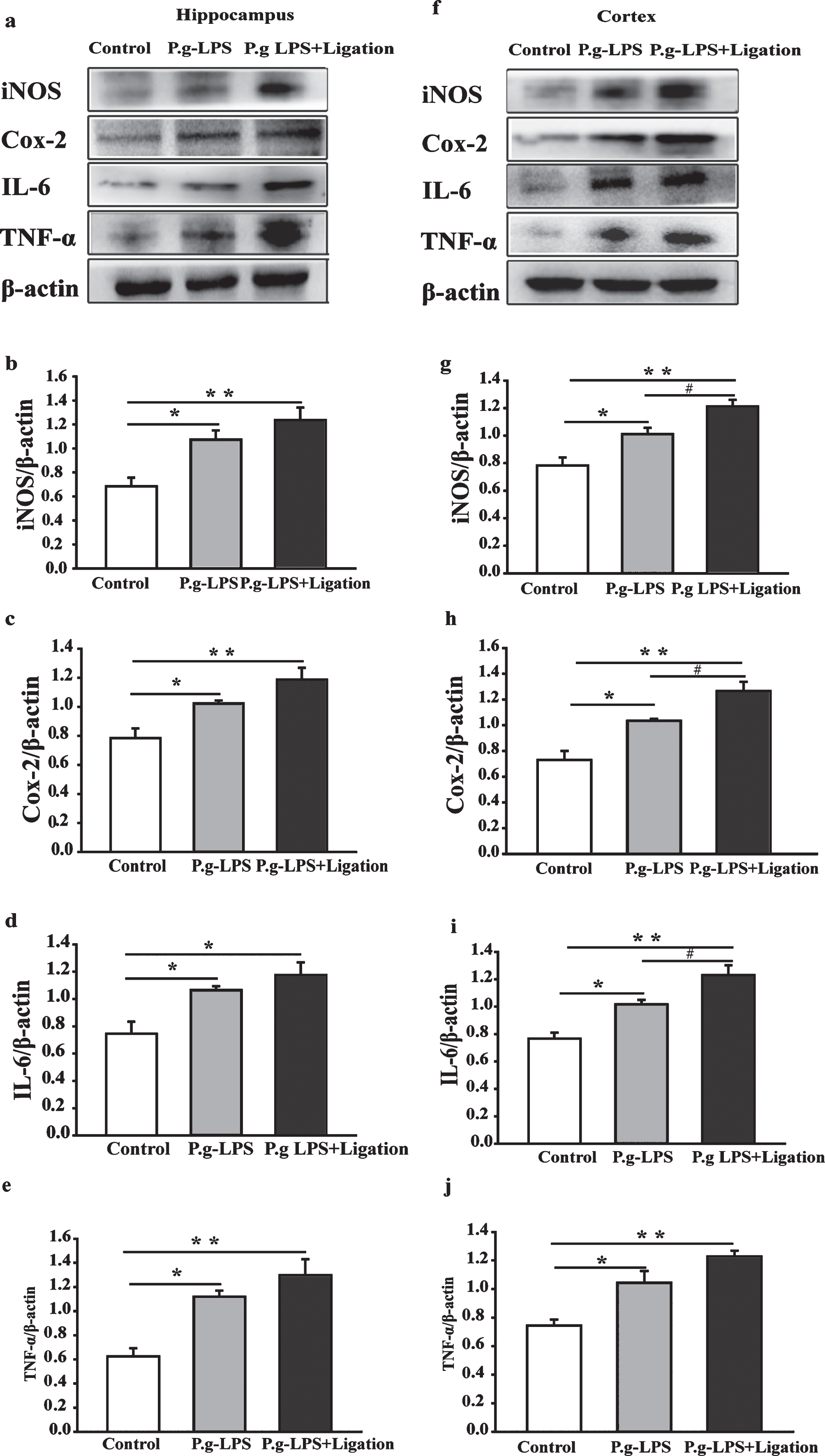
Effects of periodontitis on iNOS, COX-2, IL-6, and TNF-α expression in the hippocampus and cerebral cortex of mice. (a, f) Antibody-reactive bands of iNOS, COX-2, IL-6, and TNF-α in the hippocampus and cortex of mice, respectively. Quantitative analysis of (b, g) iNOS, (c, h) COX-2, (d, i) IL-6, and (e, j) TNF-α expression in the hippocampus and cerebral cortex. Relative optical density was normalized to β-actin. The values are expressed as mean±SEM (n = 4), *p < 0.05, **p < 0.01 versus control, #p < 0.05 P.g-LPS versus P.g-LPS + Ligation.
DISCUSSION
Periodontitis is a chronic infectious disease caus-ed by the formation of a plaque biofilm. Notably, periodontal pathogens and associated virulence factors, such as LPS, induce the secretion of proinflammatory cytokines, thereby triggering inflammatory responses that result in alveolar bone loss [37]. In the current study, periodontitis was induced by applying a silk ligature around the second maxillary molars, together with interpapillary injections of P.g-LPS, wherein insertion of the silk ligature into the gingival sulcus initiated plaque-induced marginal periodontitis, while the injections maintained an effective LPS concentration [38]. Therefore, ligation combined with P.g-LPS injection may better mimic the pathology and symptoms of periodontitis in the human mo-uth. In this study, compared to P.g-LPS injection al-one, ligation combined with P.g-LPS injection exerted increased toxic effects, suggesting that continuous LPS stimulation exerted greater toxicity under plaque colonization. Additionally, examination of the relevant proinflammatory factors in the gingival tissue, along with the micro-CT results, indicated that the periodontitis model was successfully established.
In recent years, increasing evidence has confirmed that periodontitis is remarkably associated with cardiovascular and cerebrovascular diseases, cancer, diabetes, premature birth and low birth weight, and respiratory tract infections [5, 39]. Moreover, a 2017 meta-analysis study showed that patients with periodontitis were nearly three times more likely to dev-elop AD than those without periodontitis. Moreover, the oral health of patients with AD was worse than that of patients without AD [40]. Previous studies have demonstrated that the direct inflammatory effect exerted by P.g-LPS aggravates neuroinflammation in the mouse brain [41]. Experimental studies have also shown that bacteria-induced periodontitis may be related to the exacerbation of AD pathology [42, 43]. There have also been reports that these changes may be related to age [44]. Dominy et al. identified P.g in the brain of AD patients and confirmed through mouse experiments that the entry of P.g into the brain facilitated the release of gingipain, which promoted AD. Notably, inhibition of this protein alleviated AD symptoms, thereby revealing a possible mechanism underlying the effect of P.g in AD [45]. Since it has been confirmed that P.g may be an effector of gingipain, we investigated whether P.g was also an important effector of LPS.
AβPP/PS1 mice, a widely recognized AD model, were used to verify whether periodontitis aggrava-ted the progression of dementia in AD mice. From approximately 9 months of age, AβPP/PS1 mice exh-ibited the formation of senile plaques that increased with age, in a manner similar to human Aβ stain material [46, 47]. Therefore, we used 6-month-old male AβPP/PS1 mice to detect whether intervention via periodontitis induced early AD-like pathology in AD mice. The results indicated that following 8 weeks of P.g-LPS injection combined with ligation, spatial and non-spatial learning as well as memory of mice decreased, indicating that the degree of dementia was more severe in mice with periodontitis. Although a previous study [35] showed that intraperitoneal injection of P.g-LPS might aggravate cognitive dysfunction in mice, the results of our study indicated that periodontal intervention via P.g-LPS injection alone did not cause any apparent cognitive impairment. Other studies have reported that insufficient or incomplete absorption of P.g-LPS, local anatomical structure and physiology of gingiva, or the biological effects of repeated injections of low-dose P.g-LPS over a long period leading to tolerance, may prevent significant impairment [48, 49].
Neuron loss, a pathological feature of AD, is mar-kedly associated with declining learning capacity and reduced memory. Owing to neuronal loss compounded by other factors, the brain undergoes typical changes, such as thinning of the cortex and widening of gyri, during the late stages of AD. The primary fun-ction of Nissl bodies, which are characteristic functional units in neuronal cell bodies and dendrites, is protein synthesis [50]. Upon toluidine blue staining, a Nissl body is observed as a blue granular mass. Upon neuronal damage Nissl bodies undergo morphological changes and may dissolve leading to a decrease in their number [51]. Therefore, changes in Nissl body morphology and quantity may indirectly reflect nerve cell damage. Thus, we studied the effect of periodontitis on the number of neurons by staining Nissl bodies. In animals, neurons in the cerebral cortex and the CA1 and DG areas of the hippocampus are remarkably associated with learning and memory [52]. Therefore, in this study, changes in the specific brain areas of AD mice were evaluated to assess injuries caused by periodontitis. The results showed that the number of viable neurons in the cerebral cortex as well as in the CA1 and DG reg-ions in the hippocampi of mice in P.g-LPS and P.g-LPS + Ligation groups were significantly reduced due to atrophy or dissolution of Nissl bodies, indicating that periodontitis-induced damage and loss of neurons had occurred in the brains of AD mice.
Aβ oligomers, including Aβ40 and Aβ42, have been shown to exert neurotoxic effects [53, 54]. Aβ, which is generated during AβPP metabolism, is cleared via enzymatic degradation pathways [55, 56]. Aβ deposition caused by the processing of AβPP by β- and γ-secretases, results in the accumulation of Aβ42 and Aβ40 in the hippocampus and cortical areas, thereby leading to neurotoxic effects [57–59]. Our results showed that the Aβ content in the hippocampi and cerebral cortices of the periodontitis model group was significantly increased compared with that in the control, suggesting that induction of periodontitis had effectively upregulated Aβ expression levels, via activation of the Aβ generation pathway.
AβPP is metabolized via non-amyloid and amyloid pathways under the regulation of α-, β-, and γ-secretase, wherein the amyloid pathway (comprising β- and γ-secretase) produces full-length Aβ [60]. Of note, α-secretases are members of the ADAM family of proteins, among which ADAM9, ADAM10, and ADAM17 demonstrate α-secretase activity [61]. Of these, reduced protein expression of ADAM10 is markedly associated with AD, thereby indicating that it plays an important role in the pathological process of AD [62]. β-secretase is the rate-limiting enzyme that initiates Aβ production. Of the β-secretase enzymes, BACE1, which is the key rate-limiting factor in Aβ production, plays a vital role in AD pathogenesis, according to the Aβ theory [63]. Thus, selective activation of α-secretase or inhibition of β- and γ-secretase reduces Aβ production. In our study, lig-ation-induced periodontitis increased the expression of AβPP and BACE1. Furthermore, it reduced the expression of ADAM10 in the hippocampi and cerebral cortices of AβPP/PS1 mice, suggesting that deposition of Aβ induced by periodontitis occurred via promotion of the AβPP amyloid pathway and inhibition of the non-amyloid pathway. However, the results of P.g-LPS injection alone showed no significant difference, compared to the control. Therefore, Aβ deposition caused by P.g-LPS may not be attributed to enhancement of the amyloid pathway but may be attributed to an insufficient clearance of Aβ [64].
Studies have shown that Aβ deposition is related to excessive activation of microglia and astrocytes [65, 66]. Neuropathological studies of transgenic models and subjects with AD have indicated that amyloid plaques are surrounded by multiple inflammatory proteins, such as complement factors, acute-phase proteins, and proinflammatory cytokines, along with clusters of activated microglia [67]. Similar to microglia, reactive astrocytes also tend to aggregate around fibroid amyloid plaques. When exposed to Aβ, astrocytes release cytokines and other potentially cytotoxic molecules that induce neuroinflammatory responses [68]. We examined the activation of microglia and astrocytes. A significant increase in the expression levels of Iba1 and GFAP suggested that periodontal inflammation activated immune cells in the brain, which exerted multiple toxic effects that aggravated AD. Several studies have shown that LPS induces neuroinflammation, as indicated by significant increases levels of proinflammatory mediators, such as COX-2, iNOS, IL-6, and TNF-α, in vitro in neuroglial cells in vivo in animal models [69]. In the current study, the co-expression of inflammatory proteins was higher in LPS-treated (P.g-LPS and P.g-LPS + Ligation groups) mouse brains, suggesting that microglia and astrocytes had been activated with a consequent increase in iNOS, COX-2, TNF-α, and IL-6 expression. These findings were consistent with increased amyloidogenesis.
The current study revealed that in AD mice, act-ivated glial cells and upregulated expression of infl-ammatory mediators in the brain, as well as activated neuroinflammation, were positively correlated with Aβ40 and Aβ42 deposition. As glial cells serve as the brain’s resident immune cells, the vicious circle between dysfunctional glial cells and Aβ is a crucial pathological event that accelerates the progression of AD. Excessive accumulation and aggregation of Aβ result in neurotoxicity, which eventually leads to neuronal degeneration and neuronal death [14]. Our research demonstrated that induction of Aβ deposition and neuroinflammation by P.g-LPS played an important role in the virulent effect exerted by periodontitis on the development of AD. Furthermore, combining ligation with P.g-LPS not only stimulated the clinical pathogenic effect of periodontitis more effectively but also demonstrated that the inflammatory lesions caused by synergistic colonization of periodontal pathogens were worse than those caused by LPS injection alone. This suggested that, in addition to LPS, P.g and other periodontal pathogens may function as virulence factors or resort to more complex mechanisms that promote AD. This is further substantiated by the results of a recent study conducted by Dominy et al. in which the effect of P.g gingiva on AD was investigated [45].
The current study was beset with certain limitations. Direct evidence indicating that periodontitis aggravates Aβ by affecting AβPP, as well as α-, β-, and γ-secretase may be necessary. Additional analyses of the effects of periodontitis on the amyloid and non-amyloid pathways should be conducted. Moreover, the activation of glial cells and the release of inflammatory cytokines in periodontitis, as observed in this study, may have been induced via various mechanisms. Therefore, the precise mechanism(s) underlying glial cell activation and inflammatory cytokine enhancement warrants further elucidation. Many in vitro and in vivo studies have indicated that LPS-induced inflammation is mediated by Toll-like receptor-4, which activates signaling pathways that upregulate expression of various proinflammatory cytokines and chemokines [70, 71]. Therefore, it may be useful to investigate the role played by the Toll-like receptor-4 pathway in the association between periodontitis and cognitive dysfunction in mice. Furthermore, mounting evidence indicates that gastrointestinal microorganisms may be related to AD [72, 73]. Dysbiosis of biofilm flora in the oral cavity, which serves as the portal to the digestive tract, exerts adverse effects on intestinal flora. Hence, the composition of intestinal flora as well as the intestinal environment (intestinal barrier function) and intestinal immunity are regulated by oral bacteria, either directly or indirectly [74]. Finally, all animal models are limited by the fact that no single model can represent all aspects of human periodontal disease or AD. Therefore, clarifying whether a selected model is suitable for the study of a specific hypothesis is vital [75]. Although the current study did not prove that periodontitis was a risk factor for AD, our experimental results suggested that periodontitis might exert an effect on the development of AD.
In conclusion, our experiment revealed that per-iodontitis exacerbated learning and memory impairment of AβPP/PS1 transgenic mice, and induced Aβ generation, activated glial cells, and promoted proinflammatory cytokine release.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (Grant No. 81860197, No. 81260168), Nanjing Clinical Research Center for Oral Diseases (No. 2019060009), and Jiangsu Provincial Medical Innovation Team (No. CXTDB2017014). The authors thank the Key Laboratory of Basic Pharmacology of Ministry of Education in Zunyi Medical University for providing the experimental platform.