
Abstract
Alzheimer’s disease (AD) is associated with marked atrophy of the cerebral cortex and accumulation of amyloid plaques and neurofibrillary tangles. Amyloid plaques are formed by oligomers of amyloid-β (Aβ) in the brain, with a length of 42 and 40 amino acids. α-secretase cleaves amyloid-β protein precursor (AβPP) producing the membrane-bound fragment CTFα and the soluble fragment sAβPPα with neuroprotective activity; β-secretase produces membrane-bound fragment CTFβ and a soluble fragment sAβPPβ. After α-secretase cleavage of AβPP, γ-secretase cleaves CTFα to produce the cytoplasmic fragment AICD and P3 in the non-amyloidogenic pathway. CTFβ is cleaved by γ-secretase producing AICD as well as Aβ in amyloidogenic pathways. In the last years, the study of natural products and synthetic compounds, such as α-secretase activity enhancers, β-secretase inhibitors (BACE-1), and γ-secretase activity modulators, have been the focus of pharmaceuticals and researchers. Drugs were improved regarding solubility, blood-brain barrier penetration, selectivity, and potency decreasing Aβ42. In this regard, BACE-1 inhibitors, such as Atabecestat, NB-360, Umibecestat, PF-06751979 Verubecestat, LY2886721, Lanabecestat, LY2811376 and Elenbecestat, were submitted to phase I-III clinical trials. However, inhibition of Aβ production did not recover cognitive functions or reverse disease progress. Novel strategies are being developed, aiming at a partial reduction of Aβ production, such as the development of γ-secretase modulators or α-secretase activity enhancers. Such therapeutic tools shall focus on slowing down or minimizing the progression of neuronal damage. Here, we summarize structures and activities of the latest compounds designed for AD treatment, with remarkable in vitro, in vivo, and clinical phase activities.
INTRODUCTION
Alzheimer’s disease (AD) develops with progressive memory loss and impairment of cognition, and is an important problem for public health in the world [1]. Nowadays, AD is the most common form of age-related neurodegenerative disease, and the number of persons with dementia problems is increasing, affecting families, communities, and healthcare systems. Frequently, in most cases, AD starts after age 65 years, constituting late-onset AD, but rare cases occur before the age of 65 years, termed early-onset AD, which are less than 5% of all cases. AD patients are divided into two groups: sporadic AD and familial AD [2]. Familial cases account for approximately 1% of all AD cases [3].
AD pathology is characterized by the presence of two hallmark protein aggregations: amyloid-β (Aβ) in plaques and phosphorylated tau in neurofibrillary tangles [4]. AD is associated with marked atrophy of the cerebral cortex accompanying the loss of cortical and subcortical neurons, due to cerebral deposition of Aβ peptides, especially Aβ42, forming amyloid plaques in the extracellular space of the brain. This is considered a hallmark of AD and the putative cause of AD-related neurotoxicity. Neuronal injury is not uniform, being most severe in the neocortex and hippocampus, affecting different functional regions of the brain [5], such as learning and memory process and subsequent deficits in attention, motor function, language, gnosis, and visuospatial function as well as social behavior [6].
There are many hypotheses regarding the causes of AD. Here, we focus on the amyloid hypothesis. The last results of research and clinical trials of a series of inhibitors or enhancers of secretases involved in amyloid peptide production will be presented and discussed, pointing out novel lines for drug discovery in AD therapy.
THE AMYLOID CASCADE HYPOTHESIS
In 1991, John Hardy and David Allsop sugge-sted that Aβ deposition is produced by a pathogenic mutation in the amyloid precursor protein gene on chromosome 21. This mutation results in pathol-ogical cascades, such as Aβ deposition, tau phosphorylation, neurofibrillary tangles, and ultimately neuronal death [7]. The biological function of amyloid-β protein precursor (AβPP) is yet to be fully elucidated. Primary events in AD may appear 10–20 years before the onset of dementia symptoms involving abnormal accumulation of amyloid peptides in the brain [8]. Aβ peptides are present in the central nervous system at concentrations of 10–20 ng/ml, and at much lower levels in the plasma [9]. Elderly individuals without any clinical abnormalities evidence abnormal Aβ accumulation at postmortem examination. This is associated with an elevated risk of future clinical impairment and cognitive decline [10]. Aβ peptides are derived from multiple proteolytic cleavages of AβPP. AβPP is a transmembrane protein expressed in the brain with three isoforms of interest to AD, denonoted APP695, APP751, and APP770, containing 695, 751, and 770 amino acids (aa), respectively [11]. AβPP is cleaved by α-, β-, and γ-secretases, which are the focus for drug development and will be described in this review. Further secretases are also involved in AβPP processing. The δ-secretase cleaves the APP-695 ectodomain at both N373 and N585 aa. Cleavage at N585 enhances subsequent beta-site amyloid precursor protein–cleaving enzyme (BACE)-1 processing, resulting in increased Aβ levels [12]. ϑ-secretase, the homologue of BACE1, cleaves APP-695 at the 615 aa site to yield a C-terminal fragment (CTF) with 80 amino acids (CTFθ) contributing to the generation of truncated Aβ [13]. η-secretase cleaves APP-695 after aa 504. This activity yields a soluble fragment, sAPPη, as well as an extended membrane bound CTFη. Then CTFη may act as a substrate for conventional α- or β-secretase [14]. Meprin β-secretase (MEP) may cleave APP-695 at three sites in the N-terminal region after aa 124, 305, and 308 [15]. AβPP processing by MEP leads to aggregation-prone, truncated Aβ species [16] (Fig. 1A).
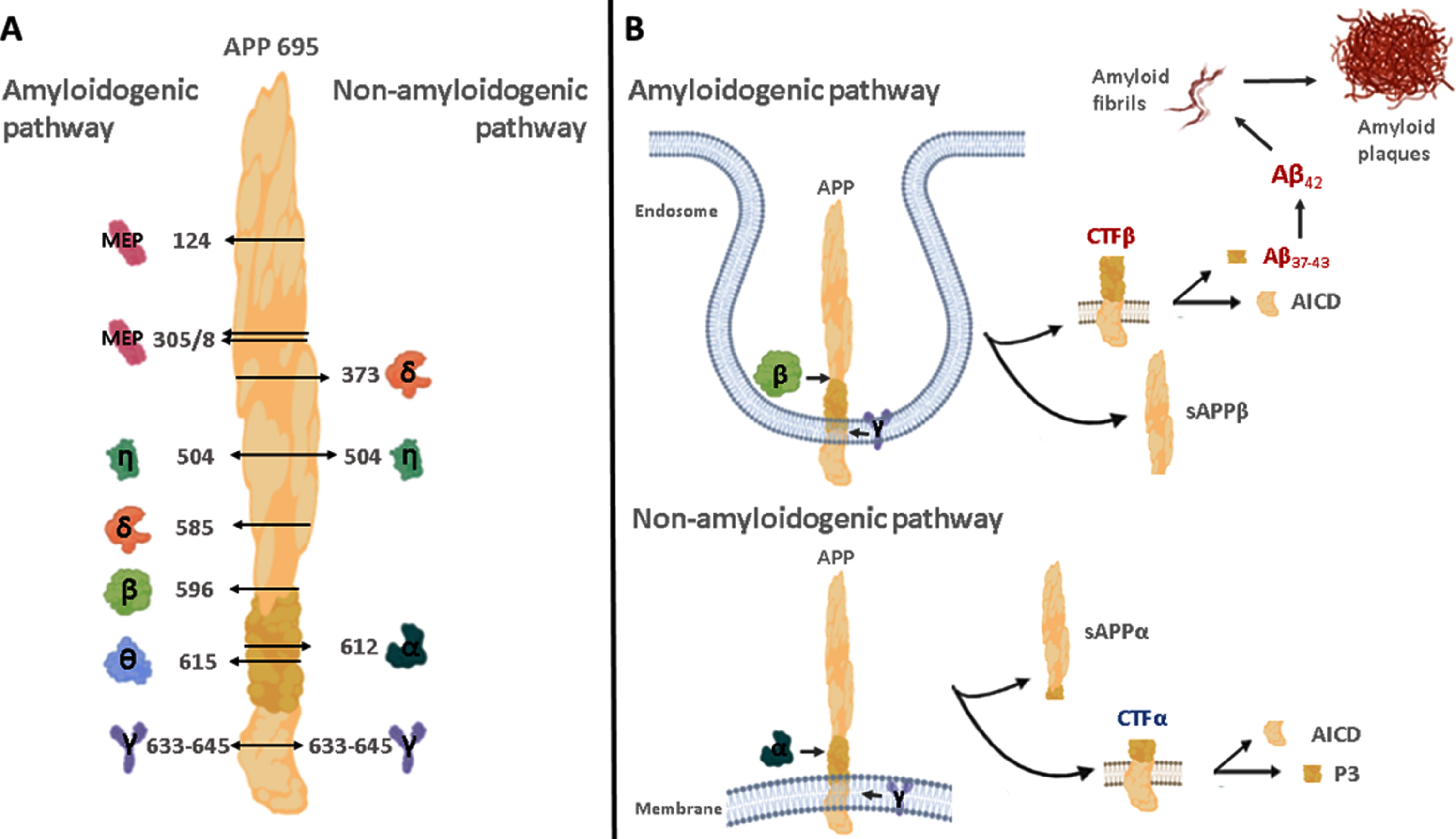
Proteolytic cleavage of the amyloid-β protein precursor (AβPP; not drawn in proportion). A) The non-amyloidogenic pathway is induced by α-, γ-, and minor secretases (δ and η) producing small fragments, such as P3 or AICD. In the amyloidogenic pathway, AβPP cleavage occurs by β-, γ-, and minor secretases (δ, η, θ, and MEP) associated with production of Aβ peptides. Numbers next to arrows indicate the cleavage sites of the AβPP amino acid sequence. B) After AβPP cleavage by α- and β-secretases, CTFα and CTFβ remain associated with the membrane and are further processed by the γ-secretase complex producing small fragments, such as P3, AICD, and Aβ peptides. Aβ42 is prone to aggregate into oligomers, forming the amyloid plaque, promoting cell death. AICD, AβPP intracellular domain; CTF, C-terminal fragments of the amyloid-β protein precursor (AβPP); MEP. Meprin β secretase. Shapes of enzymes are generic.
The α-secretase pathway hydrolyses AβPP within the Aβ sequence, which precludes Aβ formation and produces a large soluble NH2-terminal (sAβ-PPα) and a membrane-bound COOH-terminal fragments (CTFα, 10 kD). CTFα is then processed by γ-secretase originating smaller fragments, such as P3 and an intracellular cytoplasmic C-terminal domain (AICD) [17]. Different mechanisms have been described for the degradation of AICD from the non-amyloidogenic pathway, these include: the insu-lin-degrading enzyme, cathepsin B, and a proteasome-dependent mechanism. On the other hand, it has been shown that AICD generated from the amyloidogenic pathway affects transcriptional regulation, nuclear signaling, cell death, DNA repair, and cell cycle re-entry [18]. Recent work by Kuhn et al. 2020 demonstrated substantial amyloidogenic properties of the “non-amyloidogenic” P3 peptide. Their results revealed that P3 fibrils formed intermediate oligomers, which share a similar size distribution with Aβ, suggesting that P3 may not be innocuous [19].
Alternatively, in the β-secretase pathway, AβPP is first cleaved by BACE-1 at the NH2-terminus in the extracellular space to release sAβPPβ as a soluble 100 KDa NH2-terminal fragment and a 99 amino acid C-terminal fragment (CTFβ, 12 kD), which remains bound to the membrane. The C-terminal fragment is processed by γ-secretase in multiple consecutive steps, resulting in the release of Aβ peptides with different lengths, such as Aβ42 and other shorter Aβ fragments (e.g., Aβ40, Aβ38, and Aβ37), that are excreted into the cerebrospinal fluid (CSF). Aβ40 is the most abundant one in the brain, while Aβ42, is generally present in tissues and body fluids at levels 5–10% of those of Aβ40, but Aβ42 is suggested to be important in initiating Aβ aggregation, due to its higher hydrophobicity and capability of aggregation [20]. Amyloidogenic and non-amyloidogenic pathways are shown in Fig. 1B.
Moreover, patients in early AD stages showed higher baseline BACE-1 activity in the CSF compared to healthy control subjects. BACE-1 activity in the CSF has been proposed as a risk predictor in mild cognitive impairment [21]. Under physiological conditions, AβPP hydrolysis is mainly based on the α-secretase pathway, in which toxic Aβ peptides are not produced.
α-SECRETASE ENHANCERS AS THERAPEUTIC TARGETS
α-secretase is a metalloprotease cleaving AβPP between Lys-16 and Leu-17 in the middle of the Aβ domain [22], releasing a soluble N-terminal ectodomain (sAβPPα) and keeping the membrane-bound C-terminal fragment (CTFα). CTFα is further cleaved by the presenilin subunit of γ-secretase to yield a soluble N-terminal fragment (P3) and a cytosolic fragment AICD. sAβPPα has neuroprotective effects [23] and is enhancing synaptogenesis, neurite outgrowth, and neuron survival. It was shown, for instance, that sAβPPα disrupted AβPP dimers protecting neuroblastoma cells against starvation induced cell death [24] and PC12 cells against proteasomal stress [25]. sAβPPα enhances memory, has potential as a nootropic agent against age-related cognitive decline [26], and reverts behavioral, anatomical, and electrophysiological abnormalities of AβPP-deficient mice [27].
α-secretases belong to the α-disintegrin and metalloprotease family (ADAM). These proteases named ADAM9, ADAM10, and ADAM17 are involved in the cleavage of AβPP. ADAM9 has been reported to shed the heparin-binding EGF-like growth factor (HB-EGF) [28], and its expression upregulation is directly correlated with the development and progression of some cancer [29]. In the triple-negative breast cancer, ADAM9 overexpression was associated with lower survival expectation. In contrast, when ADAM9 expression had been knocked down, cancer proliferation, migration, and invasion was suppressed [30]. In brain endothelial cells, ADAM9 is regulated by expression of contactin-associated protein 1 (Caspr1) and depletion of this protein also reduce the levels of sAβPPα. Moreover, the activity of Caspr1 regulates specifically ADAM9 and not ADAM10 or ADAM17 expression [31]. Thus, proteolysis of AβPP by ADAM9 produces sAβPPα with neuroprotective effects, but its unregulated enhancement could increase the risk of cancer development. ADAM17, also known as tumor necrosis factor-converting enzyme (TACE), within the Golgi Complex is regulated by protein kinase C [32]. ADAM10 is the main α-secretase that cleaves AβPP, and its activity enhancement could be exploited for AD treatment, aiming at the decreased production of toxic peptides (Aβ42 and Aβ40) and increasing sAβPPα rates with beneficial properties [33]. However, the ubiquitous expression of this enzyme may become a problem for inhibition therapy, as ADAM17 processes substrates, which are essential for cellular functions, including Notch, PD-L1, EGFR/HER ligands, ICOS-L, TACI, MIC-A, MIC-B, and ULBPs. ADAM10 has important functions in the immune system [34] and is implicated in many pathologies, including glioblastoma, Hodgkin lymphoma, breast cancer, oral squamous cell carcinoma, rheumatoid arthritis, systemic lupus erythematosus, and psoriasis [35]. Broad substrate spectra have turned into a problem for the development of safe anti-AD drugs, which will be a major highlight in this topic.
Some drugs known for their activity against acetylcholinesterase have been studied for other targets in AD. Tacrine is a reversible inhibitor of acetylcholinesterase known as one of the main FDA approved drugs for AD. The use of tacrine was limited after its inception in therapeutic application due to hepatotoxicity generated in patients [36]. This molecule was considered a therapeutic treatment for the inhibition of the amyloid plaque formation in AD [37]. Tacrine may reduce the levels of neurotoxic Aβ42 as well as neuroprotective sAβPPα [38]. Anti-cholinesterase drugs like tacrine affect nicotinic acetylcholine receptors [39] as well as other neurotransmitter receptors, such as the NMDA-glutamate receptor [40]. Multi-factory actions with diverse downstream signaling pathways, possibly affecting gene expression patterns, may contribute to observed changes of AβPP expression and secretion, as well as to neuroprotective effects observed in vivo [41].
Due to its side effects, actually, various tacrine hybrids have been synthesized, resulting in multitarget drugs for the treatment of AD [42]. Rivastigmine is used to treat mild to moderate AD by elevating synaptic acetylcholine levels. This molecule may upregulate gene expression levels of ADAM9, ADAM10, and ADAM17 α-secretases, directing AβPP processing into the non-amyloidogenic pathway, as shown in a mouse model [43].
Cryptotanshinone is an active tetracyclic diterpene, produced by the medicinal herb Salvia miltiorrhiza. Cryptotanshinone reduces intracellular and secreted levels of Aβ40 and Aβ42, increasing the production of sAβPPα and CTF-α by upregulation of ADAM10 activity [44]. This activity is induced by the stimulation of phosphatidylinositol 3-kinase (PI3K) pathways [45]. Salvia miltiorrhiza produces a series of diterpenoids used for nervous and cardiovascular disease treatment. These also act on benzodiazepine and kappa opioid receptors with neuroprotective properties [46].
Phlogacantholide C is a natural tetracyclic diterpenoid isolated from Phlogacanthus curviflorus [47]. This compound came from the study of 69 substances from a drug library derived from traditional Chinese medicine looking for AD treatment, phlogacantholide C is the best of them, acting as ADAM10 gene expression enhancer [48].
Acitretin is a second generation monoaromatic retinoid. This analog of vitamin A is used in the treatment of psoriasis since 1997. Acitretin induced promoter activity of ADAM10 with an EC50 of 1.5μM, displaying anti-amyloidogenic actions in AD mouse models [49]. In human patients, acitretin exerts an immune stimulatory effect, which may counteract learning and memory disabilities by stimula-ting α-secretase [50]. In dermatology, acitretin therapy may have some adverse effects, such as hypervitaminosis A as well as eye, nose, and lip membrane dry out. Alopecia, desquamation of the skin, and hypertriglyceridemia occurs in 35% of patients treated with 50 mg/day acitretin, while Cheilitis is observed in almost every patients [51].
Disulfiram is a synthetic drug, clinically used for the treatment of alcohol dependence [52]. In a screening of 640 FDA-approved drugs looking for ADAM10 promoter activity enhancers, disulfiram provided best results at a concentration of 2.2μM, while concentrations higher than 5μM were toxic [53]. Side effects were observed for this drug. For instance, disulfiram (50 mg/kg/day/15days) increased acetylcholine concentration in the hippocampus of rats [54], which would be beneficial for AD treatment.
Ligustilide is a natural product of the Umbelliferae family, such as Radix angelicae sinensis and Ligusticum chuanxiong. Its lipophilic profile enables the compound of crossing the blood-brain barrier [55]. Recently, ligustilide has shown to ameliorate memory and neuroprotective properties by decreasing Aβ levels with a subsequent increase in the levels of sAβPPα by inhibition of IGF-1/Akt/mTOR signaling in AD mice and cultured cells [56, 57]. Furthermore, ligustilide might induce Aβ autophagic clearance [58].
Berberine is a benzylisoquinoline alkaloid of the protoberberine group, present in many plants as a quaternary ammonium salt of yellow color. Berberine is the main alkaloid from Berberis vulgaris exhibiting important neuroprotective effects [59], decreasing Aβ levels in the hippocampus, enhancing learning and decreasing memory deficits of transgenic mice by increasing levels of sAβPPα, ADAM10, and ADAM17 [60–62]. Figure 2 shows the structures of rivastigmine and further potential therapeutic enhancers of ADAM10 activity.
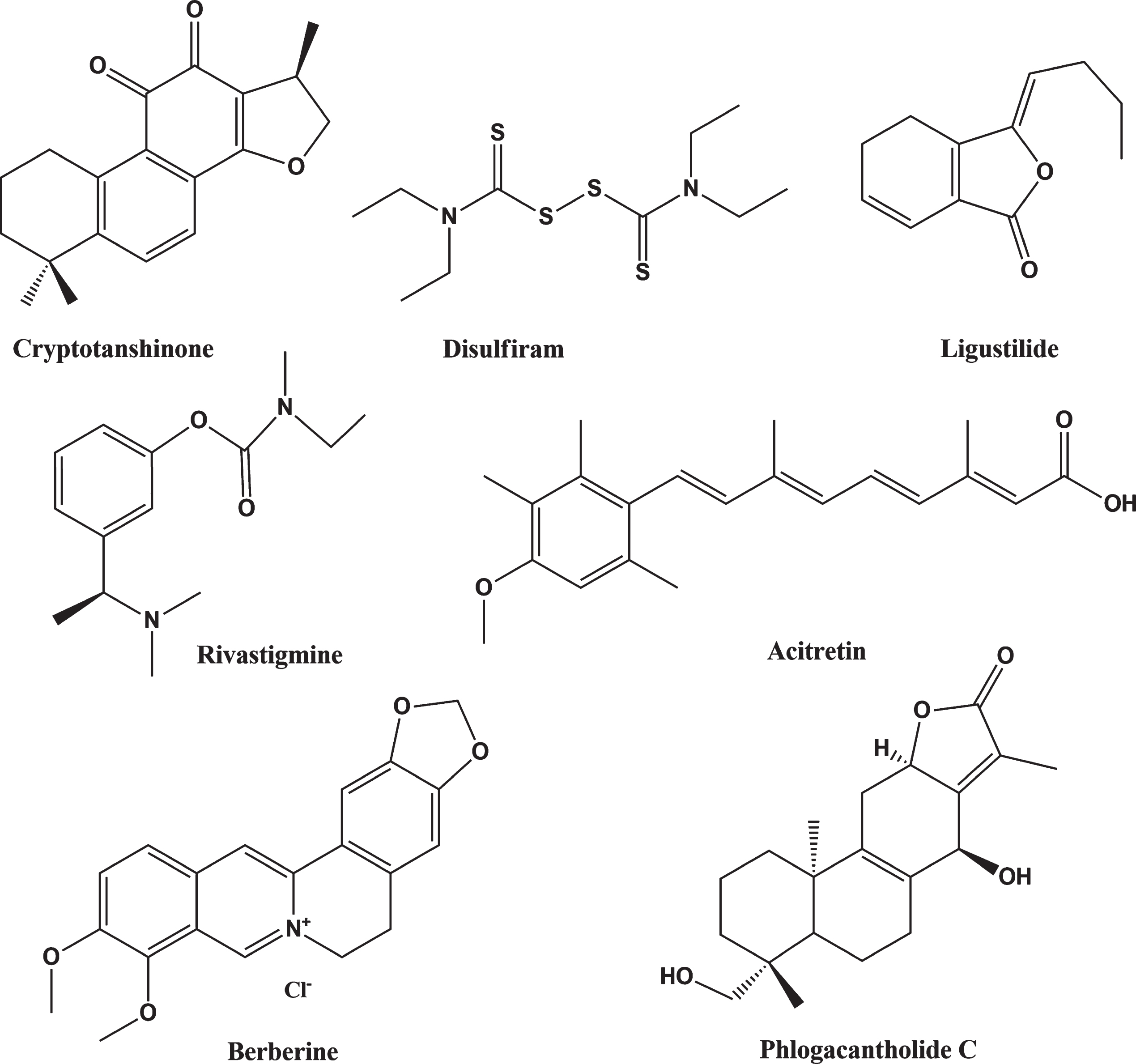
Selected ADAM10 activity enhancers with potential for AD treatment.
TARGET, β-SECRETASE INHIBITION
β-secretase and γ-secretase are the main enzymes responsible for Aβ40 and Aβ42 production in the brain. Therefore, drug development for AD treatment has focused on these targets. The development of potent and selective BACE inhibitors is a challenging task in academia and industry for avoiding aggregation of Aβ peptides [63]. Various molecules, including phenserine, have been tested for their capability of reducing Aβ concentrations. This molecule decreased secretion of sAβPPβ and Aβ into human neuroblastoma cell conditioned media by posttranscriptional regulation without cellular toxicity [64]. Furthermore, when the posiphen drug derivate of phenserine was administrated to mice, a reduction in the levels of peptides Aβ40 and Aβ42 was observed [65]. Liu et al. (2010) reported that diazoxide treatment in mice decreases the amount of full-length AβPPβ, suggesting that diazoxide inhibited amyloidogenic processing of AβPPβ by β- and γ-secretases resulting in a reduced amount of Aβ production and improving neuronal bioenergetics and increased cerebral blood flow [66]. Table 1 shows the latest developed inhibitors and their current status.
Overview and current status of BACE inhibitors
The pharmaceutical companies Janssen, Novartis, Amgen Inc., Merck, Eli Lilly, AstraZeneca, Pfizer, Biogen, and Eisai have remarkingly shared lead drugs, synthesis pathways, and preclinical and clinical results in journals and conferences. Unfortunately, most of BACE-1 inhibitors were omitted from trials, since patients developed toxic side effects to drugs or cognition impairments. However, the obtained knowledge has pointed out new strategies for AD treatment. Figure 3 shows the structures of some β-secretase blockers.
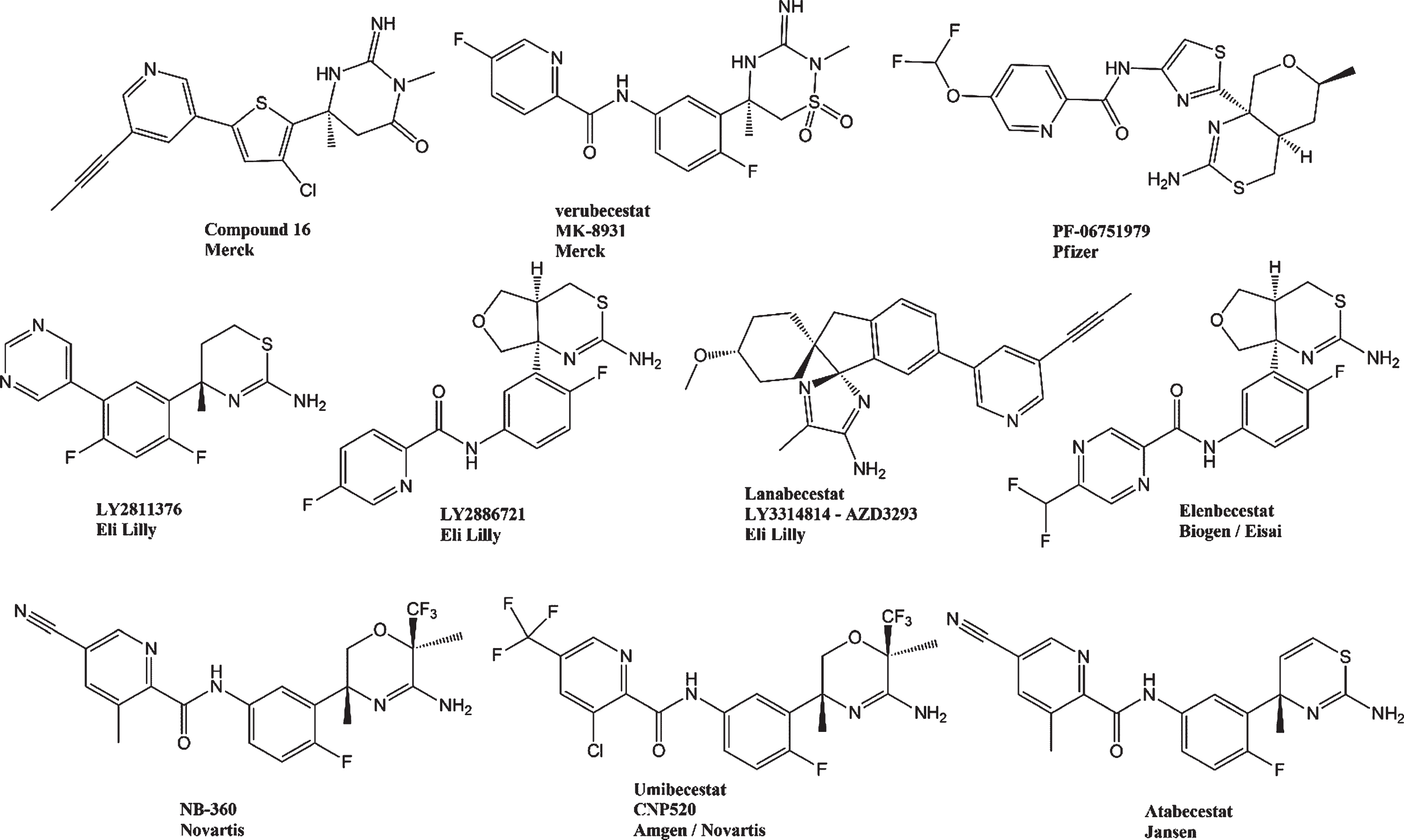
Structures of selected β-secretase inhibitors.
Eli Lilly Pharmaceutics was probably the first to design and develop a small molecule oral administered inhibitor of BACE activity in humans. The compound, named LY2811376, is an aminothiazine derivative which produced robust pharmacodynamics responses in plasma and CSF of human subjects. LY2811376 was rapidly discontinued, because chronic toxicology studies in rat showed side effects in retina and brain [67]. Based on observed mechanisms, efficacy, and side effects, the company developed a novel inhibitor denominated LY2886721, changing the phenylpyrimidine moiety to N-phenylnicotinamide. In addition, this structural feature was used in other BACE inhibitors such as verubecestat, PF-06751979, elenbecestat, NB-360, and umibecestat as well as by atabecestat, which will be discussed herein.
LY2886721 is a high-selectivity and affinity-inhibitor of key off-target proteases BACE-1 and BACE-2 with inhibition in terms of IC50 of 20.3 nM and 10.2 nM, respectively, without inhibition of further proteases, such as cathepsin D, pepsin and renin. In mice, 3–30 mg/kg doses lowered the presence of brain Aβ by 20–65%. This effect lasted up to nine hours after drug application. Reduction of amyloid formation was observed in plasma and lumbar CSF following administration of LY2886721. A single dose of 35 mg LY2886721 decreased Aβ40 and Aβ42 concentrations in the CSF with median lasting periods of 17 h [68]. The next generation of BACE-1 inhibitor with yet improved actions was lanabecestat.
Lanabecestat (LY3314814 or AZD3293) is an inhibitor of BACE-1/β-secretase. This molecule reduced Aβ40 and Aβ42 levels in the brain, CSF, and plasma of mouse, guinea pig, and dog animal models. In humans, lanabecestat reduced Aβ peptides concentration in CSF and plasma even when applied once a week.
Lanabecestat showed in the plasma a ≥64% amyloid reduction at 15 mg and ≥78% at ≥50 mg concentration, while in CSF, a decrease in amyloid production of ≥51% at 15 mg and ≥76% at ≥50 mg concentration was noted [69]. Lanabecestat at a concentration of 10μM selectively inhibited BACE-1, as revealed by in vitro radioligand binding and enzyme activity assays involving 350 targets of receptors, ion channels, transporters, kinases, and enzymes [70, 71]. Two clinical trials were sponsored by Eli Lilly & Co. and AstraZeneca. Patients were randomized and placebo-controlled in AMARANTH phase II/III (NCT02245737, 104 weeks, 539 patients completed the study) and DAYBREAK-ALZ phase III studies (NCT02783573, 78 weeks, 76 patients completed the study). As results of these clinical trials, lanabecestat treatment was well tolerated; however, no slow down of the cognitive or functional decline were noted. Furthermore, a high percentage of patients revealed psychiatric complications, weight loss, and hair color changes [72].
In 2012, Merck introduced another BACE inhibitor, called compound 16, which reduced levels of Aβ in the cortex and CSF of rats following oral administration, revealing an IC50 of 11 nM for Aβ40 accumulation [73]. From this compound Merck developed a series of related molecules, including the company’s most potent analogue verubecestat.
Verubecestat (MK-8931) is a potent and selective BACE-1 inhibitor (US 20070287692 A1 US Patent) with high permeability for the brain. Preclinical data revealed that oral-administered verubecestat is capable of crossing the blood-brain barrier and is stable in the rat brain for up to 12 h [74, 75]. This compound proved to be safe following acute and chronic administration into rats and monkeys at concentrations more than 40-fold higher than those evaluated in AD patient clinical trials. This compound at high concentrations did not elicit many of the side effects attributed to inhibition of BACE, such as interference with nerve myelination and glucose homeostasis, promotion of neurodegeneration or hepatotoxicity [76]. Pharmacokinetics and pharmacodynamics, evaluated in 24 healthy Japanese adults in a randomized and placebo-controlled phase I trial, showed safety and promising results of verubecestat for furthers trials [77]. Verubecestat has been evaluated in different clinical trials, sponsored by MERCK as: Phase I: NCT01496170 (N = 32), phase I: NCT01537757 (N12), phase II/III: NCT01739348 (N = 2221), phase III: NCT01953601 (N = 1500). Verubecestat reduced by up to 90% plasma, CSF, and brain concentrations of Aβ40, Aβ42, and sAβPPβ, as shown in a phase III clinical trial [75]. Chronic treatment with Verubecestat (12 and 40 mg/kg) dose-dependently diminished CSF Aβ levels by 40% and 80%, respectively, in AD patients [78] as well as in the Tg2576 transgenic AD mouse model [79]. Despite its potent inhibition of Aβ40, Aβ42 and sAβPPβ formation and its good tolerance, verubecestat was removed from clinical trials in February 2018. In the last trials conducted by Merck, participants treated for 13 weeks with 40 mg verubecestat scored worse when compared to the placebo group. The 12 mg treatment group performed poorly relative to the placebo group, revealing significant differences at scattered time points. Both treatment groups performed worse when compared to placebo group in a functional measure. Treated patients revealed increases in anxiety, depression, and sleep problems when compared to untreated control patients [80]. In view of that, Merck terminated clinical trials with verubecestat as potential AD drug [81].
Pharmaceutical Pfizer developed the compound PF-06751979 as selective BACE-1 blocker (IC50 of 7.3 nM for BACE-1 inhibition in contrast to an IC50 of 194 nM for BACE-2 inhibition). The inhibitor does not block related aspartyl proteases, as shown for cathepsin D (CatD) [82]. Safety, tolerability, pharmacokinetics, and pharmacodynamic properties were studied in humans in two phase I studies (NCT02509117, NCT02793232). Daily single-increasing doses up to 540 mg PF-06751979 in healthy adults and 50 mg or 125 mg multiple doses in healthy elderly subjects were well tolerated with mild-to-moderate side effects. PF-06751979 dose-dependently reduced CSF and Aβ peptide plasma concentrations. Patients treated with 275 mg QD reduced by 92% and 93% Aβ40 and Aβ42 concentrations, respectively, in the CSF after 14 days of treatment. A drug interaction study (NCT03126721) with midazolam did not detect any differences in clinical effect of multiple 100 mg PF-06751979 and midazolam doses in healthy adults. Clinical studies (NCT02509117, NCT02793232, and NCT03126721) suggested that PF-06751979 may be adequate for further development in clinics, although there is a risk of liver toxicity induction by the compound. Efficacy and side effects must be addressed in a larger study with longer application time and a higher number of patients [83].
Umibecestat (CNP520) was developed by Amgen, Inc. and Novartis Pharmaceuticals Corporation. Its development was a result of structural 3-amino-1,4-oxazine compound optimization. The compound is an oral-administered, small-molecule blocker of BACE-1 with high selectivity for this enzyme when compared to other aspartic proteases, including BACE-2 and CatD (IC50, BACE-1 : 11 nM; BACE-2 : 30 nM; CatD. 205,000 nM; CatE: 66,400 nM) [84]. CNP520 reduced Aβ concentrations in rat and dog brain and CSF, as well as Aβ plaque deposit in APP-transgenic mice [84]. CNP520 treatment was safe with no indication of retina degeneration, hair depigmentation, cardiovascular effects, or liver toxicity [84]. CNP520 was submitted to clinical phase II trials (NCT02576639, phase II/III: NCT02565511 and NCT03131453). In 2015, as part of the Alzheimer’s Prevention Initiative, a phase II/III study called GENERATION 1 was launched, involving 1,340 cognitively normal, homozygous APOE4 carriers at the age of 60 to 75. The randomized study was designed to compare a daily 50 mg CNP520 application with matching placebos and with a second group receiving injections of the investigational active immunotherapy CAD106 [85, 86]. The trial determined changes in the APICC cognitive composite [87]. In 2019, CNP520 passed the phase II clinical trial, but failed in phase III. Trials were discontinued, since CNP520 caused cognitive worsening in the treatment groups. Treated participants revealed more brain atrophy and more weight loss compared to the placebo group. These clinical data contrast previous studies, which did not associate the compound with adverse conditions or alterations in CSF AD biomarkers in healthy elderly volunteers treated for three months [88].
NB-360, developed by Novartis Pharmaceuticals Corporation, is a potent BACE-1 and BACE-2 inhibitor with an IC50 of 5.0 nM and 6.0 nM, respectively. This drug has been employed in rodent β-amyloidosis models for determining its therapeutical pharmacological efficacies in Aβ-related pathologies and BACE-1/2 blockade [89]. NB-360 efficiently halts the progression of Aβ accumulation in APP transgenic mouse brains and induces a major reduction of Aβ accumulation in rats and dogs. NB-360 revealed an IC50 of 3 nM and 33 nM in decreasing Aβ40 accumulation in wtAPP- and SweAPP-CHO cells, respectively. Further, the compound was blood-brain barrier-permeable [90]. NB-360 caused hypopigmentation phenotype in chronic mouse studies, as this compound also inhibits BACE-2. This enzyme is fundamental in producing proteolytic fragments of the pigment cell-specific melanocyte protein (PMEL17), which is essential for melanogenesis. NB-360 affected melanosome maturation and promoted hair depigmentation in a mouse model [91]. In view of that, studies with NB-360 were stopped prior to clinical trials [89].
Atabecestat (JNJ-54861911) developed by Pharmaceutical Janssen is an oral-administered BACE-1 inhibitor. In 2013, a series of phase I trials of atabecestat started with a single increasing dose application in 56 persons, followed by a second study in 70 volunteers in Belgium, and a similar study was conducted in Japan with 24 healthy volunteers. The results concluded that atabecestat is a promising drug candidate, which can reduce Aβ deposit following single or multiple doses in healthy elderly participants [21, 93].
Daily atabecestat doses of 10 to 50 mg applied for weeks reduced accumulation of Aβ40 by 67% and up to 90% in CSF of Caucasian and Japanese patients in early AD stages [93]. A multicentric, randomized, double-blind, and placebo-controlled phase IIb/III trial (NCT02569398) investigated efficiency and safety of atabecestat action in participants with elevated levels of Aβ, but not revealing cognitive impairments. The trial was discontinued in 2018 because of hepatic toxicity related adverse events [94]. Furthermore, preliminaries results stated adverse effects of atabecestat on cognition, depression, sleep, and anxiety [94]. Atabecestat in clinical phase II/III trials for people with preclinical stages of AD had to stop based on concerns of possible liver damage in some participants. In 24% of treated subjects, alanine amino transferase (ALT) levels were augmented above 1.5-fold of the upper limit of normal (ULN) and 10.9% had ALT level elevations even above 3-fold of ULN [95]. Similar results were obtained in the placebo-controlled double-blind parent ALZ2002 study, in which volunteers of age 50 to 85 years were randomized in three groups (1:1:1) treated with placebo, 5 mg, or 25 mg of atabacestat once a day for 6 months. While Aβ fragments and sAβPPβ were dose-proportionately reduced in whole brain of patients with mild cognitive impairment, elevated blood liver enzyme levels as adverse events reported in 12 participants treated with atabecestat resulted in dosage adjustment and increased monitoring frequency [96]. One case of atabecestat-mediated drug-induced liver injury showed necrosis and mononuclear infiltrate and parenchymal collapse in the centrilobular zone [95].
Elenbecestat from Biogen and Eisai is an aminothiazine derivative that in preclinical studies reduced Aβ protein levels in rat and guinea pig brain, CSF, and plasma [97], without evidence of hypopigmentation [98]. In phase I trials, the drug in a single dose of 200 mg did not have any impact on cardiac parameters in healthy Japanese and white subjects [99]. In a phase II, 18-month, placebo-controlled study elenbecestat (5, 15, or 50 mg/day) was well tolerated without liver damage. The size of the study was small, with only 43 subjects (61%) having completed the study. Neither the less elenbecestat may have attenuating effects on cognitive decline in mild cognitive impairment to moderate AD subjects [100]. Elenbecestat was studied for safety and efficacy in two large phase III trials, such as MISSION AD1 (NCT02956486) and MISSION AD2 (NCT03036280). Trials started in 2016 and compared a once-daily 50 mg elenbecestat dose to placebos in 2,100 patients with mild AD. In September 2019, Eisai and Biogen announced the discontinuation of phase III clinical trials with elenbecestat, because results indicated an unfavorable risk-benefit ratio, and, in turn, recommended termination of the trials [101].
γ-SECRETASE INHIBITORS AND MODULATORS
γ-secretase is an aspartyl protease protein complex; composed of four subunits: presenilin (PS), nicastrin (Nct), anterior pharynx-defective 1 (Aph-1), and presenilin enhancer 2 (Pen-2) in a 1:1:1:1 stoichiometry [102]. The catalytic subunit of γ-secretase is presenilin-1 (PS1) cleaving type I transmembrane proteins and having 149 reported substrates [103]. Compounds inhibiting γ-secretase (Fig. 4), targeting PS1, are potential therapeutic agents for AD [104]. γ-secretase inhibitors (GSIs) were linked to diverse side effects, such as hepatic, splenic, and cutaneous side reactions [105]. Inhibition of γ-secretase may interfere with cell-surface receptors and other proteins acting in embryonic development, hematopoiesis, cell adhesion, and further signaling events, i.e., Notch [106, 107]. Notch receptor–related nuclear signaling is crucial for developmental processes, synaptic plasticity, neural repair processes, proto-oncogene and tumor suppression [108]. Currently, GSIs were abandoned as potential AD therapies based on their toxicity and missing efficacies in clinical trials [109]. We briefly describe some GSIs, which had been under study in the last decade.
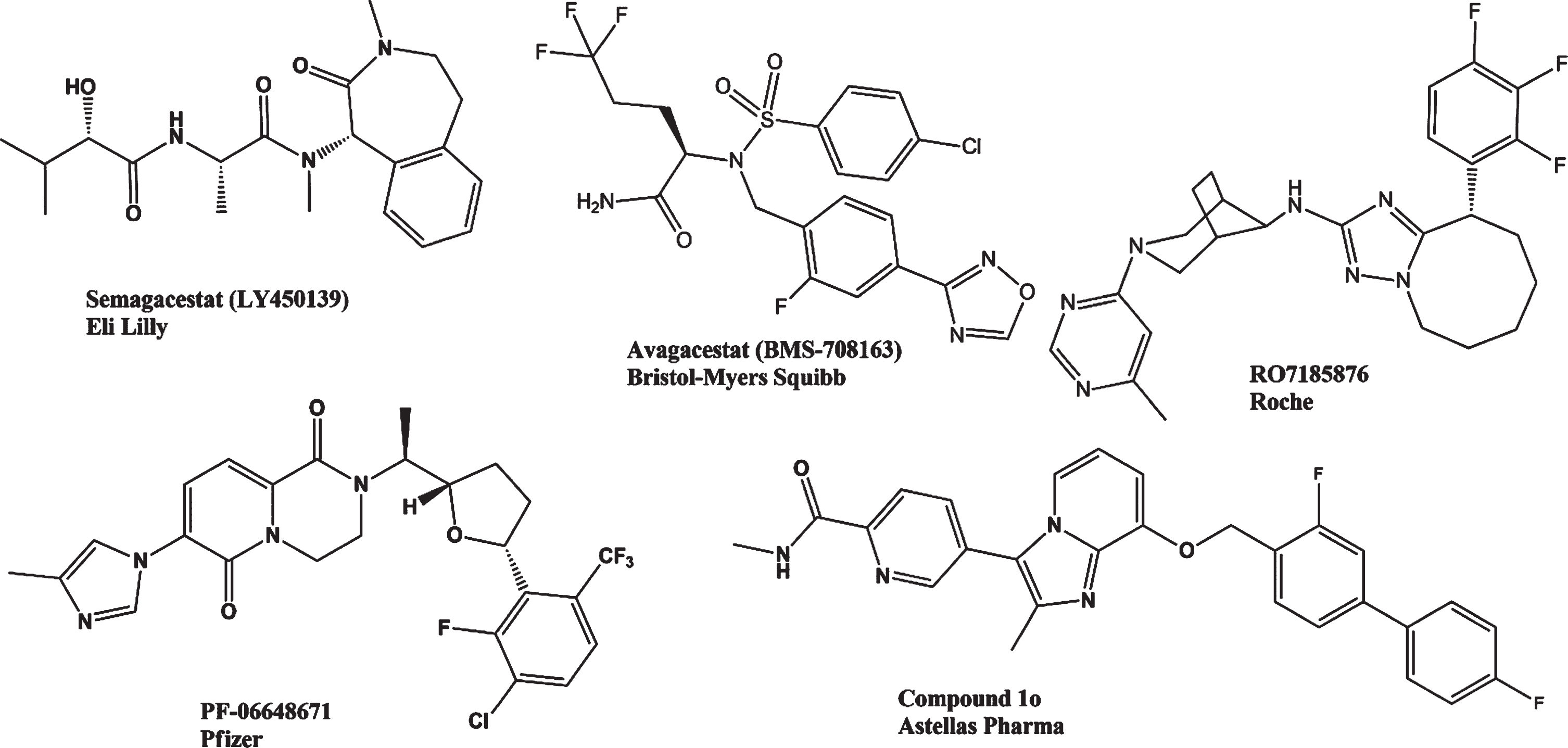
Structures of selected γ-secretase inhibitors and γ-secretase modulators.
Semagacestat (LY450139) developed by the Eli Lilly pharmaceutical company is a γ-secretase inhibitor [110] decreasing CNS Aβ production [110, 112]. However, in a phase III trial, semagacestat did not promote cognitive status improvement. Patients receiving increased doses showed a significantly worsened functional abilities. Further adverse effects were noted, including infections and skin cancers (NCT00594568) [105].
Avagacestat (BMS-708163) from Bristol-Myers Squibb is a potent and selective γ-secretase inhibitor of the arylsulfonamide family, demonstrating a 193-fold selectivity for this enzyme when compared to Notch blockade. It reduced Aβ40 production with an IC50 of 0.30 nM. BMS-708163 administration resulted in reduced Aβ40 plasma, CSF and brain levels, as studied in dogs and rats [112]. The tolerability profile of avagacestat, together with pharmacodynamic and pharmacokinetic properties of the drug, was studied by oral doses in healthy, young, male volunteers (NCT01454115). The results suggested that a single-dose range of 0.3 to 800 mg avagacestat could be suitable for further clinical development [113].
In phase II trials, avagacestat was studied in 209 outpatients with a median age of 75 years, diagnosed with mild-to-moderate AD. Patients were treated with 25, 50, 100, and 125 mg/day doses, and obtained results were compared to those of placebo treatment. Up to 50 mg/day the results were similar to those of placebos, while at higher avagacestat doses decreases in patients’ health were noted. At 100 mg and 125 mg doses, avagacestat was hardly tolerated with patients tending to cognitive capability worsening [114] (NCT00810147). In a further study conducted from May 2009 to July 2013 with CSF biomarker-negative volunteers, avagacestat treatment provided similar results. Health conditions of patients deteriorated, with the occurrence of diarrhea, nausea, vomiting, rash, itching skin, and nonmelanoma skin cancers. Avagacestat did not demonstrate desired efficacies, while promoting adverse dose-limiting effects [115].
Since GSIs showed high toxicity and side effects, a γ-secretase was selected as target for the development of activity modulators (GSMs). They are small molecules allosterically interfering with γ-secretase activity [116]. GSMs do not affect Notch and further protein substrate actions, including CD44, E-cadherin, neurexin, and ERB4. Potentially toxic AβPP C-terminal fragment (CTF) in the brain was also not detected, turning GSM into a promising tool for AD treatment [117].
RO7185876 is the first triazolo-azepines class GSM developed by Roche. This compound showed potent and selective activity in the inhibition of γ-secretase, by augmenting proportions of the smaller fraction peptides Aβ37 and Aβ38, while diminishing potential-pathogenic production of peptides Aβ40 and Aβ42, as demonstrated in vitro and in vivo. RO7185876 pharmacokinetic parameters were an IC50 of 15 nM in inhibiting Aβ42 production, 0.7μg/mL solubility and clearance in human hepatocytes of 3.1μL/min/M cells. This compound could be a potential target for decreasing the toxicity of Aβ peptides without affecting their total content and is currently in development for clinical applications [118].
PF-06648671, a GSM discovered by Pfizer (patent WO2014045156) is considered to be safe and well tolerated in healthy subjects after single oral doses up to 360 mg. Effects in reducing plasma Aβ40 and Aβ42 concentrations depended on the PF-06648671 dose [119]. Three phase I studies (NCT02316756, NCT02407353, and NCT02440100) were performed with 120 healthy subjects looking for the safety, tolerability, pharmacokinetics, and pharmacodynamics properties of PF-06648671/placebo for 14 days. The results did not indicate any serious adverse events. The drug decreased Aβ42 and Aβ40 concentrations and augmented Aβ37 and Aβ38 levels in the CSF, without changing the total Aβ concentration. However, further examination of the effects of PF-06648671 has been suggested [120].
Compound 1o (5-8-[([1,1′-Biphenyl]-4-yl) methoxy]-2-methylimidazo[1,2-a]pyridin-3-yl-N-ethylpyridine-2-carboxamide hydrogen chloride) is a potent GSM developed by Astellas Pharma Inc [121]. This compound is the result of the optimization process of a series of compounds, starting from 2-methyl-8-[(2-methylbenzyl)oxy]-3-(pyridin-4-yl)imidazo[1,2-a]pyridine (3a), inhibiting cellular production of Aβ42 (IC50 = 7.1μM) [118]. Then compound 1o was developed, reducing Aβ42 levels with an IC50 value of 0.091μM in vitro as well as mouse brain Aβ42 levels with significant efficacy [122]. Furthermore, in another work compound 1o showed excellent efficacy in the reduction of brain Aβ42 levels as well as reducing cognitive deficits in an AD mouse model [123].
CONCLUSIONS
Over the past decade, AD therapy has focused on the development of safe, potent, and specific inhibitors of Aβ. Pharmaceutical companies have developed several BACE-1 inhibitors, γ-secretase inhibitors, and α-secretase modulators, sharing their data in diverse research journals, congresses, and scientific meetings, in the hope of obtaining novel efficient drugs to face AD in the nearest future.
Many natural α-secretase enhancers show excellent profiles for AD therapy, such as cryptotanshinone, phlogacantholide c, ligustilide, or Berberine. Moreover, these compounds could be used as molecular scaffolds for the development of more potent compounds for enhancing α-secretase activity, which is a physiological pathway in the healthy organism for AβPP cleavage. More attention needs to be drawn to these molecules for advancing them clinical trials. However, the main focus of the pharmaceutical industry has been BACE-1 inhibition. Following initial reports of the BACE-1 sequence in 1999, inhibition of this enzyme seems to be key to resolve the amyloid plaque formation, and it could provide the best option to improve cognitive function in AD patients. In the last two decades, diverse generations of molecules were designed by in silico studies. As the results of chemical library screening, promising compounds raised with excellent inhibition to BACE-1 in the nM range. In the beginning, some of these compounds also inhibited BACE-2, then side effects as hypopigmentation were noted as in the case of NB-360, a 3-amino-1,4-oxazine compound of Novartis, which was quickly removed from clinical trials. Thereby, chemical modifications in the pyridine cycle of MB-360 gave the specific BACE-1 inhibitor umibecestat. Anyway, most of the compounds studied in diverse clinical trials showed toxicity or psychiatric adverse events, such as lanabecestat of Eli Lilly and AstraZeneca, or liver toxicity, such as atabecestat of Janssen, PF-06751979 of Pfizer, and LY2886721 of Eli Lilly/AstraZeneca. Toxicity was evidenced even after that, these compounds had been previously studied in animals (mice, dogs, monkeys) or in phase I trials. Although verubecestat of Merck and umibecestat of Amgen –Novartis seemed to be well tolerated by patients, both compounds were later discontinued from clinical trials due to cognitive worsening or negative outcomes.
The efforts used in the development of drugs, which are more specific against BACE-1, less toxic and more potent were successfully achieved with synthetic iminothiadiazinane dioxide derivatives, such as verubecestat, or by 3-amino-1,4 -oxazine scaffolds in the case of umibecestat. Unexpectedly these compounds did not meet proposed expectations. Underlying mechanisms have not been elucidated, why clinical trials with BACE-1 therapies showed Aβ level reductions by over 70% without any cognitive improvements. It seems that AβPP plays an important role in synaptic plasticity, and latest studies with primary cortical rat neuronal cultures suggest that only a partial BACE inhibition, by up to 50%, can be used without causing synaptic dysfunction [124], suggesting for future clinical trials a gradual reduction of Aβ concentration. Moreover, considering that Aβ levels in the brain are increased 10 or 20 years before the first symptoms of dementia appear, patients with AD signals already have chronic brain damage. In view of that, therapeutic intervention comes too late, when the disease has already progressed. Over 20 years, approximately 5 mg of Aβ peptide has accumulated in the AD brain with a rate of accumulation of 28 ng/hour [125], suggesting that therapies should start earlier in patients not showing any dementia signals. Drugs could be used in lower doses for preventing abnormal amyloid accumulation.
The γ-secretase inhibitors were the first compounds abandoned from clinical trials, because these molecules showed serious side effects and toxicity. This occurs since γ-secretase has many substrates and not only AβPP. Subsequently, γ-secretase inhibition revealed side effects in patients, as severe as nonmelanoma skin cancers. From this drawback, the new idea emerged of positive activity modulation of the enzyme in its catalytic site for AβPP, the presenilin subunit of γ-secretase. Then a new series of small molecules, GSMs, have been the focus for AD. The companies Roche, Pfizer, and Astellas Pharma Inc developed in the last years new selective compounds capable of decreasing levels of potentially pathogenic larger fractions of peptides Aβ40 and Aβ42 and increasing production of smaller fractions Aβ37 and Aβ38 in a selective way, not affecting the processing of Notch and other protein substrates relevant for development, cellular homeostasis, and signaling. These features make GSMs promising AD therapeutics. Now the effectivity of GSMs must be proven in further clinical trials. In spite of large drawbacks and challenges for the development and approval of an Aβ inhibitor drug for human treatment, the amyloid cascade hypothesis is still the best for future AD drug design and development.
Footnotes
ACKNOWLEDGMENTS
This work has been supported by a grant of the São Paulo Research Foundation (FAPESP project No. 2018/07366-4) awarded to H.U., and a FAPESP (Brazil)-Conicyt (Chile) grant awarded to H.U. and C.P (201808426-0). H.U. further acknowledges fellowship support by the National Council for Scientific and Technological Development (CNPq Project No. 306392/2017-8).