
Abstract
Obstructive sleep apnea (OSA) and Alzheimer’s disease (AD) are two common chronic diseases with a well-documented association. Whether the association is causal has been highlighted by recent evidence reporting a neurobiological link between these disorders. This narrative review discusses the brain regions and networks involved in OSA as potential vulnerable areas for the development of AD neuropathology with a particular focus on gender-related implications. Using a neuroimaging perspective supported by neuropathological investigations, we provide a new model of neurodegeneration common to OSA and AD, that we have called OSA-AD neurodegeneration in order to decode the causal links between these two chronic conditions.
Keywords
Consistent evidence links Alzheimer’s disease (AD) to obstructive sleep apnea (OSA), a sleep-rela-ted breathing disorder characterized by repeated episodes of upper airway obstruction during sleep, chronic intermittent hypoxia and sleep fragmentation [1]. In the context of emerging research, OSA can exacerbate the clinical spectrum of AD especia-lly when both disorders were severe. Additionally, AD patients may have a five times higher chance of presenting with OSA than cognitively non-impaired individuals of similar age [2].
The existence of a close relationship between these two disorders, however, brings up some fundamental questions: first, whether the relationship is causal, specifically does one disorder increase the incidence of the other or possibly the severity; and second, whether the presence of abnormal brain structures and other biomarkers in both disorders is enough to support the case for a causal relationship. It is critical to consider the perspectives from which we analyze these questions about the relationship of OSA brain pathology and AD brain pathology. From a pathogenetic perspective, it is necessary to investigate if areas of OSA neurodegeneration are the same as AD neuropathology. From a clinical perspective, it is important to investigate whether neurocognitive deficits due to OSA are similar to the neurocognitive impairment commonly occurring in AD. From a therapeutic perspective, it is crucial to know whether treatment of OSA will prevent or revert the brain alterations and clinical manifestations of AD.
In this narrative review, we used a neuroimaging approach to test the hypothesis of the existence of a common neurodegeneration between these two chronic disorders, so called OSA-AD neurodegeneration. Keeping this in mind, we review relevant neuroimaging literature to investigate whether brain regions and networks involved in OSA are the areas or targets vulnerable for the development of AD. We also highlight the neuroimaging features of AD pathology that may be neuroimaging biomarkers for detection of OSA-AD neurodegeneration.
CAUSAL RELATIONSHIP BETWEEN OSA AND AD
The hypothesis of a causal relationship between OSA and AD has strengthened over the last decades [3–5]. In keeping with the concepts of temporality, plausibility, and coherence criteria for a causal ass-ociation between OSA and AD, what are the neurobiological effects and outcomes that link OSA and AD? Second, what is the temporal sequence occurring between these two disorders? Third, are the brain areas involved in the impact of OSA similar to the vulnerable areas for AD pathology?
The neurobiological link between OSA and AD is probably mediated to an increase of amyloid-β (Aβ) deposition in the brain, hallmark of AD neu-ropathology. Indeed, accumulation of Aβ is commonly considered the final pathway of a pathogenetic process caused by an increase in the synthesis of Aβ and/or by a reduction in its clearance. In OSA, the main mechanisms underlying an increase of the amyloid synthesis are related to sleep fragmentation, sleep deprivation, slow wave sleep (SWS) disruption, and intermittent hypoxia [3]. Sleep fragmentation may be a critical pathophysiologic mechanism by which OSA contributes to AD. Indeed, increased sleep fragmentation has been associated with a higher risk for the subsequent development of AD in 6 years of follow-up period [6]. With respect to SWS disruption, the concentrations of extracellular soluble Aβ in the interstitial fluid space of the brain are related and regulated by sleep-wake cycle [7]. Finally, animal models support the contribution of intermittent hypoxia to AD pathophysiology: sustained exposure to hypoxia/ischemia increases the β-secretase activity resulting in a significant increase of amyloid production [8, 9]. Of importance is to assess the temporal relationship between OSA and AD. Does OSA occur before AD or does AD lead to OSA, so-called reversed causality. The existing literature is mainly focused on identifying the potential role of OSA in the preclinical stage of AD. This is not surprising, given that AD pathology can begin even 20 years before overt clinical symptoms and thus it is possible that both disorders may coexist. Supporting this, presence of sleep-disordered breathing disorders may be associated with an increased risk of developing mild cognitive impairment (MCI)/AD [10] especially among older woman, and an earlier age at onset [11]. Taken together, these findings suggest that OSA and AD are two closely related disorders; OSA can accelerate the Aβ deposition in the brain. Despite this, some questions remain, for instance about the areas/regions of the OSA brain that this Aβ deposition occurs.
NEUROIMAGING
In the last past decades, sophisticated and highly sensitive techniques, including PET-amyloid to det-ect Aβ deposition, have been applied to explore OSA human brain. Researchers are able to investigate structural/neuro-functional alterations occurring in OSA and explore the relationships between brain abnormalities and clinical manifestations. Neuroima-ging studies indicate that limbic and extra-limbic damage may occur in OSA. It is known that neurodegenerative changes within limbic and extra-limbic systems, including hippocampus, amygdala, cingul-ate gyrus, and insula, are the “core” of AD neuropat-hology. In order to address the hypothesis of OSA-AD neurodegeneration, we investigated which brain structures/networks within limbic and extra-limbic systems are involved in OSA. Since OSA vulnerabil-ity to neurodegeneration might vary across the different nuclei, subfields of these structures and patients, we considered the evidence within three sections: 1) lateralization and subfields; 2) gender-specific damage; 3) correlation with clinical symptoms.
NEUROIMAGING BIOMARKERS TO ASSESS THE LIMBIC DAMAGE
Hippocampus in OSA-AD neurodegeneration
There is agreement that the hippocampus is atro-phic in OSA. This is not surprising, since hippoca-mpus is a region of high vulnerability to chronic intermittent hypoxia in animal models [12, 13]. Owen and colleagues first performed histopathological investigations on human OSA brain tissue and re-ported decreased cortical thickness and demyelination in hippocampus related to the increasing severity of the disease [14]. Hippocampus is a key structure for cognitive processing since it plays a critical role in binding together item and contextual information together and processing the relationships between individual items [15].
Hippocampal roles are highly lateralized in human. The right hippocampus appears specifically involved in memory tasks requiring allocentric processing of spatial locations and the left is mainly involved in episodic/autobiographical memory [16]. Furthermore, it is not a homogeneous structure but rather it is composed of several subfields with different histological characteristics and functions: CA1 seems more sensitive than others to AD neuropathology and contains the amygdala-projecting neurons associated with emotional memory [17], while CA3/dentate is implicated in encoding spatial representations and episodic memories [18]. The questions to be considered are which side of the hippocampus and which subfields are affected in OSA and also whether there are gender-specific differences.
Lateralization and subfields
The lateralization of hippocampal deficits in OSA patients varies considerably across studies. First str-uctural evidence of gray matter (GM) loss in right hippocampus in OSA patients has been reported by Macey [19]. In line with this, a recent meta-analysis revealed significant structural atrophy in right amygdala/hippocampus of these patients [20]. There is less concordance, however, on neurofunctional changes occurring in the right hippocampus. A study using a combination of voxel-based morphometry (VBM) and arterial spin labeling perfusion, reported a decrea-sed perfusion in right hippocampus without structural alterations [21]. By contrast, significant hyperperfu-sion in this structure has been detected with SPECT imaging, a specific tool able to identify metabolic deficits due to chronic hypoxic insult and hemodyn-amic changes [22]. Metabolic abnormalities were correlated with sleep parameters such as Apnea-Hip-opnea Index, and microarousal indexes, suggesting that the increase in perfusion occurring during hypo-xia could persist also during the wakeful rest [21]. On the other hand, compared to controls, OSA patients showed significantly lower unilateral GM concentration [23] and significantly increased N-acet-ylaspartate/creatine ratio within the left hippocampus [24]. Metabolic increased levels in the hippocampal area might represent adjustments to brain bioenergetics, similar to those seen in the ischemic pre-conditioning [24]. Finally, decreased GM volume in bilateral hippocampi has been also described in OSA compared to controls [25, 26].
Overall, these discrepancies might be due to several variables such as different simple size and neur-oimaging tool, methods and setting. To date, few magnetic resonance imaging (MRI) studies are foc-used on the specific involvement of hippocampal subfields in OSA. A recent meta-analysis shows abnormalities of a region overlapping the CA1[20]. In addition, hippocampal volume increases in CA1, subiculum and uncus, and volume decrease in CA3/dentate in the posterior hippocampus have also rec-ently found in these patients [27]. Decreased cortical thinning in CA1 and dentate gyrus has been detected in OSA brain [14]. Cellular damage to spe-cific subfield-hippocampal CA1 region likely contri-butes to neuropsychological impairment among OSA patients [28].
Sex-specific damage
An important contribution in neuroimaging re-search on gender matters was provided from Macey et al., basing on the clinical observation that OSA females exhibit a different neurocognitive profile from males [27]. According their results: 1) sex differences in white matter (WM) structural integrity appeared in OSA patients, with females more affected than males and associated with specific structural changes may contribute to or derive from neuropsychological and physiological symptom differences between sexes; 2) OSA females showed volume decreases in the right hippocampus head and tail [27]. Separate neuropathologies may contribute to symptom characteristics in females and males with OSA. In line with this evidence, animal models confirmed that sex-specific changes in GABAergic neurons of hippocampus may occur following chronic intermittent hypoxia with males showing an increase of this cell population as compared to their female counterparts [29]. On a speculative basis, it is possible that males better present an adaptive neurogenesis in response to hypoxic stress.
Correlations with clinical symptoms
OSA patients commonly exhibit neuropsychological impairments ranging from vigilance decrements, attentional lapses and memory gaps which might to be explained by abnormal hippocampus function. Of clinical relevance, cerebral changes in OSA patients may precede the onset of notable neuropsychological consequence [30]. An important advance in field of OSA research has been the demonstra-tion of reversible brain damage and related clinical manifestations after continuous positive airway pre-ssure (CPAP)-treatment. Neuroimaging and neuropsychological research performed by Canessa and Castronovo clear reveal significant neurocognitive improvements involving memory, attention, and executive-functioning that paralleled GM volume increases in hippocampal and frontal structures after OSA treatment [31]. Their findings highlight the importance of early diagnosis and achieving successful treatment for this disorder.
Amygdala in OSA-AD neurodegeneration
Amygdala is one of the core areas of the emotional circuits. It is a heterogeneous structure composed of structurally and functionally distinct nuclei. Patients with damage to amygdala may exhibit aberrant facial emotions processing, emotional blunting and even aberrant sexual behaviors, dysfunctional memory, and olfactory processing [32, 33].
Lateralization and subfields
Similar to the impact of the hippocampus, the am-ygdala may show involvement of different sides and subfields in OSA patients. Tahmasian et al. recently combined VBM and functional MRI studies and reported significant structural atrophy and functional disturbances in the right amygdala/hippocampus [20]. It is remarkable that right amygdala is mainly involved in OSA neurodegeneration. Recent reports demonstrated that decreased right amygdala volumes may precede the first signs of cognitive decline in healthy elderly controls at the pre-MCI state [34].
There is concordance that of all nuclei of amygdala, the basolateral nucleus appears to be most aff-ected in OSA patients. This finding is not surprising, since basolateral nucleus is functionally connected with the parahippocampus, lateral temporal gyrus, middle occipital gyrus, and medial prefrontal cortex: it receives sensory information from the thalamus, hippocampus, and cortex and then activates or modulates synaptic transmission in target areas appropriate for the enforcement signal with which the sensory information has been associated [20].
Sex-specific damage
To date, the influence of the gender on the amygdala OSA-related damage is conflicting. A diffusion tensor imaging study [36] reported microstructural alterations in several regions including amygdala in OSA females more affected than males [35]. On the hand, Yu et al. found significantly altered resting state functional parameters between dorsal and ventral amygdala and temporal/prefrontal cortex in severe OSA male patients. The complex FC patterns found in the amygdala subregions may be the result of OSA-related selective damage to the amygdala and could partly explain the affective deficits and cognitive impairment observed in male OSA patients. These changes contribute to or derive from neuropsychological and physiological symptom differences between sexes [36].
Correlations with clinical symptoms
Kheirandish-Gozal et al. provided the first functional MRI (fMRI) evidence that cognitive and empathetic processing may be influenced by OSA in children [37]. Indeed, OSA children show greater neural recruitment of regions implicated in cognitive control, conflict monitoring, and attentional allocation in order to perform at the same level as compared to children without OSA [38]. Based on this evidence and given the clinical significance of dementia prevention, some authors investigated the associations between OSA and cortical thickness in older adults considered “at-risk” for dementia [38]. According to their results, increased volume of the hippocampus and amygdala was associated with sleep disturbance while decreased thickness in the bilateral temporal regions was associated with reduced verbal encoding. Changes in GM could reveal how OSA might contribute to neurodegenerative processes in older adults [38].
Structural/neuro-functional alterations in right amygdala, especially in basolateral nucleus appear to have been driven by contribution of neuroimaging and could explain the dysfunctions of emotional limbic processes occurring in OSA. There is not only a structural damage (atrophy) of amygdala but also an increased functional response (hyper-activation). A possible explanation is that atrophy is the first manifestation, and then the remaining sub-regions develop compensatory hyper-activation. As a result of the high functional state there is further damage to the remaining part by exhaustion [15].
Cingulate gyrus in OSA-AD neurodegeneration
Cingulate gyrus is a neural interface between emotion, sensation, and action, supported by the presence of anatomical connections linking this structure with brain areas closely associated with each of these functions [39].
Lateralization and subfields
Cingulate gyrus may be distinct in at least two regions with different neurocognitive functions: anterior and posterior. The anterior cingulate is crucial in humans for integrating cognitive and emotional processes in support of goal-directed behavior whereas the posterior support internally-directed cognition. Despite this, the existing literature is mainly focused on the role of anterior cingulate gyrus in OSA, since it participates in attention and executive processes, and its atrophic nature may partially explain the cognitive deficits often reported in these patients [40].
According to the results of a recent multimodal meta-analysis, anterior cingulate gyrus was decrea-sed in volume with hyper-activation [15]. To explain the neurobiological mechanisms underlying their findings, the authors speculated that: 1) the damage of anterior cingulate gyrus might appear before functional alterations accompanied by a compensatory hyper-functionality of the remaining GM; 2) hypoxemia, low brain blood flow, or other factors which are characterized in OSA lead directly to hyper-ac-tivation [15]. A decrease of volume in anterior cingulate gyrus might be an irreversible deficit in OSA since it appeared reduced before and after surgical treatment as compared to controls [41].
Sex-specific damage
To date, few neuroimaging studies are focused on the sex-related damage of cingulate gyrus in OSA. A fMRI study explored the spontaneous brain activity in untreated males with severe OSA compared to male good sleepers [42]. Compared to the controls, OSA patients showed significant lower parameters in the right precuneus and bilateral posterior cingulate gyrus. This evidence suggests that OSA may involve dysfunction in the default mode network and an adaptive compensatory response in the frontal lobe, which reflect the underlying pathophysiology of cognitive impairment [42].
Correlations with clinical symptoms
Several fMRI studies [43, 44] have demonstrated that, compared to the controls, OSA group showed reduced performance especially involving memory and attention functions, during cognitive tasks. For instance, during a Go-NoGo task, OSA patients showed more false positives (error of commission) than controls, accompanied by decreased brain activation in the left postcentral gyrus, cingulate gyrus, and inferior parietal lobe, as well as right insula and putamen [43]. A decreased brain activation in cingulate, frontal, and parietal regions typically involved in attention tasks has been also reported in OSA patients in association with altered sleep parameters such as arousal index [45]. Finally, after CPAP treatment, OSA patients showed decreases in activation with treatment in the left inferior frontal gyrus and anterior cingulate cortex, and bilaterally in the hippocampus associated with significant improvement compared to baseline [45].
NEUROIMAGING BIOMARKERS TO ASSESS EXTRA-LIMBIC DAMAGE
Insula in OSA-AD neurodegeneration
The insula is a cortical structure connected with many areas of the cortex and limbic system, specifically the amygdala [15] and plays a crucial role in emotional processing. Insular changes have been consistently demonstrated in a range of anxiety disorders [46].
Lateralization, subfields, and correlation with clinical symptoms
Right anterior insula is an important node of the salience network and serves to switch between two major cognitive-related functional brain networks, the central executive network and the default mode network [47]. Left anterior insular cortex is associated with speech, emotion, and affective-cognitive deficits. Structural and functional abnormalities have been described in right anterior insula of OSA pat-ients with functional neuronal changes considered to result from the underlying structural changes: GM concentrations of males with OSA were significan-tly decreased in several areas including right insular gyrus [25], functional disconnections between this structure’s prefrontal cortex were also correlated with the severity of the OSA [48] as well as selective impairment of the functional connectivity between the right anterior insula and the cortical network, which may be a candidate substrate for cognitive impairment in OSA patients [48]. Finally, in a multimodal neuroimaging meta-analysis, right insula showed hyper-activation but no structural damages, thus suggesting the existence of unknown mechanisms leading to insular adaptive response [15].
DISCUSSION
Here, we propose the hypothesis of a common neurodegenerative process between OSA and AD, so called OSA-AD neurodegeneration. To test the hypothesis, we investigated whether brain regions and networks involved in OSA are the same as the vulnerable areas or targets for the development of AD and whether there are neuroimaging biomarkers with the potential to detect OSA-AD neurodegeneration.
Hippocampal involvement in OSA brain pathology provides scientific evidence in favor of our hypothesis (Fig. 1A-3). Firstly, the hippocampus is atrophic in OSA patients and hippocampal atrophy is a hallmark of AD-neurodegeneration. Not only is there atrophy, but AD neuropathology has also been recently reported in the autopsy tissue from the hippocampus and brainstems of 34 Icelandic people with clinically-verified OSA. A positive correlation between the Aβ burden and OSA has been also detected suggesting that the disease severity might be a significant predictor of Aβ plaque burden in the hippocampus [49]. Neuropathology and neuroimaging studies have identified at least three AD biological subtypes: 1) typical AD displays neurofibrillary/tau-related pathology and atrophy both in hippocampus and association cortex; 2) limbic-predominant AD predominantly in the hippocampus; 3) hippocampal-sparing AD with counts predominantly in the association cortex [50, 51]. Results from a recent meta-analysis suggest that limbic-predominant AD was characterized by specific clinical pattern with a late-onset, prevalence of female sex, APOE ɛ4 carrier and anamnestic syn-drome [51]. Based on this information, it is reasonable to speculate that OSA might be linked to the limbic-predominant subtype of AD. Secondly, there is consistency in the preliminary evidence that CA1/CA3 hippocampal subfields are involved in OSA neurodegeneration. CA1/CA3 subfields and dentate, however, are commonly altered in AD patients [52, 53]. Atrophy of CA1 appears to be associated to the increased risk of conversion from MCI to AD [54] whereas a smaller CA3/dentate volume has been found in amnestic MCI patients [52]. It is interesting to note that the CA3/dentate subfield appears to be decreased in volume in both disorders, while CA1 volume was increased in OSA but decreased in AD. Conventionally, volume decrease or atrophy revealed with VBM analysis may be the result of neuronal death or damage to cells whereas volume increase suggest inflammation and glial activation. It can be speculated that CA1 region has been investigated at two different time points in OSA and AD, initial inflammation with increased volume in OSA and late neuronal death with atrophy in AD. Thirdly, hippocampal involvement in OSA is sex-specific related, with females more affected than males. It is well-established that the incidence of AD is greater among women than men and this discrepancy increases with advanced age [55–57]. A meta-analysis of 13 population studies also indicates that women are at significantly greater risk of developing AD, though not other dementias [58]. Findings from brain imaging, postmortem analyses, hormone therapy, and genetics suggest that AD affects men and women differently [59]. There is cumulative evidence that there is poorer cognitive visuospatial, verbal, episodic memory, and semantic memory outcome for women with AD compared to men [60] in keeping with the greater impact of AD in females. Sex differences in the development of MCI/AD development can be predicted by changes in the hippocampus [61]: hippocampal volumes predict the progression to or odds of probable AD more so among women than men [61]. Overall, these findings indicate that hippocampal atrophy might be a neuroimaging biomarker in OSA for early identification of patients at risk of developing AD, especially of limbic predominant subtype. Since there is greater risk of AD for females compared to males, initial attention should be early focused on the alterations of CA1/CA3 hippocampal subfields in OSA females. As research agenda, longitudinal investigations are needed in order to evaluate whether OSA females with hippocampal changes at baseline develop MCI/AD over the time.

Key points on the involvement of hippocampus (A) and amygdala (B) in OSA-AD neurodegeneration.
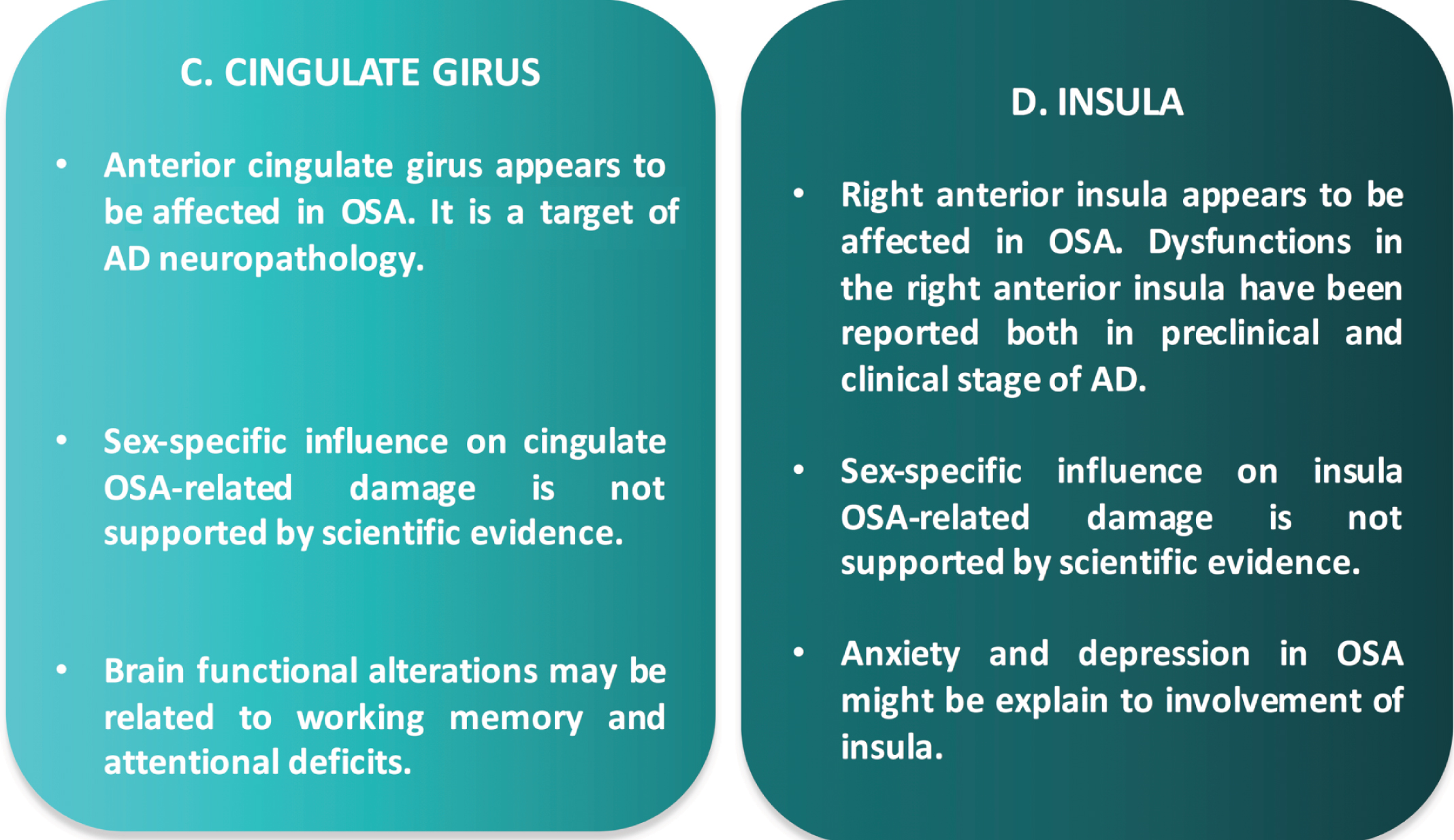
Key points on the involvement of cingulate gyrus (C) and insula (D) in OSA-AD neurodegeneration.
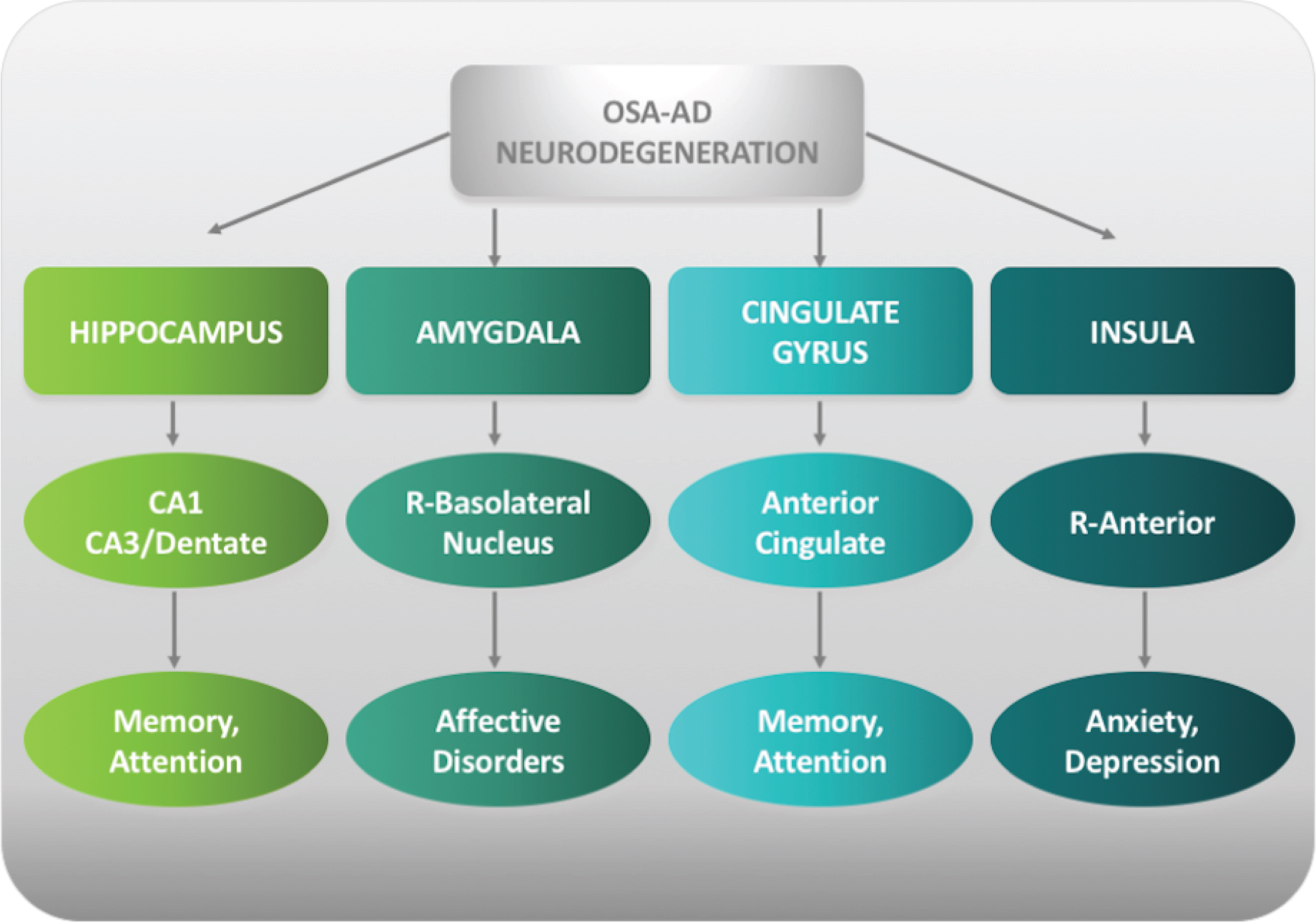
Neuroimaging biomarkers of OSA-AD neurodegeneration. Schematic representation of main OSA brain structures linked to AD neuropathology (OSA-AD neurodegeneration) and related cognitive impairments. OSA, obstructive sleep apnea; AD, Alzheimer’s disease; R, right.
The involvement of other brain structures in OSA patients supports our hypothesis and provides insight to the possible pathophysiological mechanisms of OSA-AD neurodegeneration (Fig. 1B-2; Fig. 3). There is agreement that of all nuclei of amygdala, bas-olateral nucleus appears to be most affected in OSA which could explain the affective deficits and cognitive impairment that occurs often in these patients. Basolateral nucleus of amygdala, however, is a key structure in AD neuropathology. Indeed, AD patients show disrupted connectivity patterns in this structure as compared to controls [62]. It is also not surprising that the anterior cingulate gyrus is involved in OSA and that neuroimaging and neuropathologic investigations have demonstrated that anterior cingulate gyrus is affected by AD neuropathology. Neurofibrillary tangles, characteristic pathological findings of AD, have been found in the anterior cingulate gyrus of pathologically confirmed AD in association with agitation [63]. A significant increase in the numbers of Fluoro-Jade stained cells in layers I-III of the anterior cingulate gyrus has been reported in pat-hologically confirmed AD, demonstrating that asymmetries in different cortical regions can be used as another marker in the post-mortem diagnosis of AD [64]. Furthermore, structural and metabolic alterat-ions in the anterior cingulate cortex seems to be associated with a diminution of awareness in patients with early-stage AD [65]. Finally, of interest, the ant-erior part of right insula is involved in OSA brain pathology with metabolic changes contributing to au-tonomic and neuropsychological dysfunction, especially anxiety and depression in these patients [66, 67]. In keeping with the OSA-AD neurodegeneration hypothesis, the right insula is considered the “core re-gion” in association with neuropsychiatric symptoms (hallucinations) in neurodegenerative diseases such as AD [68]. Structural and functional alterations of right insula have been reported in AD patients in association with neuropsychiatric symptoms both in clinical and preclinical stage of the disease [69, 70].
To strengthen the hypothesis of a common OSA-AD neurodegeneration, here we discuss some sti-mulating aspects as the opposite direction of this relationship AD versus OSA, and the pathological mechanisms underlying AD-neurodegeneration alt-ernatively to the amyloid model. An important contribution on the first research topic comes from the works of Bubu’s group. These authors first quantified the effect of sleep problems/disorders on cognitive impairment and AD. Results from their meta-analysis revealed that individuals with sleep problems had a 1.68 times higher risk for the combined outcome of cognitive impairment and/or AD [71]. They also suggest that approximately 15%of AD in the population may be attributed to sleep problems [71]. Basing on these preliminary trajectories, they subsequently tested the hypothesis of a potential association between OSA severity and longitudinal increase in amyloid burden in cognitively normal elderly (NL). As expected, an increased amyloid burden OSA-related over the 2-year follow-up has been detected in these subjects [72]. Their third study determined the effect of self-reported clinical OSA diagnosis on the longitudinal biomarkers changes in the brain amyloid PET and several cerebrospinal fluid (CSF) biomarkers in NL, MCI, and AD elderly [4]. Of interest, they found a faster annual increase in florbetapir uptake and decrease in CSF Aβ42 in participants OSA+(with OSA) compared to those OSA- (without OSA). It is noteworthy that the longitudinal biomarkers changes have been detected in subjects with early/prodromal AD whereas no significant variations over time were seen in manifest AD [4]. Finally, an analysis of the AD Neuroimaging Initiative (ADNI) cohort revealed that the presence of OSA was associated with an earlier age onset (11 years earlier than OSA-) of cognitive decline and that CPAP treatment delayed the progression of cognitive impairment [73, 11]. Thus, we can summarize that presence and severity of OSA may anticipate the onset of cognitive impairment and influence the amyloid burden/accumulation rate in the brain of NL/MCI subjects. Considering the clinical relevance of this research topic, future investigations mainly focused on the early identification of brain structures/areas shared between AD neuropathology and OSA brain pathology are necessary.
Interpreting the AD-risk basing solely on the amyloid hypothesis might result incomplete. Although tau-pathology in OSA has been less studied and needs further confirmation, some preliminary evidence sup-ports the role of tau-related mechanisms in contributing to cognitive impairment and development of AD. In a recent population-based Mayo Clinic Study of Aging, some authors identified a significant association between witnessed apneas in cognitively unimpaired adults and elevated tau-PET signal in tau-susceptible brain regions as the entorhinal cortex and inferior temporal cortex [74]. These neuroimaging findings are in line with previous biological evidence suggesting that compared to OSA-, individuals with OSA+may have elevated plasma concentrations of total-tau [75] and phosphorylated-tau [76]. This is to be kept in mind, since plasma total-tau level is considered a predictive biomarker for dementia [77]. Moreover, Bubu et al. also reported an increase in CSF total-tau and phosphorylated-tau associated with increases in amyloid burden over the time, in NL and MCI subjects with OSA [4]. Taken together, these findings suggest that amyloid and tau pathological changes might be synergistic in contributing to development and progression of AD-like cognitive impairment in OSA patients. The chronopathological window in which this can really make the difference is the preclinical/prodromal AD-stage in which the amyloid/tau pathological changes are present in the brain in absence of evident clinical manifestations or frank dementia.
Whether is legitimate to consider OSA pathology through the lens of neurodegenerative disease processes, given the reversibility of the brain deficits after the treatment, is matter of discussion. Recent findings by Owen and colleagues [14, 71] complete a circle of evidence confirming that AD-neuropathology may occur in human brain OSA (in particular hippocampus) and support a plausible neurodegenerative model for this chronic disease. Conversely, the reversibility of brain deficits suggests that the structural changes seen in OSA patients might be not all permanent, which points to some adaptive or pathological changes short of neuronal death [78]. To match these contradictory aspects, the most likely scenario has been recently proposed by Macey et al., as OSA-related brain damage is a combination of irreversible cell death and adaptive/reactive changes in the cerebral cellular environment [78]. Thus, the adaptive/reactive changes related to inflammation and more specifically hypoxia-induced astrocyte activation could be reversed with CPAP-treatment [78, 79]. On this basis, we strongly believe that the timing of treatment is crucial for reverting or preventing the brain tissue damage in OSA patients. Indeed, in an early phase in which the initial inflammation and adaptive/reactive changes occur but neuronal death with atrophy and secondary neurodegeneration is not yet occurred, the reversibility of the brain OSA-related damage might be until possible. An ideal clinical trial in which OSA treatment begins several years before of onset of cognitive symptoms could be useful to determine whether OSA and AD are associated by a causal link resulting in a neurodegenerative process and what is the direction of this causality, or rather are these two chronic coexisting disorders exercising a “double hit” like effect on the brain.
CONCLUSION
The evidence that OSA can accelerate Aβ deposition in the brain complements the information from neuroimaging studies that OSA brain pathology can lead to AD. We strongly believe that OSA and AD share common areas of neurodegeneration and neuropathology in the brain indicating that the hypothesis of an OSA-AD neurodegeneration is credible. Specific changes in specific limbic structures, especially hippocampus, can be considered biomarkers of this novel condition. Research focused on the early identification of AD brain biomarkers in OSA patients should be encouraged. As challenges and future directions, the treatment of OSA might slow cognitive decline and reduce the risk of AD and be a successful therapeutic strategy for AD.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-1066r2).