
Abstract
The cerebral hypoxia-ischemia can induce a wide spectrum of biologic responses that include depolarization, excitotoxicity, oxidative stress, inflammation, and apoptosis, and result in neurodegeneration. Several adaptive and survival endogenous mechanisms can also be activated giving an opportunity for the affected cells to remain alive, waiting for helper signals that avoid apoptosis. These signals appear to help cells, depending on intensity, chronicity, and proximity to the central hypoxic area of the affected tissue. These mechanisms are present not only in a large list of brain pathologies affecting commonly older individuals, but also in other pathologies such as refractory epilepsies, encephalopathies, or brain trauma, where neurodegenerative features such as cognitive and/or motor deficits sequelae can be developed. The hypoxia inducible factor 1α (HIF-1α) is a master transcription factor driving a wide spectrum cellular response. HIF-1α may induce erythropoietin (EPO) receptor overexpression, which provides the therapeutic opportunity to administer pharmacological doses of EPO to rescue and/or repair affected brain tissue. Intranasal administration of EPO combined with other antioxidant and anti-inflammatory compounds could become an effective therapeutic alternative, to avoid and/or slow down neurodegenerative deterioration without producing adverse peripheral effects.
Keywords
INTRODUCTION
The degenerative processes in the central nervous system (CNS), also known as neurodegeneration, are characterized by progressive loss of neural functions associated with intellectual and/or motor impairment. In several diseases that mainly affect older people such as Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), stroke, Huntington’s disease, Creutzfeldt-Jakob disease, multiple sclerosis, and many others, hypoxia-ischemic play a central role in the triggering of neurodegeneration. Similarly, other pathologies such as refractory epilepsy, brain infections, or hereditary diseases that involve neurodegeneration with brain iron accumulation also lead to chronic brain inflammation with loss of neural cells and severe consequences on brain function. In all these neurodegeneration processes, brain iron accumulation can affect normal functions of neurons, oligodendrocytes, astroglia, and microglia cells, as well as vascular endothelial cells [1–6].
The neurovascular unit is perhaps the main target of the severe hypoxic-ischemic insult where a wide spectrum of cellular alterations can be developed. Furthermore, during brain hypoxia-ischemia the autophagy machinery will slowly accumulate high amounts of iron, copper, and zinc as well as various toxins over the course of several decades, which constitutes the main characteristic of some of these neurodegenerative diseases [7, 8] as observed in the amyloid plaques of AD [9].
Chronic inflammation is an important feature of neurodegenerative diseases, which exacerbates the pathology by activating microglia, releasing pro-inflammatory cytokines as IL-1 and IL-6, and producing reactive oxygen species (ROS). All together, these events contribute to the spread of oxidative stress (OS) with a concomitant breakage and dysfunction of blood-brain barrier (BBB) and alteration of brain microenvironment [10].
ROS, including superoxide anion, can contribute to vascular endothelial cell dysfunction in the BBB during hypoxic conditions, and under OS, high levels of the superoxide anion produced overcome the metabolic capacity of superoxide dismutase (SOD) [11]. In neurodegenerative diseases, the production of free radicals and OS is usually accompanied by the accumulation of misfolded proteins, such as Aβ, lipofuscin, ferritin, hemosiderin, or intracellular aggregation of mut-SOD1, huntingtin, α-synuclein oligomers (Lewy bodies), etc. [12, 13].
In some of these neurodegenerative processes, iron and/or copper homeostasis can be altered, and interestingly, experimental iron or copper brain overload is directly associated with development of brain dysfunction related with high OS [14, 15]. Primary brain vascular endothelial cells of rat cultured with hydrogen peroxide induced ROS generation and increased P-glycoprotein (P-gp) in a concentration-dependent manner, and this upregulation was related with several signaling effectors including ERK1/2, Akt, and JNK, which in turn activated nuclear factor-κB (NF-κB) [16].
In this regard, a combined action of ROS, hypoxia, and inflammation was suggested as a determinant factor in the cancer initiation and progression [17], as well as in chronic inflammation in arthritis [18]. The triad “hypoxia - oxidative stress –inflammation” is a vicious circle where any one of them can initiate the activation of the others and sustain in time the progressive cerebral deterioration that characterizes neurodegenerative processes. In this context, clinical and experimental studies have widely demonstrated that brain hypoxia-ischemia can induce seizures or epilepsy, the seizures induces hypoxia-ischemia, and in both condition inflammation is involved [19–21]. Furthermore, inflammatory stimulus are also enough to develop not only excitotoxicity or seizures [22], but also hypoxic conditions [23], where hypoxia inducible factor (HIFs) and prolyl-hydroxylase domain enzymes, play isoform-selective roles in an environmental context of hypoxia and inflammation [24]. Despite a wide spectrum of clinical manifestations, seizures are associated with ictal or postictal hypoxia, which stabilizes hypoxia inducible factor 1a (HIF-1α), and induces the expression of several HIF-responsive proteins, as the erythropoietin receptor (EPO-R), and P-gp, among others [25, 26].
Moreover, status epilepticus, as well as repetitive severe seizures, can induce not only cerebral but also systemic hypoxia-ischemia events, significantly affecting peripheral organs such as heart in both adults and pediatric pharmacoresistant epileptic patients [27, 28]. In this regards, the brain-heart connection has been described to be related with sudden unexpected death in epilepsy (SUDEP) where an important “neurocardiac” list of genes have been suggested as candidates of genomic biomarkers of SUDEP risk [29]. In this context, a progressive heart and brain overexpression of P-gp playing a role in the neuronal and cardiomyocytes depolarization was observed, concomitant with an altered cardiac depolarization during and between seizures, which was also related with inwardly rectifying potassium channels dysfunctions. All these results suggest that after repetitive pentilenetetrazol (PTZ)-induced seizures or status epilepticus respectively, mechanisms of gene induction and repression may contribute to simultaneous brain and heart depolarization, increasing the severity and drug resistance of seizures, and leading to prone the fatal heart failure [30, 31].
OXIDATIVE STRESS
OS processes in brain are associated with neurodegenerative disorders such as AD, PD, ALS, and vascular dementia (VaD). Histopathology markers are characteristic of each one: amyloid deposits with amyloid-β (Aβ) aggregates are present in AD [32] and α-synuclein is characteristic of PD [33]. However in all cases, free radical molecules and ROS as superoxide anion (O2-), hydrogen peroxide (H2O2), singlet oxygen (1O2), nitric oxide, lipid hydroperoxides (ROOH), HO, and peroxynitrite (ONOO), contribute to neuronal death [34, 35], with increased processes of lipid peroxidation, protein oxidation, nucleic acid oxidation, and depletion of antioxidants [36]. Causes of neuronal death in neurodegenerative disorders are multifactorial (OS, protein deregulation and aggregation, chronic inflammation, mitochondrial dysfunction, metal dyshomeostasis, depletion of intracellular thiols, redox and signaling deregulation) and develop as complex networks of pathological events that are interconnected and generate processes leading to cell demise, that is neither classical necrosis nor apoptosis and that may be triggered during the terminal phase of the chronic progressive disorder [37–40].
The reactive species O2–, H2O2, and ROOH are normal metabolites continually produced as secon-dary products of respiration and oxidative meta-bolism in aerobic cells, but are increased in OS and in pathological situations [35–40]. An intracellular oxidizing environment leads to increased formation of HO• radical through the Fenton/Haber-Weiss reaction and contributes to neuronal oxidative damage and to disease progression [36].
While OS is a reversible “emergency” situation in cell homeostasis, it can also initiate damaging processes that ultimately lead to cell death [41, 42]. OS is considered as an imbalance between oxidant production and antioxidant defenses that may lead to oxidative damage to biomolecules and drive to cell death [43]. The “redox hypothesis” updated the classic concept of OS considering that it is due to an oxidative process that alters the redox balance of the thiol groups of low molecular weight components, such as glutathione (GSH) and proteins involved in signal pathways and regulation of physiological functions [44, 45].
AD is associated with inflammatory processes in the brain, characterized by the presence of astrocytes and cells of the activated microglia and the increase of IL-1 and IL-6 in plasma and in areas near the deposits of Aβ [46]. Cellular production of H2O2 may trigger signal transduction or may be detoxified by antioxidant enzymes as catalase or glutathione peroxidase. However, the relationship between inflammatory processes in the brain and the inflammatory response in peripheral blood is still unclear.
According to the concept of hormesis [47], iron (Fe) and copper (Cu) are required for normal aerobic life, but at high levels they are toxic for brain biochemistry and physiology [48], associated to neuronal damage, OS, protein misfolding, and aggregation [49, 50] incidence and progression of neurodegenerative disorders and of the cognitive disorders associated with aging [51]. The mechanism of toxicity seems afforded by Fe- and Cu-catalyzed production of HO• radical, which damages neuronal macromolecules and promotes neuronal oxidative injury and death. Neuronal Fe (II) reacts with α-synuclein in PD. Excessive Fe was found in PD in substantia nigra pars compacta, where dopaminergic neurons are exposed to high levels of O2– and H2O2 from dopamine oxidation, and favors the generation of amyloid plaques in the brains of patients with AD, because it joins with Aβ [52]. Neuronal Cu facilitates the formation of amyloid plaques in AD and increases the formation and accumulation of Aβ from the amyloid-β protein precursor (AβPP). Cu binds to Aβ and to AβPP, where it is reduced and contributes to protein cross-linking and aggregation of amyloid plaques [53]. The amyloid plaques in postmortem brains of AD patients have elevated levels of Cu, Fe, and Zn [54], and experimentally, a Cu acute overload impaired the mitochondrial ATP formation, decreasing state 3 O2 uptakes and respiratory control in liver and brain [55]. Cell death is mediated by Cu cytotoxicity and OS in vitro in a time and concentration dependent manner. Cu interacts with proteins and promotes aggregation and cellular toxicity when it accumulates in cytosol, due to the cross-linking covalent interaction between Cu (II) and thiol groups of proteins [56].
Neuroimaging, postmortem histology, and cerebrospinal fluid studies are the most common brain neurodegenerative biomarkers used. Additionally, systemic detection of OS markers in neurodegenerative disorder patients can be useful indicators of diagnosis and/or disease progression. By-products of free-radical reactions in the brain, such as lipid peroxidation compounds (measured as thiobarbituric acid reactive substances, TBARS) and carbonyl groups in small peptides are the most evaluated biomarkers in blood, plasma, serum, and cerebrospinal fluid. Small molecules as H2O2, NO, ROOH, and MDA diffuse from the intracellular media to extracellular fluids and blood. Blood and plasma reflect the rate of a free-radical mediated chain reaction in the vascular space or oxidative damage occurring in target organs, where inflammatory response results in the systemic disease of the adaptive control of redox homeostasis, generating as a consequence, antioxidant deprivation and dysfunction. In previous research, biomarkers of OS and damage were evaluated in blood of neurodegenerative disorder patients. In plasma, the markers gave significant indication of systemic OS compared to healthy subjects with TBARS values that were increased 35%(AD, p < 0.001) and 21%(PD and VaD, p < 0.05 and 0.01 respectively) and protein carbonyls (higher than 100%, p < 0.05), TRAP (Total Reactive Antioxidant Potential) levels that were decreased 34%for AD, 28%for PD, and 39%for VaD (p < 0.001) in plasma and higher t-butyl-hydroperoxide initiated chemiluminescence in erythrocytes values that were 50%in AD and VaD and 72%in PD [41, 57–59].
The redox homeostasis was evaluated in erythrocytes of neurodegenerative disorder patients measuring GSH content, the index GSH/GSSG, activity of glutathione reductase and antioxidant enzymes. GSH decreased in AD (45%) and VaD (38%), p < 0.05 respectively; GSH/GSSG decreased: 89%in AD, 57%in PD, and 95%in VaD (p < 0.05). GR increased 55%in AD and 64%in VaD (p < 0.05), but did not showed changes in PD. These results indicate that OS is associated with neurodegenerative disorders. The physiological intracellular O2– concentration is 10–9 M, controlled by cytosolic Cu, Zn-SOD1, avoiding O2– damaging reactions. SOD activity was found increased 80%in AD patients, 60%in PD, and 70%in VaD (p < 0.001), indicating and adaptive response to the increased levels of O2– [41, 57–59]. Furthermore, it has been demonstrated that Aβ needs of redox reactions mediated by metal ions to acts a toxic compound [60].
AD, affecting 50%of people aged over 80 years, is the most prevalent neurodegenerative disease. Usually cognitive functions are significantly declined, and memory loss, paranoia, loss of reasoning powers, and confusion are observed, but these features will be evident too late to have any opportunity for an effective treatment [61–63].
Several theories about the potential causes of AD have been reviewed [64]. One of them is related with high concentrations of copper cations found in senile plaques in AD patients’ brain that proposes a gain-of-function of Aβ after binding Cu2 + [65, 66]. In this regard, a more recent hypothesis suggests that Aβ play a critical protective role within the brain, against the excess of toxic metals including iron and copper [67]. Two very recent and complete revisions, showed controversial results where both the copper deficiency and overload are potential risk factors involved in AD development or other neurodegenerative disorders [9, 68] (Fig. 1).
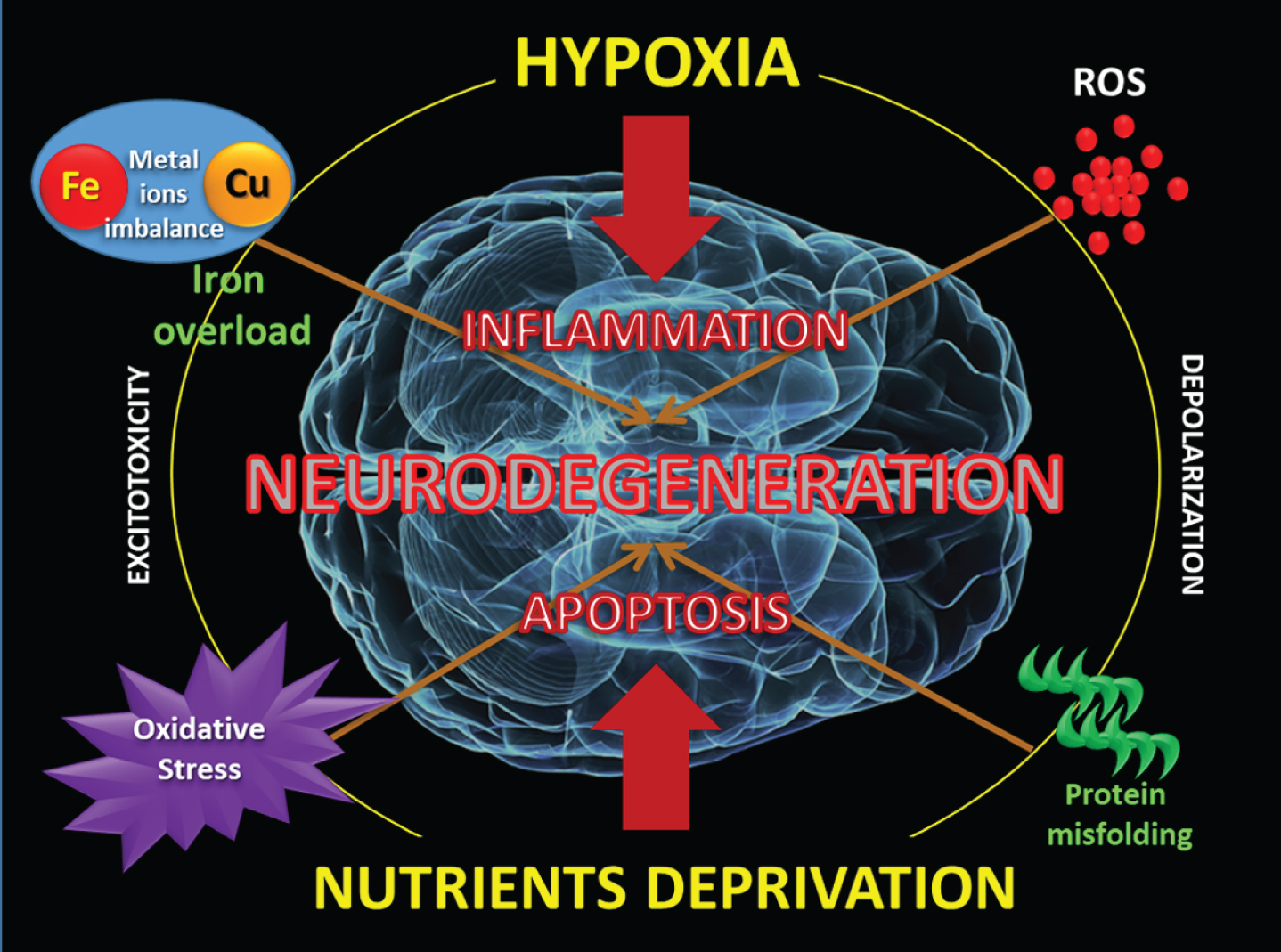
Both hypoxia and nutrients deprivation can trigger aberrant responses that generate depolarization, protein misfolding, metal ions imbalance, oxidative stress, with brain iron accumulation and apoptosis.
The mysterious forbidden love between of the Roman Gods “Mars” (iron) and “Venus” (copper) seems to explain the eternal mutual need between them, for a correct adaptation to the continuous oxi-reduction imposed by atmospheric O2. Only in this way, the organism can achieve a metabolism with high energy performance, and adequate control of OS and the generation of free radicals. Cooper and iron are involved into the early chemistry of life as water-soluble ferrous iron while copper was in the water-insoluble Cu (I) state as highly insoluble sulfides. The advent of oxygen changes water-soluble Fe2 + to water-insoluble Fe3 +, and inversely, water-insoluble Cu (I) to water-soluble Cu (II) [69]. Perhaps we should interpret a “functional deficiency” and a “molecular overload” of both metals as simultaneous mechanisms responsible for neurodegenerative disorders.
HYPOXIA
Cerebral hypoxia, either as global hypoxia or as focal cerebrovascular accident or stroke, is a severe neurological disorder with heterogeneous characteristics and multiple etiologies, which represents the second cause of death and the disease with the greatest sequelae of disability in the world [70, 71]. About 10%of deaths are due to this disorder, and the World Health Organization (WHO) was able to determine that around 270,000 deaths occurred in 2002 in Latin American countries for this reason [72].
The causes of this disorder encompass a broad set of clinical syndromes including heart conditions (e.g., cardiac left-right interconnection, myocardial insufficiency), arteriosclerosis, hypertension, disease of the small penetrating vessels in CNS, or genetic disorders, which in turn may be aggravated by additional factors such as smoking, diabetes, lack of exercise, etc.
In this regard, the perception of nutrient deprivation can activate transcription signals similar to those induced by O2 deprivation, such as sirtuin-1 (SIRT1), AMP-activated protein kinase (AMPK), and HIFs (especially HIF-2α), responsible for regulating hundreds of genes and proteins capable of maintaining homeostasis and cell survival, in the face of the adversity of the microenvironment [73].
Metabolic stress by nutrient deprivation or pharmacological inhibition of mTOR (mechanistic target of rapamycin kinase), robustly activates autophagy in astrocytes and neurons. Similarly, mTOR can be regulated during mild or acute hypoxic exposure giving rise to neuroprotection, but under severe hypoxia, will be overactivated inducing to neuronal death [74].
Moreover, during inflammation cellular oxidative phosphorylation can redirect to an increased aerobic glycolysis condition known as Warburg’s effect, initially described in cancer cells, and associated to nutrient deprivation [75, 76].
This mechanism was also observed in diabetes and several neurodegenerative processes such as AD. In fact, Aβ can triggers acute microglial inflamma-tion accompanied by this metabolic reprogramming in mTOR-HIF-1α pathway dependent manner. Under these conditions, mTOR regulates glucose metabolism sensing the ATP status of the cell, where AKT acts as coupler of extracellular signals to increase phosphorylation of mTOR. Simultaneously, AMP-activated protein kinase (AMPK) acts as a sensor of AMP and ADP increasing levels, representing metabolic exhaustion, to inhibit phosphorylation of mTOR. Finally, phosphorylated mTOR increases the expression of HIF-1α, the master transcriptional regulator of glycolysis also activated during hypoxia [77].
Furthermore, several other situations, such as anemic hypoxia, deficit of pulmonary ventilation or obstruction defects, or neonatal asphyxia, hypobaric altitude hypoxia, or toxic situations such as CO inhalation, etc., are the causes most common of a lower contribution of O2.
We assume that cerebral hypoxia is produced when a loss (partial or complete) of oxygen supply to the CNS occurs, not only because of decreased blood flow but even in the presence of adequate blood flow. Consequently, this concept is often confused with that of cerebral ischemia, a situation that is specifically produced by a decrease in cerebral blood flow, which will not only produce a consequent loss of oxygen supply, but also of nutrients and factors from the blood.
We must consider all those systemic pathologies, which generate a loss of the ability to deliver oxygen to the CNS as simultaneously affecting other tissues. In these cases, hypoxia will be a systemic phenomenon, and as such will induce systemic responses in search of improved oxygen supply. The best example of this situation is the renal overproduction of erythropoietin (EPO) and the consequent increase in the count of circulating erythrocytes. Instead, the reduction in the contribution of O2 in the CNS due to a decrease in the speed of blood flow or “ischemia” [from the Greek “isceiu” (BRAKE) +“aima” (BLOOD)] will not have the same systemic responses.
This concept was initially described by Rudolf Virchow in 1858, resulting from an imbalance between tissue demand for O2 and blood flow, a phenomenon characterized by alterations in the microvascular permeability barrier, reduction in flow with the vascular bed and/or thrombosis at the microvasculature of brain-supplying arteries, known as Virchow’s Triad [78]. This lower contribution of O2 in the brain parenchyma will be translated into a loss in energy performance dependent on aerobic mitochondrial metabolism [79].
Acute ischemia induces an important cascade of molecular events dependent on both the duration of the insult and the progression time of its immediate effects and long-term consequences [80–82].
These mechanisms basically begin immediately in response to energy deprivation and can induce neurotoxicity. So, the overstimulation of glutamatergic receptors will produce excitotoxicity excessive intracellular accumulation of Na+ and Ca2 +, and membrane depolarization, mitochondrial damage, and inflammatory responses ending in neuronal apoptosis. The consequences of cerebral hypoxia-ischemia can be understood as a brain “traumatic” phenomenon, which will be directly linked to the intensity of the insult, its durability and the compromised brain region, whatever the cause (Table 1).
List of some different causes of cerebral hypoxia
Depending on the brain region involved, different sequelae will be observed. In addition, the predominant disability will be related to the different brain areas affected, generating sequelae with motor, language, or visual compromise, of understanding and/or behavior, or the sensitive area, or any of its combinations (Table 2).
List of the aftermath of cerebral hypoxia in both children and adults
Hypoxia is a biological stimulus capable of promoting both rescue and survival mechanisms, as well as triggering a sequence of irreversible events that lead to cell, tissue, and even individual death. Ischemic symptoms due to obstruction of blood vessels generate a wide spectrum of biological effects but only limited to the tissue, organ, or part of the organ compromised by the interrupted irrigation, and this tissue will seek to adapt to the new situation, pending the restitution of the normal supply of O2 and nutrients. Eukaryotic cells have developed a highly sensitive and complex mechanism capable of identifying the hypoxic condition and giving rapid responses for rescue as well as for induction of cell death. These ambivalent responses to acute or chronic hypoxia are mediated by the transcription factor HIF-1α, described originally by Greg L. Semenza who won the Nobel Prize 2019 for this discovery [83, 84].
If hypoxia is important and long-lasting, HIF-1α will activate apoptosis via p53, generating a zone of infarction with a high rate of cell death. In the tissue surrounding that critical area, HIF-1α will translocate to the cellular nucleus, to stimulate a long list of genes intended to restore the O2, nutrients, and energy required to the affected tissue [85]. In this area named as “penumbral zone”, the hypoxic cells that are still viable will try to recover their normal metabolism; failing that, they will enter a late apoptosis increasing the size of the original infarction.
Although the brain comprises only 2%of total body mass, it consumes up to 20%of the energy produced in the body and depends exclusively on cardiac work to incorporate O2 and glucose as sources of ATP. Beyond the damage that global hypoperfusion or hypoxia can cause to other organs or systems, brain damage secondary to lack of O2 constitutes a wide spectrum of disorders that may include coma, seizures, and various motor and cognitive disorders.
Hypoxic damage on brain triggers molecular processes that culminate in neuronal damage. If ATP production falls more than 50%, neurons lose the ability to maintain the membrane potential, and will develop a progressive depolarization [86], characterized by failure of Na+/K+-ATPase activity with intracellular increase of Na+ and extracellular K + . The process will lead to a massive influx of Ca2 +, the production of free radicals, OS and cell death. This depolarization favors the “sensitivity” to the glutamate-stimulating excitotoxicity that exacerbates the deterioration and increases the area of the infarction [87].
Although this mechanism involves progressive damage with edema, inflammation, apoptosis, and finally necrosis, it is also a process that could be avoided or stopped, and would even be potentially reversible. In cerebral ischemia, ATP reserves are rapidly reduced due to lack of input and continuous expenditure by the Ca2 +, Na+, and K+ pumps that continue to function for some time. Physiological levels of free cytosolic Ca2 + at a value close to 100 nM last a short time, and the massive entry of Ca2 + into the cytosol with loss of the ionic gradient, will produces a permanent membrane depolarization and excitotoxic action of glutamate by activation of NMDA receptors. Thus, the intracellular increase in Ca2 + and ROS fluctuates in a range of concentrations capable of promoting apoptosis and cell necrosis, as well as rescue and survival responses [88].
Brain trauma, defined as primary or secondary brain injuries, have high prevalence in the world and it is one of the main causes of disability in young adults [89]. Primary damage refers to the destruction processes immediately caused by direct traumatic damage and defined in the “core area” from the hypoxic-ischemic insult. The secondary damage is the cell injury that predominantly occurs after the initial damage in areas that were less damaged or undamaged and defined as “penumbra area” [90]. It has been postulated that the duration of hypoxia correlates with the period of time during which HIF-1α remains active in the cells. So, the called “memory of HIF” [91] can choose between the decision to sustain survival or induce cell death. In both situations, a time-course of these primary and secondary conditions will reflex the same processes as necrosis, excitotoxicity with depolarization, inflammation, and apoptosis. Excitatory glutamate-signaling is directly related with different pathogenic mechanisms as chronic neurodegenerative disorders such as Huntington’s disease and AD, hypoxia-ischemia, epilepsy, P-gp overexpression, production of ROS, COX2 induction, and inflammation which will induces more glutamate-dependent excitotoxicity [92–98].
Perhaps, we must to consider chronic neuroinflammation with hypoxic conditions as the best scenario to develop neurodegeneration processes such as AD [99, 100]. In this regard, these sequential events have been also described after seizures that produce brain acute hypoxia-ischemia [101, 102] (Fig. 2).
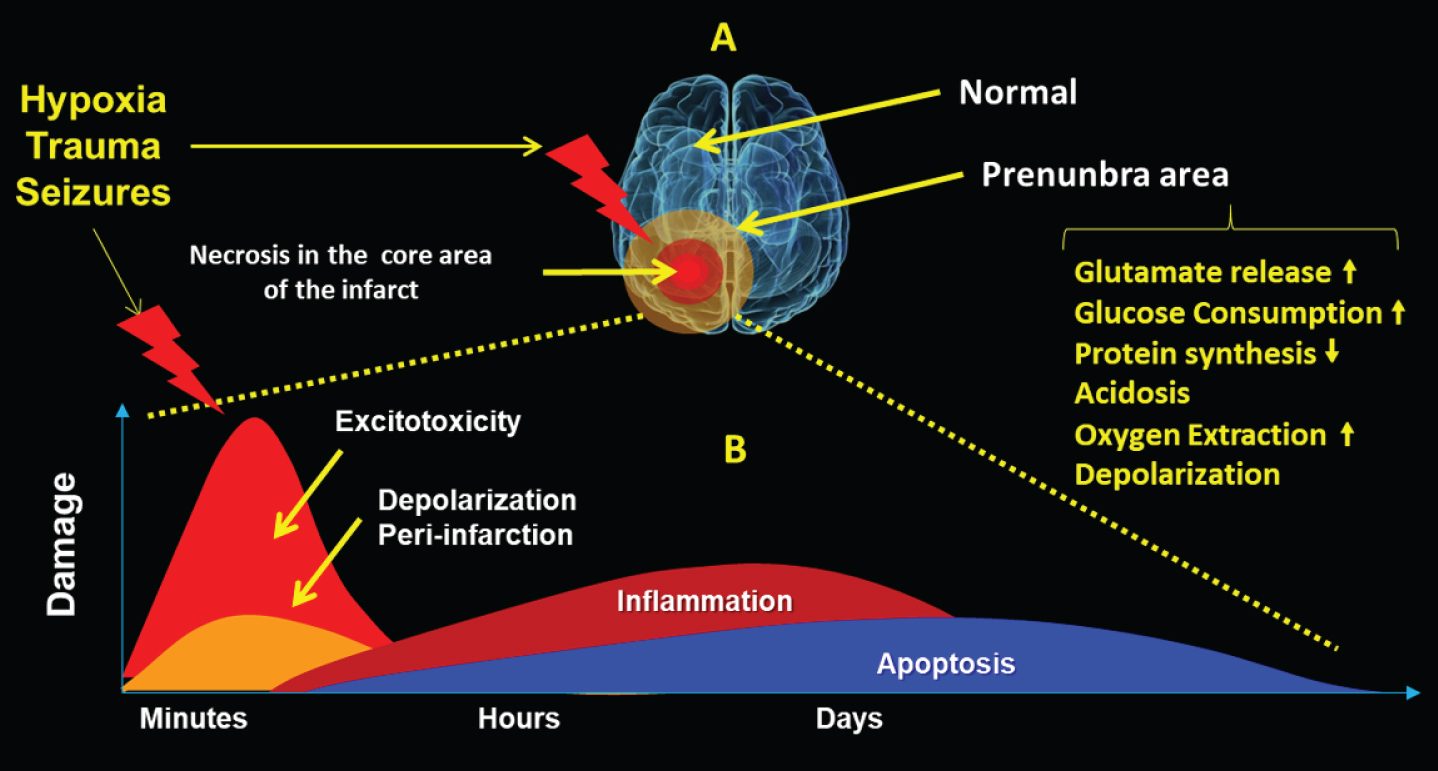
A) Hypoxia, trauma, or severe seizures will generate a necrotic core zone, and a surrounding penumbral area where the cells activate pro and antiapoptotic mechanisms with metabolic changes. B) Evolution in time of sequential mechanisms that include exitotoxicity, depolarization, inflammation, and apoptotic death.
In this regard, it was also demonstrated in ischemic experimental studies that AQP4 mRNA is decreased in the core of the ischemic brain tissue, whereas it is maximally increased in the surrounding area three days after the ischemic stroke, starting a cytotoxic edema with water accumulation in astrocytes and neurons [103].
Very recently, we demonstrated experimentally, that repetitive seizures and glutamate-induced excitotoxicity in vitro, develops hypoxic responses by HIF-1α activation and high brain expression of P-gp, the product of the Multiple Drug Resistance (MDR-1/ABCB-1) gene, in according with pharmacoresistant phenotype observed in refractory epilepsies. However, the co-expression of EPO-R was also observed. In these experiments, human recombinant EPO (rHu-EPO) can inhibit the P-gp-dependent efflux activity, suggesting that under hypoxic-ischemic, traumatic, or convulsive insults exogenous administration of EPO not only can to offer a therapeutic chance surviving, but also could reverse the MDR phenotype [104–106].
Under experimental conditions of cerebral hypoxia-ischemia, a tissue-specific neuroprotective response to the HIF-induced secretion of EPO by astrocytes has been demonstrated. Furthermore, EPO demonstrates its ability to provide neuroprotection because it reduces neuronal death from oxygen glucose deprivation, glutamate toxicity, and nitric oxide–induced death [107]. In spite of these properties, spontaneous self-activation of the Epo/Epo-R system does not guarantee neuroprotection. High levels of Epo-R expression with an pharmacological amount of exogenously administered EPO, will required to achieve its activation and exert an effective neuroprotective action [108, 109].
Several genes also induced by HIF-1α, such as BNIP3 and COX2 among others, will be associated with stabilization of p53, inducing a pro-death situation by activation of the apoptotic pathway and proportionally related to intensity and durability of HIF-1α stabilization. The increased ROS produced synchronically with decreased levels of pO2 may also trigger secondary transcription factors also responsible for cell death.
This damage will be also developed in the penumbra area neighboring to the core site of the impact of the primary damage. There cells must choice between joining the apoptotic process or surviving thanks to other also HIF-1α responder’s antiapoptotic genes, and in this penumbra area, the fate of the affected tissue will be the results of simultaneous action pro and anti-apoptotic mechanisms.
Under this scenario a pharmacological action is required to avoid the slow but progressive neurodegenerative process that will develop, whose expansion will be worsening of the consequences of the initial insult with increased cell death. Different therapeutic approaches, including calcium channel blockers, corticosteroids, free radicals scavengers, N-methyl-D-aspartate (NMDA) receptor antagonists, EPO, and hypothermia, have been suggested as drug therapies that can be employed to treat this type of injury [110] (Fig. 3).

The hypoxic insult will establish a necrotic core (NC), and an area of penumbra with neurodegenerative changes, in which the intensity of the damage will decrease as we move away from the NC.
The role of HIF-1α is key for the stimulation of a long list of genes such as Vascular Growth Factor (VEGF), EPO, and their respective receptors (VEGF-R and EPO-R), glycolytic enzymes, transporters of both glucose and Fe (transferrin and its receptor), all involved in anti-apoptotic mechanisms of survival and energy and O2 restitution [111]. Similarly, HIF-1α induces the P-gp related with pharmacoresistant phenotype as well as membrane depolarization on neuronal cells, cardiomyocytes or doxorubicin-resistant osteosarcoma cells [112–114].
ABC-TRANSPORTERS AND NEURODEGENERATIVE DISORDERS
The BBB plays a most important role in the brain protection, by limiting the penetration of many exo-genous agents. Several mechanisms including the ABC-transporters activity are involved in this protective task. Some of them as P-gp, BCRP (Breast Cancer Resistant Protein) and MRPs (Multidrug resistance-associated protein), are highly expressed in BBB as part of these complex protective mechanisms. All of them are in charge of expelling both potentially dangerous substances and drugs from the interior to the exterior of the cells, and always directed toward the exterior of the organism. By virtue of these properties, the overexpression of the ABC-transporters will be related to different dysfunctional behaviors, mainly drug resistance as described in cancer, hematologic neoplasm’s and in refractory epilepsies [97, 115–117], but their loss of expression or function, are related with accumulation of Aβ in AD as described below.
An interesting feature of conditions of OS relapses that ROS may promote endothelial cell survival and induces an increased P-gp expression. However, ROS may also lead to increased lipid peroxidation inducing BBB disintegration, with a net loss of P-gp activity and/or expression. In this regard, the expression or functionality loss of each ABC-transporter can be involved in the accumulation of different substances that the body normally metabolizes and excretes outside its different organs and tissues, including brain. Over the last decade, a number of reports have shown that P-gp encoded by the ABCB1 gene, actively mediates the transport of Aβ peptide. It is an very important information that explain why in the advanced stages in different neurodegenerative processes such as AD as well as PD, a progressive loss P-gp expression or function has been reported [118, 119].
It is not clear even if the ABC-transporters can export each and every one of the compounds particularly accumulated in the different neurodegenerative pathologies, neither has it been identified which compounds generated in them can inhibit the function and/or the expression of these transporters. The truth is although an initial increase in the expression of P-gp has been reported in these pathologies, the most important evidence indicates that the amount of Aβ deposited in the brain is inversely related to the expression of cerebrovascular P-gp, thus suggesting a key role of P-gp in its elimination [120, 121].
Because expression and activity of P-gp can be induced pharmacologically, administration of rifampicin (a potent inductor) improved Aβ clearance from brain of patients with mild to moderate AD, and showed a lower cognitive decline, after 12 months of treatment with this antibiotic [122]. Additionally, to the mentioned about AD, P-gp could also play a role in the development of PD, characterized by the loss of dopaminergic cells in the substantia nigra and the presence of protein aggregates known as Lewy bodies (LBs), composed mainly of insoluble oligomers of α-synuclein. In this regards, P-gp can efflux of some pesticides and other environmental toxins responsible for PD onset [123, 124].
A negative correlation between ageing and the P-gp function in the BBB has been suggested in an age-dependent manner, occurring also at the level of olfactory bulbs [125]. In Huntington’s disease, it was speculated with a possible dysfunction of P-gp as a disease-causing factor, while in Creutzfeldt-Jakob disease the increase in prion protein concentration in the brain is associated with a decrease expression of P-gp at BBB vascular endothelial cells level, and both diseases share some features of neurodegenerative disorders [126].
At present, 48 different ABC-transporters have been identified in humans, which were classified into 7 different subfamilies (A to G) playing an important role in maintaining the body’s homeostasis by extruding metabolites and limiting uptake of xenobiotics. At the BBB level, they play a physiological function in tissue protection by reducing or limiting the brain accumulation of neurotoxins. Furthermore, overexpression of some of these transporters are closed associated with the pharmacoresistance to CNS acting drugs, because they contribute to the decrease bioavailability or access of drugs to brain [127].
In this regard, altered ABC transporters activity (increased or decreased) at protein expression and/or functionality level, has been associated with many neurological diseases, including epilepsy, multiple sclerosis, ALS, and AD.
The familial AD (5%of cases) is due to the over-production of Aβ from mutations in the amyloid precursor protein (APP) gene or in the AβPP processing enzymes, and consequently, a reduced clearance of Aβ from the brain as the most compelling hypothesis of non-genetic AD is purposed [128]. Additionally, a detailed review describes the role of ABC-transports in AD, where both decreases and increases of different ABC-transports as well as polymorphic genetic variants in several on these genes, were related to the severity of dementia affecting these patients. In this article, the authors suggest that a better understanding of the specific role of each ABC-transporter in AD can contribute to select the specific medication that can reverses or at least halts cognitive decline, preventing disease onset [129].
Some controversial data have been reported in reference to the relationship between P-gp and AD. Changes in ABCB1-gene expression and/or function at the BBB may not only alter the expression and function of other molecules at the BBB but also affect brain environment. The appearance of Aβ plaques in the extracellular compartment of the brain parenchyma plays a central role in the AD pathogenesis, associated with the presence of extensive OS. The exact mechanism by which a redox disbalance is producing free radicals in the AD brain is not fully understood. A vicious cycle that promotes the progression of AD has been described based on that Aβ induces ROS formation via PI3K/Akt/GSK3 and MAPK/ERK1/2 pathways, that increase Aβ production/aggregation and tau phosphorylation [130].
Additionally, the discovery of cellular prion protein (PrPC), a protein with high affinity for soluble Aβ oligomers (Aβo), demonstrated that the Aβ-PrPC coupling activates the intracellular tyrosine kinase Fyn that phosphorylates tau at residues near the amino terminus. This interaction has been postulated to impact AD pathogenesis. In neuronal culture, extracellular Aβ signaling induces the phosphorylation of intracellular Fyn in a PrPC-dependent fashion [131, 132]. PrPC is ubiquitously expressed and highly abundant in the CNS and the Aβ-PrPC complex also interacts with the metabotropic glutamate receptor (mGluR5) to activate intracellular Fyn kinase [133]. In this regard, experimentally it was recently demonstrated that reduction of Fyn activity ameliorates the development of tau-related pathology, and it might be an effective therapy for tauopathies [134].
It is well established that NMDA receptors play a central role in synaptic neurophysiology, plasticity, neuronal homeostasis, and neurodevelopment, as well as in the neurobiology of several brain diseases. It is also known that activated Fyn phosphorylates both NR2A and NR2B subunits of the NMDAR, but selectively increases NR2B trafficking and membrane stabilization resulting in increased synaptic expression and enhanced receptor transmission [135]. One interesting aspect of NMDA receptors activation is the induction of iron molecules movements into neurons mediated by the small GTPase Dexras1 via the divalent metal transporter 1 (DMT1). So, neuronal iron homeostasis likely has functional relevance after activation of NMDA receptor by Fyn. The functional relationship between iron and NMDA and AMPA receptors, as well as a potential protective role of PrPC with a regulatory iron neuronal acquisition role has been reported [136–139]. Furthermore, on speculative ground, we might consider that these data suggest an important highlight of potential new not yet described mechanisms for EPO, perhaps acting as protective factor on excitotoxicity from glutamatergic neurotransmission, and also capable to avoid iron brain accumulation.
EPO AS POTENTIAL PROTECTIVE FACTOR AGAINST NEURODEGENERATION
EPO binds to the EPO-R inducing the auto-trans-phosphorylation/activation of JAK2 kinase and starting erythropoietic and antiapoptotic mechanisms. This EPO-R signaling pathway through JAK2/STAT5/Bcl-xL have been also described to play a central role in neuroprotection against apoptosis induced by Aβ25–35 [140].
On the other hand, this signaling cascade, include the above-mentioned Fyn pathway, is also an additional kinase involved in EPO signaling pathway, by targeting STAT5 activation related with EPO-R. This closed relationship was demonstrated in genetically lacking Fyn–/–mice, that exhibit reduced Tyr-phosphorylation of EPO-R and decreased STAT5-activity [141]. Additionally, one set of ion channels as the transient receptor potential channels of canonical type (TRPC channels), as TRPC4/C5 that mediate Ca2 + entry are activated by EPO [142–144].
The amyloid hypothesis is one of the predominant hypotheses for the pathogenesis of AD including hyperphosphorylated tau protein accumulation, calcium homeostasis disruption, etc. Recently, it was suggested that TRPC channels may have a function in AD development, especially TRPC6 acting against mentioned accumulation [143]. According to this, neurons displaying higher levels of Aβ and phospho-tau had lower levels of TRPC6, than those of control neurons [145]. Furthermore, a reduced TRPC6 mRNA levels in blood was observed in patients with AD and mild cognitive impairment [146]. Interestingly, this PRTC6 channel has been regulated by recombinant human EPO pretreatment in acute renal tubular injury against ischemia-reperfusion [147]. This last result suggests that the neuroprotective effects of EPO in AD would not be mediated by its action on the role of TRPC6 and Aβ or tau-related pathology, but by its antiapoptotic mechanisms related to the hypoxic condition. A recent report showed EPO can be active on mature elytroid cells without EPO-R expression. During red blood cells death (“eryptosis”), the erythrocyte shrinkage and phosphatidylserine (PS) exposure mimic some features observed during of apoptosis in nucleated cells, activating the mechanism “find me and eat me” that activate the phagocytosis by macrophages. This PS exposure could be avoided by EPO and additionally partially prevented the activation of the macrophage system [148].
COULD EPO TO INHIBIT HYPERPHOSPHORYLATION OF TAU?
Very little information is available that can answer this question. The abnormal hyperphosphorylation of the microtubule-associated protein tau generates its destabilization that affects axonal transport and can lead to neuronal death. It was demonstrated that EPO prevented tau hyperphosphorylation through the PI3K/Akt-GSK-3β pathway in SH-SY5Y cells exposed to the Aβ peptide [149], a mechanism that is also induced by hypoxia and that activate erythropoiesis and glycolysis [150]. In a recently reported clinical study, developed in the SRM Medical college Hospital and Research Centre (SRM Institute of Science and Technology) from India, 30 patients with chronic kidney disease (CKD) and cognitive dysfunction patients and other 30 patients with CKD but without cognitive deterioration were treated with rHu-EPO by their anemic condition. All cases intellectually affected had increased Aβ, total and P-tau protein levels, and interestingly the rHu-EPO administration retrieved significantly these protein abnormalities, as well as improved neuropsychological tests scoring [151]. Additionally, the ior-EPOCIM, a Cuban variant of rHu-EPO, not only was safe, but also showed positive results in motor function, cognitive status, and mood in patients with PD [152].
Because the rHu-EPO treatment was indicated by their renal and hematological condition, it is not recommended for non-anemic patients. In this regards, in an above mentioned experimental study in rats, it was demonstrated that the altered motor function after focal cortical lesion due to CoCl2 injection, has been recovered by nasal administration of rHu-EPO, and this administration method had no hematological effects [25].
FUTURE PERSPECTIVES
From the above, some controversial situations arise that will require more study for a better understanding of the mechanisms that govern neurodegenerative processes. It is interesting to note that similar mechanisms such as hypoxia, exitotoxicity, inflammation, and OS are present in most of the mentioned pathologies, but in some there is an overexpression of ABC transporters and in others, a significant drop in its expression with accumulation of pathological compounds in the brain. These functional differences have been documented using 11C-verapamil and positron emission tomography (PET) imaging, showing increased activity in refractory epilepsy patients [153], and a significant reduction in AD patients compared to cognitively normal subjects [154].
A contradictory situation arises when analyzing that the absence of ABC-transporters in above mentioned neurodegenerative diseases such as AD, PD, etc., should have an enormous pharmacokinetic advantage, because all therapeutic drugs will have free access to the brain; however, no effective treatment has been developed yet. In turn, but with similar contradictions, it was suggested that pharmacological induction of ABC-transporters expression could eliminate the compounds accumulated in these neurodegenerative diseases, but this induction will be itself drug-resistant due to the transporter overexpression obtained.
In the opposite way, inhibition of ABC-transpor-ters, mainly P-gp, has a sensitizing effect to the usual therapies, as in the case of refractory epilepsies. The adjuvant uses of cannabidiol (CBD) to-gether with common anti-epileptic drugs has improved the pharmacological responses in refractory epilepsy patients. One of the mechanisms to explain these effects is that CBD can inhibit the P-gp mediated drug-efflux activity [155, 156]. However, recently it was reported that CBD-enriched endogenous cannabinoid extract reduced the dementia-like phenotype but not the increased vulnerability to epileptic seizures in an animal model Lafora disease [157, 160].
Interestingly, as mentioned above, we demonstrated that both CBD and EPO, two natural compounds with multi-target properties, were able to inhibit the multi-drug efflux activity of P-gp [105, 159]. In this regard, it was recently suggested that overexpression of endocannabinoid system receptors could exert neuroprotection against PD and reduced neuroinflammation in AD [158]. All these evidences are indicating that more than one mechanism could be targeted by CBD and EPO, suggesting that these compounds could have high capacity to reduce inflammation, excitotoxicity, and cell apoptosis.
In the other hand, beneficial effects of rHu-EPO, as anti-apoptotic and anti-inflammatory therapy, and potentially in SUDEP were described [159]. Additionally, severe convulsive stress mimics hypoxic conditions, and rHu-EPO not only could have a protective role, but also it has the ability to inhibit P-gp efflux activity [105]. But this potential benefic effective use of rHu-EPO could have an unfavorable effect on those pathologies that need to eliminate accumulated pathologic compounds from the brain of patients with neurodegenerative diseases. Continuous evidences are reported on the beneficial effects of EPO in neuroprotection, of which we refer to the last 15 most significant experimental studies published in 2020 (Table 3).
Selected reports of experimental evidences of neuroprotective effects of Erythropoietin reported in 2020
Taken together the information reviewed here, the hypoxia-oxidative stress-inflammation axis can generate a progressive and self-perpetuating mechanisms leading to neurodegenerative processes, characterized by neuronal depolarization, and apoptosis, also associated with progressive and severe cognitive and motor losses. In this regards, nasal administration of rHu-EPO, antioxidants, and iron chelators could be the new stratagem to neuroprotection avoiding neurodegenerative [6, 160].
The administration of drugs targeting intracellular signals that upregulate BBB P-gp in the early stages of AD has the potential to increase Aβ clearance from the brain and reduce Aβ brain accumulation [161]. Additionally, it was demonstrated that P-gp downregulation in BBB by Aβ was mediated through RAGE–NF- κB signaling pathway where the contact between astrocytes and endothelial cells is an important factor in the regulation of P-gp expression [162]. Furthermore, in a very elegant study, it was demonstrated that exposing brain capillaries to Aβ40 triggers ubiquitination, internalization, and proteasomal degradation of P-gp, explaining the loss expression of P-gp in the BBB of patients with AD [163].
Therapy by iron reduction could be a useful tool for chronic diseases associated with iron excess, such as nonalcoholic steatohepatitis, atherosclerosis, hereditary hemochromatosis, and thalassemia. In this regard, it was proposed that calibrated phlebotomy can reduce stored iron without anemia development, slowing neurodegenerative change, brain iron overload, and improving the cognitive and behavioral functions in AD [164].
According to the aforementioned, the evidence that the damage exerted on the CNS, by the triad “Hypoxia-Oxidative Stress-Inflammation” is a char-acteristic common to most neurodegenerative diseases (Fig. 4).
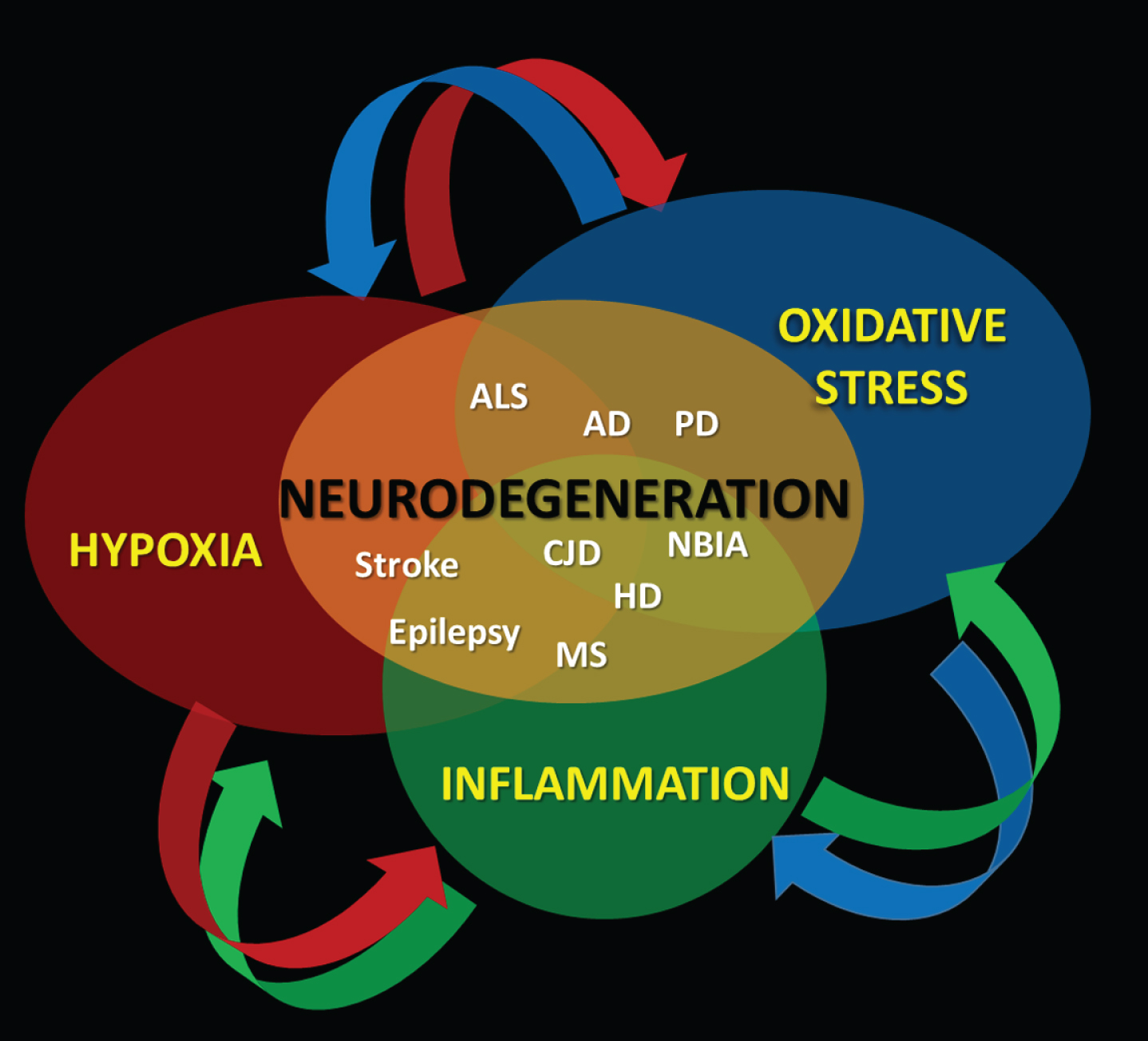
A mutual direct cause-effect relationship between hypoxia, oxidative stress, and inflammation sustains the development of neurodegeneration over time.
Finally, two recently published articles show the promising antioxidant action of ozone [165], as well as a trivalent iron chelator deferasirox [166, 167], which, added to the mentioned intranasal EPO therapeutic strategy, could slow down cognitive and/or motor impairment and improve the quality of life of these patients (Fig. 5).

Different mechanisms can lead to neurodegenerative disorders, where gliosis and apoptosis are common features.