
Abstract
Background:
Cognitive impairment (CI) is a key feature of late life depression (LLD), but the contribution of underlying neurodegenerative pathology remains unclear.
Objective:
To evaluate cognitive dysfunction in LLD relative to a sample of nondepressed (ND) older adults with matched levels of memory impairment and amyloid-β (Aβ) burden.
Methods:
Participants included 120 LLD and 240 ND older adults matched on age, education, sex, Mini-Mental State Exam, mild cognitive impairment diagnosis, and PET Aβ burden.
Results:
LLD showed higher rates of impairment relative to ND with 54.6% of the LLD sample demonstrating impairment in at least one cognitive domain compared to 42.9% of controls (H = 7.13, p = 0.008). LLD had poorer performance and higher rates of impairment on Rey Auditory Verbal Learning Test learning and memory compared to controls. In the overall sample, Aβ positivity was associated with worse performance on Logical Memory I (p = 0.044), Logical Memory II (p = 0.011), and Trail Making Test –B (p = 0.032), and APOE ɛ4 genotype was associated with worse performance on Logical Memory I (p = 0.022); these relationships did not differ between LLD and ND.
Conclusion:
LLD showed higher rates of CI driven by focal deficits in verbal learning and memory. Alzheimer’s disease (AD) biomarkers were associated with worse performance on timed set-shifting and story learning and memory, and these relationships were not impacted by depression status. These findings suggest that AD may account for a portion of previously reported multi-domain CI in LLD and highlight the potential for AD to confound studies of cognition in LLD.
INTRODUCTION
Late life depression (LLD) is a highly prevalent and disabling psychiatric disorder in older adults. Up to 5% of community-dwelling older adults meet criteria for major depressive disorder (MDD) and 8–16% report clinically significant depressive symptoms [1, 2]. LLD is associated with a host of negative outcomes in aging, including increased rates of cognitive impairment (CI), disability, incident dementia, suicide, and all-cause mortality [3–6]. CI has been identified as a key feature of LLD that contributes to increased functional disability and mental health care costs, poor treatment response and compliance, and higher rates of relapse [7–10]. Cognitive deficits in LLD have been reported in multiple domains, including executive functioning, information processing speed, episodic memory, language, and visuospatial skills [11]. However, determining the incidence of clinical CI in LLD is complicated by significant variability in methods of normative comparison and the complex relations between LLD and neurodegenerative diseases of aging, which both feature CI as a primary symptom. It remains unclear to what extend CI in LLD is related to underlying concurrent neurodegenerative pathologies.
While previous studies have explored the pattern, course, and neural substrates of cognitive dys-function in LLD, few have reported these findings using clinically meaningful thresholds for impair-ment or demographically corrected normative comparisons. Commonly cited estimates suggest that roughly 25–60% of individuals with LLD demonstrate impaired performance in at least one domain of cognitive functioning [12–15]. However, others have reported much lower estimates in the range of 0–7% [16, 17]. This variability in the literature may be accounted for, in part, by several methodological factors. There is a relative dearth of published studies that report rates of impairment using clinically meaningful thresholds. While most, if not all, widely cited manuscripts on cognitive dysfunction in LLD use the term “impairment,” most do not apply clinical standards for interpreting cognitive test performance as impaired based on normative comparison standards. Of the few studies that do utilize normative comparisons, there is significant variability in the threshold for clinical impairment, ranging from 1-2 standard deviations below the reference group mean [14–18]. Further, published impairment rates are based on relative comparison to healthy control samples, which are often small, poorly characterized, and free of common medical comorbidities that increase risk for CI. To our knowledge, there are no published rates of CI in LLD that utilize demographically corrected normative comparisons beyond age-adjusted scores, which are commonly used in clinical practice to account for variability in cognitive performance due to factors such as age, race/ethnicity, gender, and education. Additionally, no published studies of an LLD sample have applied regression-based norming procedures, which allow for more precise correction across multiple demographic factors relative to traditional norms [19].
Cognitive dysfunction in LLD often persists despite treatment and remission of other depressive symptoms, suggesting that observed impairments are more than simply state-dependent effects of depression, but rather reflect multiple neurobiologi-cal pathways including cerebrovascular, inflammatory, and neurodegenerative etiologies [14, 20–22]. Prior research examining determinants of cognitive dysfunction in LLD has focused largely on vascular [23] and inflammatory pathways [24], while relatively less is known about the contribution of APOE ɛ4 genotype and cerebral amyloid-β (Aβ) deposition, two widely recognized Alzheimer’s disease (AD) biomarkers that are associated with increased CI and decline in non-demented older adults [25, 26]. APOE ɛ4 genotype and Aβ positivity are estimated to be present in up to 30% [27] and 30–43% [28, 29] of cognitively normal older adults depending on age, respectively, and the relationships among APOE, Aβ burden, and cognitive dysfunction may not be apparent early in the course of AD pathological changes. Thus, a significant proportion of cognitively normal older adults with and without LLD likely have undetected elevated risk for CI and future decline due to AD risk factors, which are not routinely accounted for in studies of cognitive functioning in LLD.
The full impact of APOE and Aβ on CI in LLD is further obscured by the use of cognitively normal nondepressed (ND) comparison groups in the existing literature [30–32]. Given that up to 48% of LLD samples meet criteria for mild cognitive impairment (MCI), typically based on the presence of memory impairment, mild functional difficulties, and preserved global cognition [18], this approach limits our understanding of how AD risk and other age-related neurologic changes (e.g., white matter hyperintensities, cerebral atrophy) may contribute to CI in LLD samples. In the absence of comparison groups with equivalent levels of global cognitive functioning and MCI diagnosis, it is difficult to determine the degree to which previously described patterns of CI in LLD could be accounted for by AD pathology. Instead, comparison of individuals with LLD to a ND normal control sample with equivalent rates of MCI and levels of global cognitive functioning allows for more precise assessment of factors that contribute to CI in LLD and ND older adults.
The current study was conducted with two primary aims. The first aim was to assess the specific profile and rates of neuropsychological impairment in LLD relative to ND controls using regression-based demographically corrected norms and a clinically meaningful cutoff for impairment. We hypothesized that, in a sample matched on key demographic (age, sex, education) and clinical characteristics (MCI status, global cognition, and Aβ positivity), LLD participants would demonstrate a greater proportion of impairment overall and higher rates of impaired performance on tasks of executive functioning, information processing speed, and episodic memory relative to ND participants. The second aim was to assess the relative contributions of Aβ burden and APOE ɛ4 genotype on cognitive performance using a well-characterized sample of LLD and participants without LLD matched on key demographic and clinical characteristics, including MCI status and global cognitive functioning. We hypothesized that higher Aβ burden and APOE ɛ4 genotype would be associated with poorer performance on measures of global cognition and episodic memory in the overall sample.
METHODS
Participants
Participants included 120 older adults with clinician-diagnosed MDD enrolled in the Alzhei-mer’s Disease Neuroimaging Initiative –Depression (ADNI-D; ida.loni.usc.edu/login.jsp?project=ADNID) project and 240 non-depressed individuals participating in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) study. The ADNI-D project was launched in 2013, led by Principal Investigator R. Scott Mackin, Ph.D. The primary goal of ADNI Depression is to characterize the mechanisms contributing to CI and accelerated cognitive decline in LLD. The ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early AD.
The ND sample was matched two-to-one based on age, sex, education, Aβ positivity, Mini-Mental State Exam (MMSE), and MCI status (cognitively normal versus MCI). MCI status was defined by ADNI criteria described below. All participants provided written informed consent upon their enrollment in the study. The study was conducted in accordance with the Declaration of Helsinki for protection of human subjects, with procedures approved by the institutional review boards of each study site (University of California, San Francisco and University of Pittsburgh).
For LLD participants, inclusion criteria included current diagnosis of MDD, unipolar type, without psychotic features, with reported symptom severity of≥15 on the 17-item HDRS [33], and current episode lasting at least six weeks. Diagnoses of MDD were made by a licensed clinical psychologist using the Structured Clinical Interview (SCID) for the Diagnostic and Statistical Manual of Mental Disorders –IV [34], with allowable comorbidities of Generalized Anxiety Disorder or simple phobia. Other Axis I disorders or significant current neurologic illness such as epilepsy, Parkinson’s disease, traumatic brain injury, or cortical stroke, excluded individuals from participation. Those with evidence of dementia (< 25 on the MMSE) were excluded from participation as well. If inclusion criteria for the study were met, and consent was given, participants underwent a blood draw for DNA and RNA banking (with APOE genotyping), MRI imaging, and florbetapir PET imaging.
For the ND comparison group, data for the current study was obtained from the ADNI database. The ADNI study is conducted in accordance with the Declaration of Helsinki and procedures were approved by the institutional review boards of all participating sites. All participants provided written informed consent at enrollment. Exclusion criteria at baseline included: 1) the presence of MDD or significant symptoms of depression (Geriatric Depression Scale 15-Item score > 6) within the past year; 2) modified Hachinski ischemia score > 5; 3) significant neurological or psychiatric illness; 4) high dose of neuroleptics or chronic sedatives or hypnotics, antiparkinsonian medication, and use of narcotic analgesics. A lifetime history of depression (i.e., more than one year prior to enrollment) was not exclusionary. Criteria for cognitively normal participants included a MMSE > 24, Global Clinical Dementia Rating Scale (CDR) score = 0.0, and no evidence of memory impairment based on WMS-R Logical Memory II raw score adjusted for education. Clinician-rated MCI criteria for ND and LLD participants included a MMSE > 24, a Global CDR score of 0.5, and scoring below an education-adjusted cutoff score on WMS-R Logical Memory II Story A [35]. All ND participants were selected from the ADNI-2 dataset, which further delineated early (EMCI) from late MCI (LMCI) based on memory scores. For the purposes of this study, all MCI participants met criteria for ADNI-2 LMCI (Logical Memory II cutoff = 8 for those with = 16 years of education; cutoff = 4 for those with 8–15 years of education; cutoff = 2 for those with 0–7 years of education). Clinician-rated MCI subtype (amnestic versus non-amnestic) was also obtained from the ADNI dataset and was based on the presence or absence of memory impairment [36].
Procedures
After an initial screening phone interview where inclusion criteria were assessed and demographic data obtained, eligible LLD participants were referred to their nearest ADNI clinic—either University of Pittsburgh Medical Center or University of California San Francisco, Mission Bay, where they underwent core ADNI study protocol (including some cognitive tests, a physical, blood draw for DNA and RNA, and MRI and PET imaging). Measures of cognitive functioning, and neuroimaging were obtained in a single, baseline assessment period for all participants. After completing the core ADNI protocol, LLD participants were assessed at an affiliated psychiatry site, either at the University of Pittsburgh or the University of California San Francisco, Parnassus campus, for additional clinical assessment.
Regression based normative procedures
The core ADNI study protocol includes a comprehensive battery of neuropsychological assessments described below. Raw scores from primary outcome variables for each neuropsychological test were regressed on age, sex, education, and race to create demographically corrected scores using an independent sample of baseline data from cognitively normal ADNI participants (n = 821). The normative sample was 56% female, 89.4% Caucasian, and had an average age of 72.91 and 16.54 years of education. Of the 550 participants with available PET imaging, 36.9% were Aβ positive. Of the 753 participants with APOE genotyping, 30.4% were E4 carriers. Because a small proportion of the sample was non-white, all other racial/ethnic categories were combined (n = 87) to create a dichotomous variable (i.e., white versus non-white). Impairment was defined as > 1.5 standard deviations below the estimated average score of cognitively normal ADNI participants (i.e., below the 7th percentile). This threshold for impairment was selected in accordance with guidelines for labeling of performance test scores put forth by the American Academy of Clinical Neuropsy-chology [37].
Measures
Global cognition
Global cognitive functioning was assessed using the Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog). The ADAS-Cog consists of 13 subtests designed to measure cognitive dysfunctions characteristic of AD, including delayed word recall, following commands, constructional praxis, naming, ideational praxis, orientation, word recognition, auditory comprehension, word finding ability, and spoken language ability [38]. The primary outcome variable was the ADAS-Cog total score, which ranges from 0–85 with each point representing a performance error and lower scores reflecting better performance.
Memory
Verbal learning and memory were assessed using the Wechsler Memory Scale –Revised (WMS-R) and the Rey Auditory Verbal Learning Test (AVLT). The Logical Memory test was used to assess learning and memory for stories, with number of story elements recalled immediately following presentation (Logical Memory I) and after a 30 min delay (Logical Memory II) as the outcome measures [39]. The AVLT assesses learning and memory for a 15-item word list presented across five learning trials [40]. AVLT outcome variables included the sum of immediate recall for learning trials, immediate recall, and recall of the target list following a 30 min delay.
Language
Language was assessed using the Boston Naming Test (BNT) and the Category Fluency Test. The BNT is a measure of visual confrontation naming, with total number correct as the outcome variable [41]. Semantic fluency was assessed using the Category Fluency Test (Animals), with the total number of correct items generated as the outcome variable [42].
Attention/processing speed/executive functioning
Attention and psychomotor processing speed were assessed using the Trail Making Test –Part A. Executive functioning was assessed using the Trail Making Test –Part B and letter fluency. The Trail Making Test –Part B measures rapid set-shifting, with time to completion as the outcome variable [43].
Depression severity and history
Severity of depression symptoms at baseline was assessed using the 15-item Geriatric Depression Scale [44]. Depression history was collected with a self-reported retrospective measure that was verified in clinic with research coordinators. Lengths of individual depressive episodes were coded in months. The depression history retrospective measure was developed utilizing the basic structure of NIMH’s life-chart method [45].
Amyloid burden
All participants underwent florbetapir PET amyloid imaging as described previously [46]. Four 5 min frames were acquired at 50–70 min post-injection. Florbetapir images were coregistered to a concurrently acquired structural MRI scan. Regional and cortical summary standard uptake value ratios (SUVRs) were calculated using anatomical regions defined in FreeSurfer 5.3 [47]. The cortical summary SUVR was a composite of frontal, cingulate, temporal, and parietal regions relative to the whole cerebellum. Aβ positivity was defined as SUVR > 1.11 [48].
APOE ɛ4 genotype
For the purpose of this study, genotype was analyzed as a dichotomous variable defined as presence or absence of 3/4 or 4/4 genotypes, referred to as ɛ4 allele, commonly associated with increased AD risk [49].
Statistical analysis
Demographic and clinical features of LLD and ND groups were compared using Wilcoxon rank sum tests for continuous demographic and clinical characteristics and Fisher’s Exact test for categorical variables. To evaluate cognitive functioning between the two groups linear regression models were estimated and tested. Model fits were evaluated through analysis of the residuals. Skewed measures were log transformed (Trail Making Test –Part A, B). Linear regression models were used to model factors associated with cognitive performance. These factors included age, education, sex, MCI status (i.e., MCI versus cognitively normal based on ADNI criteria), Aβ burden, APOE status, depression severity, and global cognition. p-values were adjusted for multiple comparisons with a Holm correction [50] with a p-value of < 0.05 considered statistically significant. Analyses were conducted using R [51].
RESULTS
Demographic and clinical characteristics
The mean age for the overall sample was 71.15 (SD = 6.13), the mean level of education was 16.43 (SD = 2.31), and the mean total MMSE score was 29.04 (SD = 1.15). The overall sample was 65.6% female and 90.0% Caucasian. Twenty percent of the overall sample was Aβ positive using a cutoff of 1.11 SUVR, 28.7% were APOE ɛ4 + , and 33.6% met criteria for MCI. The ND group had a higher proportion of amnestic MCI (97.5%) compared to the LLD group (39.6%, p < 0.001). The LLD group had higher total CDR (p = 0.001) and reported more depressive symptoms on the GDS (p = 0.001) compared to the ND control group. The study groups did not differ on any other demographic or clinical characteristics (see Table 1).
Demographic and clinical characteristics of LLD and ND groups (n = 360)
*Matching variable; CRD, Clinical Dementia Rating Scale; WMH, white matter hyperintensity; MCI, mild cognitive impairment; MMSE, Mini-Mental State Exam; GDS, Geriatric Depression Scale; MDD, major depressive disorder; LOD, late onset depression.
With regard to group comparisons of raw cognitive performance, the LLD group performed significantly worse on measures on AVLT learning (t = –4.06., p = 0.001) and immediate recall (AVLT Immediate Recall, t = –2.90, p = 0.040). Raw performance on TMT-B and AVLT Delayed Recall were numerically lower in the LLD group, though these effects did not differ after correction for multiple comparisons. Raw performance on other neuropsychological tests did not differ between groups (see Table 2).
Neuropsychological test performance
ADAS, Alzheimer’s Disease Assessment Scale; AVLT, Auditory Verbal Learning Test; LM, Logical Memory; BNT, Boston Naming Test; TMT, Trail Making Test.
Rates of impairment using normative comparison
When CI was examined as a categorical variable (i.e., percentage of individuals falling at or below the 7th percentile for each measure), 56.4% of the LLD group was impaired on at least one cognitive domain compared to 42.9% of the ND group. The distribution of CI differed between groups, with a greater proportion of the LLD group impaired on more domains compared to the ND control group (H = 7.13, p = 0.008). A significantly greater proportion of the LLD group was impaired relative to ND controls on AVLT Learning (21.8% versus 11.2%, p = 0.006), AVLT Immediate Recall (19.5% versus 8.4%, p = 0.003), and AVLT Delayed Recall (17.8% versus 9.6%, p = 0.023, see Fig. 1). Rates of impairment did not differ between groups on any other measures (all ps > 0.05).
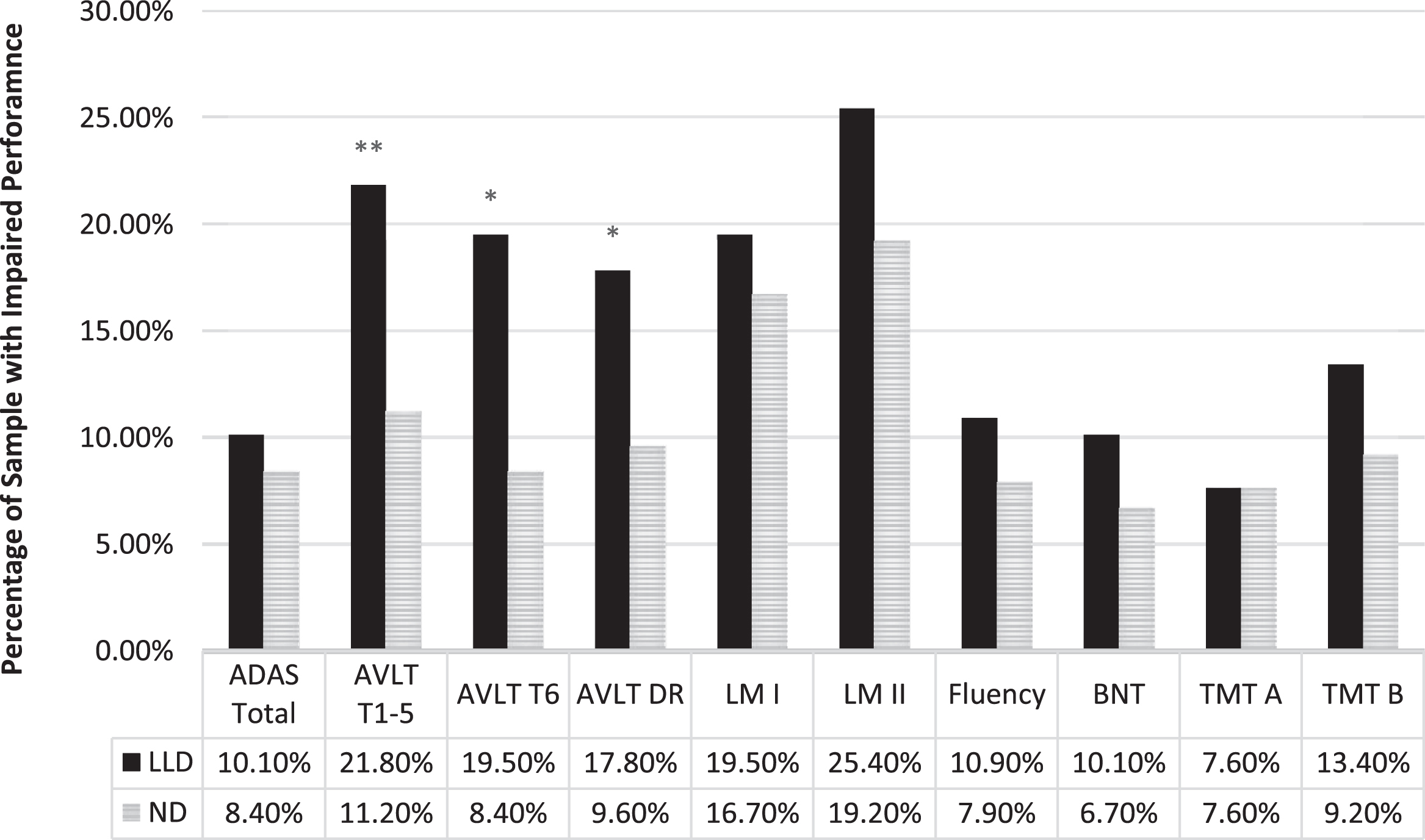
Proportion of participants with clinically significant impairments on neuropsychological test performance at –1.5 standard deviations. LLD, late life depression; ND, non-depressed; ADAS, Alzheimer’s Disease Assessment Scale; AVLT, Auditory Verbal Learning Test; LM, Logical Memory; BNT, Boston Naming Test; TMT, Trail Making Test; *p; < 0.05; **p; < 0.01; ***p; < 0.001.
Contribution of amyloid and APOE
Multiple linear regressions models were used to assess the impact of APOE genotype and Aβ burden on neuropsychological test performance in the whole sample (Table 3). After accounting for relevant demographic (age, sex, education) and clinical characteristics (MCI diagnosis, MMSE, depression severity), Aβ SUVR was associated with poorer performance on Logical Memory I (β = –3.55, S.E. = 0.18, t = –2.02, p = 0.044), Logical Memory II, (β= –0.50, S.E. = 0.19, t = –2.57, p = 0.011), and Trail Making Test - Part B (β= 0.05, S.E. = 0.02, t = 2.15, p = 0.032), while APOE genotype was associated only with poorer performance on Logical Memory I (β=–0.92, S.E. = 0.40, t = –2.30, p = 0.022). The relationship of Aβ, APOE, and these cognitive measures did not differ between LLD and ND groups. There was no difference in overall rate of CI between Aβ positive and APOE ɛ4 positive LLD participants compared to biomarker negative LLD participants (all ps > 0.05).
Summary of clinical and demographic predictors of neuropsychological test performance
ADAS, Alzheimer’s Disease Assessment Scale; AVLT, Auditory Verbal Learning Test; LM, Logical Memory; BNT, Boston Naming Test; TMT, Trail Making Test; DF, degrees of freedom; *p < 0.05; **p < 0.01; ***p < 0.001.
DISCUSSION
The present study was conducted to determine the incidence of CI in LLD using regression-based demographically corrected norms, as well as to assess the impact of Aβ burden and APOE genotype on cognitive functioning in a sample of individuals with LLD and ND controls matched on key clinical and demographic variables. Our results showed that, after matching for AD risk factors and global cognitive status, 54.6% of the LLD sample and 42.9% of the ND control sample demonstrated impairment in at least one cognitive domain. We found that LLD was associated with focal deficits in list learning and memory relative to nondepressed controls, and that increased Aβ burden and APOE ɛ4 carrier status were independently associated with poorer cognitive functioning on tests of story learning and memory and rapid set shifting in the sample as a whole, but these relationships did not differ for LLD and ND participants. Each of these results will be discussed below.
Our finding that 56.4% of the LLD group demonstrated CI is ostensibly consistent with the extant literature, though it should be noted that our threshold for impairment was more conservative (i.e., performance < 7th percentile) than that used in widely-cited estimates (< 10th percentile) [12, 14]. This result is important for several reasons. First, to our knowledge, this is the first study to use regression based, demographically corrected norms to classify CI in LLD. While others have established impairment in LLD participants via relative comparisons to standard scores derived from performance of healthy control participants [12, 14–17], this approach is vulnerable to confounding effects of uncontrolled or unknown demographic and clinical factors in the comparison sample. Thus, while the rate of impairment we report is consistent with previous studies, our findings suggest that using traditional metrics of control group performance as the standard for relative comparison may result in artificially inflated impairment rates. Second, our results demonstrate that, when matched on key demographic and clinical variables, over 40% of the ND control group also showed clinically meaningful CI in at least one domain. This is likely driven, in part, by AD biomarkers, as nearly 35% of our ND control group met criteria for MCI, 20% were Aβ positive, and 30% were APOE positive. Taken together, these findings illustrate how defining CI relative to a limited sample of healthy control participants may misrepresent the true incidence of CI in LLD, especially when neurodegenerative disease burden is unknown.
When comparing rates of impairment across neuropsychological tasks using regression-based norms we found that individuals with LLD showed greater rates of impaired performance on learning, immediate recall, and delayed recall trials for a word list episodic memory task (AVLT Trials 1–5, Immediate Recall & Delayed Recall) compared to ND participants. For each of these metrics the rate of impairment was approximately twice as high in the LLD group relative to the ND group even after matching for MCI status and overall cognitive functioning. These results highlight a particularly salient dysexecutive learning process in LLD characterized by inefficient learning that negatively impacts performance on subsequent memory trials. In contrast, when comparing cognitive performance using raw scores without regression-based normative correction, individuals with LLD showed worse performance on learning and immediate memory only (AVLT Trials 1–5 & Immediate Recall), suggesting that reliance on scores that are not corrected for demographic variables to characterize cognitive dysfunction may obscure clinically meaningful memory impairment unique to LLD. Contrary to our hypothesis, we did not find evidence for reduced performance or increased CI in the LLD group on standalone measures of executive functioning or information processing speed, which are thought to represent core features of cognitive dysfunction in LLD [12, 53], though there was indirect evidence of a dysexecutive memory profile. Given that our sample was uniquely matched on multiple AD risk factors, these more focal findings suggest that early effects of AD pathology may account for some previously described multi-domain cognitive deficits in LLD.
With regard to the contribution of AD biomarkers to cognitive performance in the whole sample, we found that Aβ SUVR predicted worse performance on measures of story learning and memory (LM I & II) and rapid set-shifting (TMT-B). APOE ɛ4 carrier status was significantly associated with worse performance solely on story learning (LM I). These findings are consistent with existing literature showing associations between Aβ, APOE, and measures of episodic memory in nondemented older adults [26, 27] and highlight the importance of accounting for the impact of AD biomarkers when evaluating aging populations, even in samples that are largely cognitively intact. Given that APOE ɛ4 genotype is present in 30% of nondemented older adults depending on age and Aβ status [27] and Aβ burden increases with age, with up to 60% of adults over the age of 85 meeting criteria for Aβ positivity [54], there is potential for AD to function as a significant confounding factor in geriatric psychiatry research samples. Though the relation of Aβ SUVR and TMT-B performance was unexpected in a sample that was largely cognitively normal, it is consistent with the executive dysfunction commonly present in clinical AD and may reflect early cognitive changes secondary to elevated AD pathology. We also report that the relationship of Aβ and cognitive dysfunction did not differ between depressed and non-depressed older adults. This importantly suggests that elevated rates of CI in LLD are independent of Aβ deposition.
Participants in our analyses were matched on global measures of cognition and proportion of MCI with the intent to make the most conservative comparison of specific CI and relationship of Aβ to cognition. While we recognize that matching the groups on MCI impacts our ability to detect memory differences between these groups, particularly on the Logical Memory subtest, we did report more impairment in LLD on a test of list learning. We interpret these results to largely reflect an impaired learning process in earlier trials demonstrated by the LLD group that may be more evident in list learning paradigms than for story learning. However, we acknowledge that based on our matching criteria a similar deficit in Logical Memory may have been obscured. Additionally, we found that in the ND sample, clinician diagnosed MCI was more likely to be rated as amnestic than in the ADNI-D sample despite identical criteria for MCI diagnosis. In the ADNI, the MCI participants are almost always rated as amnestic, in part because the ADNI was designed to capture memory impairment characteristic of early AD, as opposed to other cognitive phenotypes (e.g., dysexecutive, multi-domain). However, as our results show, multi-domain and non-amnestic MCI were present in the ADNI ND sample but not captured by clinician rating. Even in participants categorized as cognitively normal by ADNI criteria, there is still significant CI in non-memory domains. These findings highlight the high degree of variability in clinician ratings of MCI, even when following established diagnostic criteria, which has the potential to negatively impact studies of aging and cognition. Given evidence from prior studies showing improved sensitivity, specificity, validity, and reliability of more comprehensive neuropsychological diagnostic methods with actuarial decision-making relative to ADNI MCI criteria [55–57], future research is warranted to examine the potential impact of MCI diagnostic criteria on geriatric psychiatry research.
Though these results are robust, there are some limitations to state. The cross-sectional nature of the data makes it impossible to speculate about longitudinal change. For example, this highly educated sample may have already experienced cognitive decline but remains within a normative range thus appearing unimpaired. The cross-sectional design also is uninformative with respect to future cognitive decline in either group. Further, it is possible that the relative contribution of AD risk to CI in this sample will increase over time as underlying neurodegenerative processes progress. Thus, it will be important for future research to examine longitudinal trajectories of cognitive change in LLD. Although matching on key demographic and clinical variables is considered a strength of this study, matching on MMSE and MCI status, which is based in part on memory performance, may have obscured or minimized memory impairments secondary to LLD. Finally, our study sample and normative reference sample were both highly educated and predominantly Caucasian, which limits the generalizability of our findings to racial/ethnic minorities and individuals with lower levels of education. Ideally, a normative reference sample would include equal proportions of representation across all levels and categories of variability; however, we were restricted to broader, less specific categories (e.g., white/non-white) due to limited representation in low education and racial minority groups.
To our knowledge this is the first study to assess rates of neuropsychological impairment in LLD relative to ND controls using regression-based demographically corrected norms and first to investigate the impact of in vivo Aβ burden and APOE on cognitive functioning in a large sample of clinically depressed and ND older adults matched on key demographic and clinical variables, including AD biomarkers. When controlling for AD biomarkers, LLD was associated with a ∼30% higher rate of CI relative to ND participants. Though the overall rate of CI in LLD was consistent with previous findings, our results suggest that the profile of LLD neurocognitive dysfunction may be more focal than previously reported. We found increased rates of impairment on measures of list learning and memory that support a pattern of memory impairment in LLD primarily characterized by a dysexecutive, retrieval-based memory profile. However, we acknowledge that controlling for MCI and controlling for multiple comparisons may have limited the results to the most prominent differences.
Our results also highlight the confounding role of AD processes in cognitive functioning in LLD, which have not been well characterized in prior studies and may account for more diffuse patterns of impairment reported in the extant literature. Our findings underscore the potential for AD risk to impact results of clinical trials in LLD and other geriatric psychiatry research examining cognition as an outcome. Collectively, these findings offer important clarification on the relative contributions of depression, Aβ burden, and APOE genotype to CI in nondemented older adults and serve as an important step in disambiguating the role of AD as a confound in the assessment of CI in LLD.
Footnotes
ACKNOWLEDGMENTS
We acknowledge Avid Radiopharmaceuticals for providing Florbetapir for this study.
This research was also supported in part by the Ray and Dagmar Dolby Family Fund.
Data collection and sharing for the ADNI Depression Project was funded by the National Institute of Mental Health (NIMHR01MH098062-01A1), and through contributions from Eli Lilly and Company. The grantee organization is the University of California, San Francisco, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI Depression data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.;Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (![]() ). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.