
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by the presence of neuritic plaques and neurofibrillary tangles. The impaired synaptic plasticity and dendritic loss at the synaptic level is an early event associated with the AD pathogenesis. The abnormal accumulation of soluble oligomeric amyloid-β (Aβ), the major toxic component in amyloid plaques, is viewed to trigger synaptic dysfunctions through binding to several presynaptic and postsynaptic partners and thus to disrupt synaptic transmission. Over time, the abnormalities in neural transmission will result in cognitive deficits, which are commonly manifested as memory loss in AD patients. Synaptic plasticity is regulated through glutamate transmission, which is mediated by various glutamate receptors. Here we review recent progresses in the study of metabotropic glutamate receptors (mGluRs) in AD cognition. We will discuss the role of mGluRs in synaptic plasticity and their modulation as a possible strategy for AD cognitive improvement.
Keywords
INTRODUCTION
Glutamate, as a critical excitatory neurotransmitter and primary player in the synaptic plasticity, exerts its physiological functions through binding to its specific receptors [1]. Four classes of receptors have been shown to mediate glutamate transmission: N-methyl-D-aspartate receptors (NMDAs), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAs), kainate receptors, and metabotropic glutamate receptors (mGluRs). NMDAs and AMPAs, as well as less abundant kainate receptors, belong to ionotropic glutamate receptors (iGluRs), which are ligand-gated ion channels and require glutamate for conducting rapid excitatory neural transmission [2, 3]. mGluRs require a cascade of signals to transmit a glutamate stimulus from the cell surface to the nucleus [4]. It has been well documented that the levels of glutamate present at any given time in the brain must be optimally maintained for a normal function of the nervous systems [5]; coordination of various iGluRs and mGluRs controls the major and vital functions of glutamate synapses. Because of its indispensable roles in learning and memory as well as synaptogenesis, even the slightest dysregulation in glutamate signaling can inadvertently lead to cognitive dysfunction [6, 7]. Reduced glutamate neurotransmission has been found in association with impaired learning and memory in Alzheimer’s disease (AD) patients [8, 9]. Both iGluRs and mGluRs are found to mediate synaptic failures in AD [10–14]. Memantine, known to modulate NMDA receptor function, is currently prescribed for AD treatment while Ampakines, known as positive allosteric modulators of AMPA receptors, were explored for memory storage and consolidation in AD. In this review, we focus on the action of mGluRs in modulating synaptic transmission in AD, an area that is under investigated.
mGluRs STRUCTURE AND ACTIVATION MECHANISM
mGluRs belong to the class C of G-protein coupled receptors (GPCRs), which have a large extracellular ligand-binding domain, a typical heptahelical membrane structure, and variable lengths of a C-terminal tail. Membrane-bound GPCRs are activated by extracellular or allosteric binding of a ligand following which the coupled G-protein signaling ensues, relaying signals to various downstream effector molecules [15]. Akin to other class C GPCRs, mGluRs structurally comprise of an extracellular domain, bearing the orthosteric binding site for glutamate, and can dimerize constitutively [16, 17]. The conformational changes, occurring in the extracellular domain in response to ligand binding, are transmitted to the helices in the intracellular transmembrane (TM) domain of the receptor that facilitates receptor binding to a G-protein, a process that is popularly known as G-protein coupling of mGluRs [18, 19]. In addition to the orthosteric binding of glutamate, mGluRs can also bind certain molecules allosterically. The allosteric sites are more receptor subtype-specific and can sensitize or desensitize the receptor. Therefore, allosteric binding molecules can be either allosteric agonists, allosteric antagonists, or allosteric modulators [positive allosteric modulators (PAMs) and negative allosteric modulator (NAMs)] [20, 21]. While allosteric agonists can activate the receptor even when there’s no glutamate binding, allosteric modulators fail to do so. Upon binding at the allosteric site of the receptor, PAMs and NAMs indirectly modulate the effects of glutamate transmission [22].
CLASSIFICATION AND EXPRESSIONS OF mGluRs
There are 8 different types of mGluRs, which can be further classified into 3 groups (Table 1) based on their signaling events, sequence similarity, and pharmacological effects [23–25]. The group I mGluRs consist of two members, mGluR1 and mGluR5. The group II also has two members, mGluR2 and mGluR3. The group III has four members: mGluR4, mGluR6, mGluR7, and mGluR8.
Three groups of mGluRs and their signaling pathways
Group I mGluRs are predominantly present at the excitatory synapse (Fig. 1), but their locations may vary based on the type of different isoforms [26]. The distribution of mGluR1 in the hippocampus and cerebellum has been extensively studied, and its expression correlates with the establishment of the glutamatergic neurotransmission system and synaptogenesis in these brain regions [27]. mGluR5 receptor is expressed in the hippocampus, amygdala, olfactory bulb, dorsal striatum, nucleus accumbens, and lateral septum [28, 29]. At the synapse, both mGluR1 and mGluR5 appear to be mostly located at postsynaptic terminals, and Homer family proteins regulate the positioning of these receptors close to second messengers [30]. Upon activation, they follow the Gq route of G-protein signaling and activate phospholipase C-β (PLC-β), thus generating inositol triphosphate (IP3) and diacylglycerol (DAG) to trigger intracellular calcium mobilization and PKC activation [31]. Group I mGluRs can sometimes act independent of G-protein-coupled signaling by phosphorylating other target proteins such as transcription factors and ion channels [32]. MAPK/ERK and mTOR/P70 S6 kinase may also be activated by type I receptors and contribute to synaptic plasticity [33].
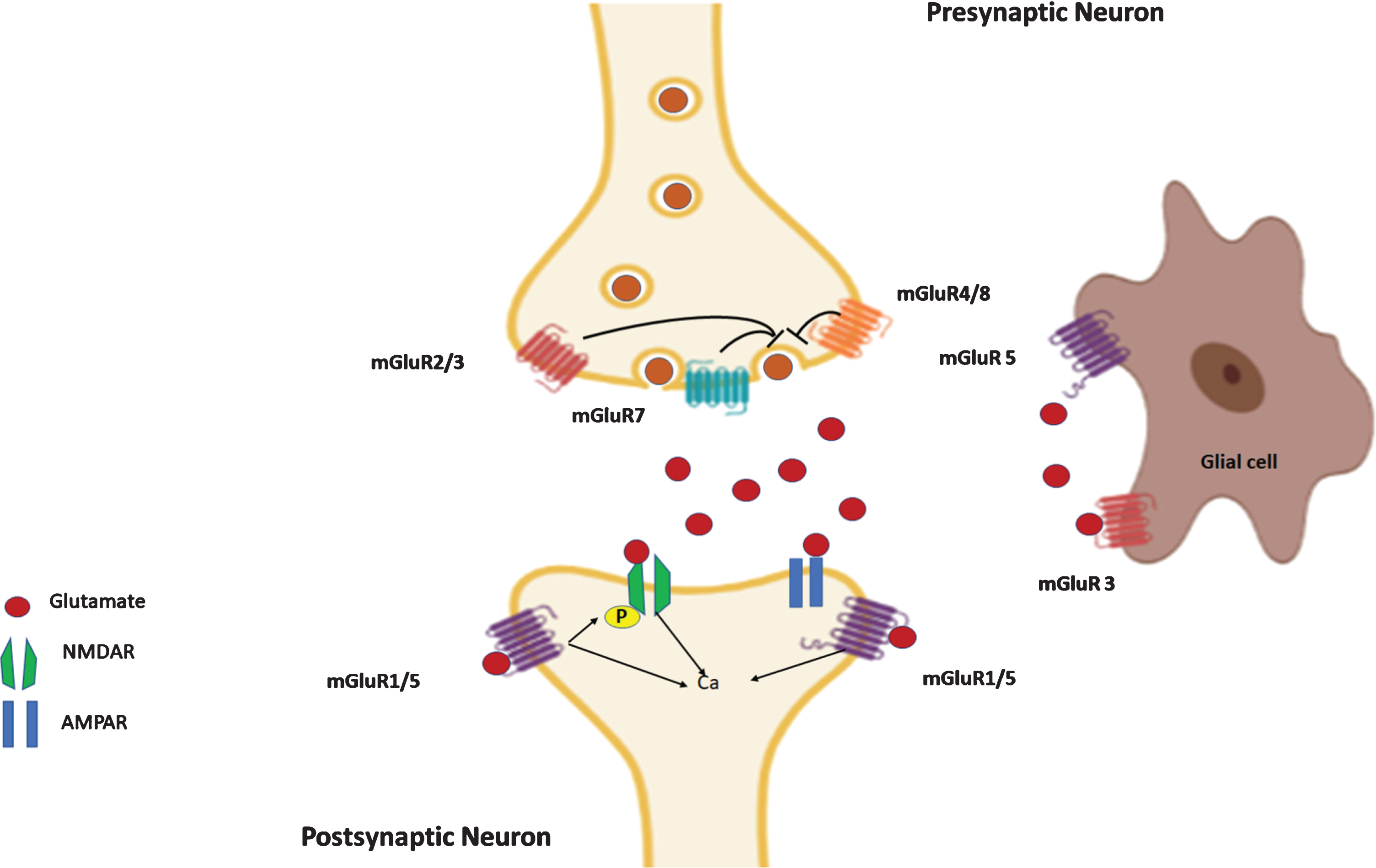
Distribution of different groups of mGluRs at glutamatergic synapse. Group I mGluR1/5 are mostly localized postsynaptically. mGluR2/3 are mainly in presynaptic site, located perisynaptically, but in certain brain regions group II mGluRs are localized in postsynaptic sites. mGluR4/7/8 are found near presynaptic active zones. The presynaptic group II and III mGluRs have an inhibitory effect on the release of glutamate from the synaptic vesicles as well as on calcium signaling. The postsynaptic group I mGluRs promote a rapid calcium influx upon their activation and contribute toward LTP induction. The glial cells (astrocytes, microglia, oligodendrocytes) also reportedly express mGluR1/5, mGluR2/3, and mGluR4/7/8 that play a significant role in glutamate neurotransmission, glial cell proliferation and release of cytokines.
Neuronal group II mGluRs are found both pre- and postsynaptically in broad brain regions including the limbic system, sensory neurons of cortex, dorsal and ventral striatum, amygdala, and thalamus [34–36]. Presynaptic group II mGluRs are often localized to the preterminal region of axons of glutamatergic neurons, away from the active zone, and are viewed as autoreceptors controlling the release of glutamate in response to excessive glutamate release [37, 38]. Presynaptic group II mGluRs have also been found on GABAergic neurons, and their activation negatively feeds back on GABA release [39]. In layer 4 of primary sensory cortical neurons, mGluR2 has a postsynaptic localization and functions as a negative regulator of glutamate transmission [40]. Immuno-electron microscopic results show that most of mGluR3 is located in presynaptic elements and not closely associated with glutamate and GABA release sites, but about 20% neuronal mGluR3 is found in spines of dentate neurons, away from glutamatergic synapses [38].
Group II mGluRs regulate hippocampal synaptic plasticity by inhibiting glutamate release and exerting a suppressive effect on LTP. This attenuation of glutamate release is due to activation of their coupling to Gi/Go proteins, in which the activated receptor acts as a guanine nucleotide exchange factor by replacing GDP with GTP in Gαβγ trimeric complex [41]. GTP-bound Gα, after being dissociated from Gβγ, separately activates their respective downstream signaling enzymes. They negatively regulate the expression of adenylate cyclase (AC) thus reducing intracellular cAMP levels and inhibiting PKA, or decreasing voltage-gated Ca2 + channel activation [15, 42], thereby inhibiting glutamatergic neurotransmission. They are activated by significant synaptic glutamate release or astrocytic glutamate [39].
Group III mGluRs have been found predominantly in presynaptic active zones, with the exception of mGluR6, which is exclusively present at postsynaptic sites in the retina and transmits signals to cGMP phosphodiesterase [43]. Among the group III mGluRs, mGluR7 is most prominently expressed in neuronal cells of cortex, olfactory bulb, hippocampus, thalamic nuclei, cerebellum, and is located within the presynaptic active zone of both glutamatergic and GABAergic synapses, acting to inhibit glutamate or GABA release [44, 45]. mGluR7 has 5 spliced forms with mGluR7a most broadly expressed. In globus pallidus, striatal, and retinal synapses, mGluR7 is not only present in pre-synaptic synapses but surprisingly found in postsynaptical sites as well [46, 47]. In the CNS, mGluR4 is mostly present in the cerebellum, but is also detected in the cerebral cortex, olfactory bulb, hippocampus, lateral septum, septofimbrial nucleus, striatum, thalamic nuclei, lateral mammillary nucleus, pontine nuclei, and dorsal horn [48]. mGluR4 shares a similar expression profile to mGluR7, albeit in reduced or comparable levels. Expression of mGluR8 is more restricted in the sense that it is only found in some cortical areas and the thalamus with reduced expression in olfactory bulb, neocortex, hippocampus, amygdala, and cerebellum, and its expression level is normally lower than that of mGluR4 and mGluR7 [49].
Sharing about 70% sequence homology with Group II mGluRs, group III mGluRs also signal through a distinct pathway by coupling to Gi/o proteins [50]. Besides maintaining an equilibrium between their active and inactive states, these mGluRs also exhibit a constitutive activity independent of agonist binding. Consequently, they negatively impact excitatory neurotransmission [51].
While most mGluRs are mainly localized in either pre- or postsynaptic neuronal sites, multiple mGluRs are expressed by non-neuronal cells. For example, immunohistochemical staining shows expression of mGluR1 in astrocytes, microglia, and oligodendrocytes while mGluR5 is mainly found in microglia [52–54] and mGluR5a in developmental oligodendrocyte precursor cells (OPCs). Among group II mGluRs, mGluR3 is more expressed by glial cells than neurons [55]. Early studies showed that mGluR3, but not mGluR2, is found postsynaptically and on astrocytes [38, 57]. Glial mGluR3 activation regulates excitatory amino acid transporters (EAATs) [58], such as increasing the production of glutamate transporters GLAST and GLT-1 [59]; glial mGluR3 also controls the production of neurotrophic factors like TGF-β [60], and is involved in the astrocytic-neuronal communication [55]. More recently, mGluR3 was also detected in microglia, OPCs, and oligodendrocytes [52]. Moreover, low levels of mGluR2 are detected in mature MBP+ oligodendrocytes in early developmental stages but downregulated in the adult [61–63]. Group III mGluR4, mGluR7, and mGluR8 have also been found in microglia, reactive astrocytes, and oligodendrocytes [64, 65]. Functionally, mGluRs expressed by glial cells contribute to synaptic function in the brain, particularly in the context of glial uptake and synthesis of glutamate. It has been shown that the deletion of mGluR5 only in astrocytes impairs high-frequency glutamate uptake [66]. Activation of certain mGluRs in glial cells is shown to alter synaptic function and protect against neurodegeneration [63].
PHYSIOLOGICAL ROLE OF mGluRs
Synaptic plasticity is the neural process in which neuronal networks and their connections are either strengthened or weakened at appropriate synapses through calcium waves. Synaptic strength can be reflected through measures of long-term potentiation (LTP), referring to improved and extended signal transmission, and long-term depression (LTD), which is the inhibition of excitatory neurotransmission [25]. LTP and LTD largely govern motor control, memory, and learning in most of the brain areas studied so far. Acting at either pre- or postsynaptically, mGluRs regulate synaptic plasticity by controlling neuronal excitability or neurotransmitter release (Table 2). For instance, deficiency in mGluR1 in mice decreases LTP and cognitive functions [67–69], while deficiency in mGluR5 decreases CA1 LTP by altering a component of NMDA receptors and restraining synaptic flexibility [70, 71]. mGluR7 is also required for LTP induction at hippocampal SC-CA1 synapses through decreasing GABAergic synaptic transmission in CA1 pyramidal cells [72]. Their sustained effects regulate neuronal plasticity, maintain CNS function, and contribute to pathophysiological conditions related to anxiety, fear extinction, and spatial working memory [73]. It was also noted that mGluR2 is actually required for the development of mossy fiber synapses [74].
mGluRs in the control of synaptic plasticity
Postsynaptic functions
By localizing at dendritic fields or peri-synaptic sites surrounding the ionotropic receptors, group I mGluRs (mGluR1/mGluR5) are shown to modulate synaptic strength through redistribution of AMPA and NMDA receptors at excitatory synapses, but they are not normally needed for the initiation of LTP [75]. In subiculum areas, mGluRs are shown to independently induce LTP based on the sliding-threshold model of synaptic plasticity [76]. It is shown that induction of LTP and LTD in CA1-subiculun synapses depends upon the relative activation states of NMDARs and mGluRs. In regular spiking cells, this threshold shifts toward mGluR activation in response to low-frequency afferent stimulation that triggers calcium signaling and masks NMDAR-mediated LTD [76, 77]. Reduced mGluR activity decreases LTP amplitude and prevents neuronal excitotoxicity under normal physiological conditions [78]. Postsynaptically localized mGluRs appear to trigger LTD by enhancing endocytosis of ionotropic AMPA receptor subunits (i.e., GluR1 and 2 in CA1 and GluR2 only in cerebellar Purkinje cells), leading to a long-term reduction in the number of postsynaptic surface AMPAR [79]. In striatal regions, postsynaptically localized type I mGluRs are found to mediate LTD initiation but not its maintenance [80].
Presynaptic functions
At the presynaptic site, mGluRs can regulate synaptic plasticity by affecting intracellular calcium levels to modulate neurotransmitter release, mostly by altering membrane potential and internal storage of calcium ions [81]. In many published studies using mGluR agonists, activation of presynaptic group II or III mGluRs inhibits excitatory synaptic transmission at specific synapses by reducing glutamate release [48]. 4C3HPG, a group II agonist (and group I mGluR antagonist), was found to inhibit LTP expression in the dentate gyrus in vivo. However, transgenic mice and more specific drugs must be used to tease apart the individual contributions of mGluR2 and mGluR3 to synaptic changes. mGluR2 knockout mice exhibit normal basal synaptic transmission and LTP, but impaired LTD, at mossy fiber-CA3 synapses in the hippocampus [83]. In vitro, activation of mGluR3 receptors by group II agonist NAAG leads to inhibition of hippocampal LTP [84], and induction of chemical LTD [85]. mGluR3 antagonism via β-NAAG blocked LTD but not LTP in the dentate gyrus in vivo. It has been suggested that presynaptic and postsynaptic mGluR3 have distinct roles in synaptic plasticity: postsynaptic mGluR3 are necessary for LTD while presynaptic mGluR3 modulates both LTP and LTD [86].
Activation of group II and III mGluRs (presynaptically localized) can also lead to the development of LTD through G protein-coupled receptors by dissociation of Gαi with Gβγ. The released Gαi and the second messenger nitric oxide are necessary steps in the presynaptic LTD cascade, while Gβγ directly bind voltage-dependent Ca2 + channel to reduce calcium influx to decrease transmitter release [87, 88]. It has also been shown that Gβγ binds directly to the C-terminus region of the SNARE protein SNAP-25, and this binding induces LTD of vesicular glutamate release [89]. On the other hand, inhibition of group III mGluRs promotes NMDA and protein synthesis-dependent induction of long-lasting LTP at the otherwise LTP deficient (SC) CA2 synapses [90]. These receptors can also activate other signaling pathways, including mitogen-activated protein (MAP) kinase and PI-3 (phosphatidylinositol-3) kinase pathways [15, 92].
Group II mGluRs have been shown to associate with a number of interaction partners, including calmodulin [15], β-arrestins [93], glutamate receptor-interacting protein [94], protein interacting with PRKCA 1 [94], Na+/H+ exchanger regulatory factor 1 and 2 [95], protein kinase A [96], protein phosphatase 2C [97], tamalin [98], and Ran binding protein in the microtubule-organizing center [99]. In addition, group II mGluRs directly and indirectly interact with members of other mGluR classes. Recent spectroscopic and biochemical studies show that mGluRs can heterodimerize, which has important functional implications. mGluR2 can form dimers of equal affinity to members of group II mGluRs, and with lower affinity with group III mGluRs [100]. mGluR2/4 heterodimeric complexes are found to differentially regulate the responses to various positive or negative allosteric modulators of mGluRs [101]. In the hippocampus, mGluR2 heterodimerizes with mGluR7, and this complex exhibits both fast kinetics and cooperativity that boosts the affinity and efficacy of each subunit, priming the receptor for activation even in absence of agonist. These properties appear to make mGluR2/7 uniquely suited to activate in response to synaptic glutamate [102]. Finally, mGluR3 and mGluR5 interact synergistically through cross-talk of signaling pathways, where it was found that mGluR3 activation is required for maximal mGluR5 signaling, and mGluR5 activation is required for the induction of mGluR3-dependent LTD in the PFC [103].
Other regulatory functions
In addition to the effect on synaptic plasticity, mGluRs present in the hypothalamus and pituitary can also regulate neuroendocrine functions by controlling the secretion of various hormones [104]. For example, gonadotropin-releasing hormone (GnRH) neurons receive input from GABAergic afferents via GABAA receptors, and presynaptic mGluRs can regulate GABAergic neuronal GABA release to GnRH neurons and control GABAA receptor-mediated postsynaptic currents in GnRH neurons for generation/modulation of rhythmic GnRH release [105, 106]. In the spinal cord, all mGluRs except mGluR6 are expressed within the nociceptive pathways: group I mGluRs are pro-nociceptive while group II and III mGluRs are anti-nociceptive [107]. mGluRs also play a role in the auditory system by enhancing the currents emanating from voltage-gated K+ channels in the neurons responsible for time coding in the auditory brainstem. The cochlear regions show an intense expression of group I mGluRs and a region-specific expression of the other two groups [108]. While many comprehensive reviews have discussed these aspects [109–113], this review will focus on mGluRs in AD cognitive functions.
PIVOTAL ROLE OF mGluRs IN AD PATHOPHYSIOLOGY
The brain usually shrinks to some degree in healthy aging, but surprisingly, does not lose large numbers of neurons under healthy condition [114, 115]. In AD, erroneous synaptic activity precedes memory loss, corroborated by the fact that the synaptic density at hippocampal and cortical regions is reduced. A plethora of literature have documented various synaptic dysfunctions in AD brains [116–121]. Synaptic decline in AD animal studies is often reflected by a significant reduction of LTP or increase in LTD in AD mouse models [122–124]. A remarkable and early synaptic decline in AD has also been associated with altered glutamatergic systems [125]. Glutamatergic synapses, mainly involving NMDA and AMPA receptors, are directly relevant to LTP/LTD changes. Upon LTP induction, AMPA receptors are recruited at synapses, and dendritic spines are increased in number while LTD leads to synaptic loss and spine shrinkage [126, 127].
AD, as one of the most prevalent forms of age-dependent progressive neurodegenerative disorders in patients experiencing cognitive deficits, behavioral changes, and personality alterations, is best recognized as a significant memory loss, occurring even in the mild cognitive impairment stage [128]. Gradual structural and functional damages to the hippocampus and cortical regions, resulting from excessive Aβ aggregation and deposition, neurofibrillary tangles (NFT), and progressive neuronal loss, are believed to cause synaptic dysfunctions in AD brains [14, 129]. Neuronal loss could stem from glutamate excitotoxicity, which is a process by which abnormal glutamate levels exert an excitatory shock to neurons and can cause cell death. When glutamate receptors are overactivated, a massive surge in calcium influx occurs and perturbs normal cellular physiological processes, causing changes in mitochondria function, intracellular and extracellular signaling cascades, and oxidative stress [130, 131]. Depending upon the cellular stage and state of neurons and external factors, the contribution of different types of iGluRs and mGluRs towards excitotoxicity may vary relative to each other [132]. Under normal physiological conditions, excitotoxic levels of glutamate at synaptic clefts can be prevented by rapid conversion of glutamate to glutamine in glial cells. Presynaptic neurons then uptake glutamine, after which it is converted to glutamate by intracellular glutaminase, and normal synaptic signaling events can thus be maintained [133]. Abnormal Aβ levels in the brain have been shown to regulate glutamate levels at synaptic clefts [134, 135].
Aβ peptides, namely Aβ40 and Aβ42, are generated from large transmembrane amyloid-β protein precursor (AβPP), sequentially cleaved by β- and γ-secretases. Aβ exists in the monomeric form, but can self-aggregate to form dimer and oligomers, and even higher order structures called Aβ fibrils with aging [136, 137]. Amyloid plaques mainly consist of Aβ soluble oligomers and insoluble aggregates. Under physiological conditions, Aβ is released at synapses of neurons and may promote normal memory and synaptic plasticity, especially LTP at picomolar levels [138]. Augmentation of LTP by increase of cGMP levels may also require Aβ at physiologically-controlled levels [139]. Aβ peptides are shown to stimulate presynaptic α7 nicotinic receptors to induce synaptic plasticity [140]. APP KO mice exhibit deficits in memory and LTP, emphasizing the fact that Aβ may have a modulatory effect on synaptic plasticity and neurotransmission [141, 142]. On the other hand, under pathological conditions, the soluble form of Aβ oligomers is shown to be highly neurotoxic [143]. This is likely because pathologically increased Aβ oligomers block uptake of glutamate thereby leading to elevated glutamate concentrations in synaptic clefts, and cause neuronal hyper-excitation due to overstimulating NMDA receptors [144]. This event inhibits LTP by desensitizing NMDA receptors, resulting in synaptic depression and the eventual loss of synapses and neuronal death [145]. This Aβ-mediated synaptic loss can occur early in the course of AD and is also accompanied by oxidative stress and tau phosphorylation. Examination of postmortem brains of AD patients reveals excessive free radical generation and altered nitric oxide cascade in correlation with loss of synapses [146–148]. Hence, a precise regulation of Aβ production and release at synapses is required for maintenance of synaptic plasticity [149–152].
Relevance of mGluRs in AD synaptic dysregulation
Active synaptic transmission requires not only functional iGluRs (mostly glutamate and AMPA receptors) but also mGluRs [153]. Aβ oligomers have been shown to contribute to synaptic deficits, either directly or indirectly, by interacting with iGluRs as well as mGluRs [154]. The effect of Aβ oligomers on iGluRs in AD synaptic deficits has been well documented [155–158] and this review focuses recent publications on mGluRs in this aspect. Altered mGluR levels or activities have been associated with AD pathogenesis by changing glutamatergic synaptic functions [10]. Each mGluR has been explored in various conditions and we will dissect their contributions to AD synaptic dysfunctions (Table 3). Following sub-sections will summarize our current knowledge.
mGluRs in Alzheimer’s disease
Group I mGluRs inAD
In AD research, the role of group I mGluRs (mGluR1 and mGluR5) has been studied to a good extent, and their levels are found to be reduced in AD brains. Studies using postmortem brain samples of AD patients have revealed reduced levels of these two mGluRs in the hippocampus and cortex, and changes are region- and receptor subtype-specific. For example, mGluR1 levels are reduced in the hippocampal CA1 region in AD brains [159]. Interestingly, a study in Japanese population using positron emission tomography (PET) imaging did not find any significant difference in the mGluR1 availability between AD and control groups, especially in the early stages [160]. However, they did not rule out possible changes associated with the progression of AD, which will require further longitudinal follow-up. Indeed, in a separate study, both mGluR1 and mGluR5, quantified by radioligand binding assays, were found to be significantly decreased in pure AD cerebral cortex [161]. In particular, the decrease in mGluR1 levels occurred in cases with early AD changes. Moreover, mGluR1 levels in pure AD cases were lower than those obtained in patients with Lewy body dementia and its reduction is correlated with progression of illness, indicating a strong correlation to neuropathological changes. Expression levels of the phospholipase Cβ1 (PLCβ1) isoform, which acts as the effector of group I mGluRs, is also significantly decreased in AD [161]. mGluR5 levels have been found to be significantly decreased in 16-month-old Tg-ArcSwe (an AD mice model) when compared with control mice [162]. After the compound [18F]FPEB was developed for specific binding to mGluR5, [18F]FPEB-PET was conducted in 16 mild AD patients and 15 age-matched normal controls, and reductions of mGluR5 binding in the hippocampus of early AD was revealed [163]. Aβ oligomers form a complex with cellular prion protein (prp) and mGluR5, and this complex alter signaling involving Fyn and PyK2. Consequently, neuronal functions including reduction of dendritic spines and synaptic memory are disrupted, and blocking this interaction is explored as a potential strategy for AD treatment.
Since Group I mGluRs/PLC signaling is downregulated and desensitized in AD cases and worsen with progression of AD pathologies, it is not surprising to speculate that group I mGluR dysfunction is likely one culprit for the cognitive impairment and dementia in pure AD patients. Activation of group I mGluRs may need to be explored as a potential strategy for reversing pathological changes in these dementia patients. Encouragingly, a couple of studies show that activation of group I mGluRs accelerates non-amyloidogenic processing of AβPP by α-secretase, potentially protecting from Aβ-mediated neurotoxicity [164, 165]. Thus, dysfunction of group I mGluRs is likely associated with AD disease progression and contributes to AD cognitive failure.
Group II mGluRs in AD
In the hippocampus of AD patients, mGluR2 expression is increased in the CA1 and CA3 pyramidal neurons compared to age-matched controls, and immunostaining of mGluR2 exhibited significant overlap with hyperphosphorylated tau-positive neurons [166], implying a role of mGluR2 in the pathogenesis of AD. The dual mGlu2/3 receptor agonist LY379268 was found to induce ERK activation in rat primary cortical neurons [92], and this activation directly increased tau phosphorylation in primary neurons [167]. Since ERK is chronically elevated in AD neurons and is shown to phosphorylate tau in regions of the AD brain that harbors NFT [168], activation of mGlu2/3 receptor may facilitate tau pathology. However, the ERK pathway functions in cell proliferation, differentiation, and survival—indeed, it is commonly implicated in human cancers—and thus while NFT have historically been seen as detrimental to neurons, some authors suggest that tau phosphorylation represents a neuronal compensatory response to stress and is associated with survival [169, 170].
On the flip side, group II mGluR activation is protective for neurons against excitotoxicity via the inhibition of glutamate release [171–173]. Recent efforts have been directed toward teasing apart the individual contributions of mGluR2 and mGluR3 to neuronal survival and synaptic function and dysfunction in the setting of AD. Overall, AD appears to be associated with reductions in mGluR3 but increases in mGluR2 function. activation of mGluR2, but not mGluR3, inhibits GABA release and becomes harmful to neurons subjected to an excitotoxic insult; in mixed cortical cultures, the addition of group II agonist LY379268 reduced K+ depolarization-evoked GABA release in neurons cultured from wildtype or mGluR3 KO, but not mGluR2 KO mice [174]. Also, LY379268 treatment protects cultured neurons from NMDA-mediated excitotoxicity only when astrocytic mGluR3 is present, an effect that can be amplified by the absence of neuronal mGlu2 receptors [174]. Furthermore, the mGluR2 activation by the PAM LY566332 potentiates Aβ toxicity in vitro regardless of whether glial mGlu3 receptors is present, an effect that is abrogated when neurons lack mGlu2 receptors [175]. It is further shown that group II agonist LY379268 is neuroprotective against Aβ toxicity in mixed cultures, likely through a paracrine mechanism mediated by TGF-β1; Anti-TGF-β antibodies also eliminate the neuroprotective action of LY379268 in cultures challenged with Aβ25–35 or Aβ1–42 [175]. LY379268 treatment abrogates its neuronal protection if co-cultured astrocytes lack mGluR3. The selective mGluR3 NAM LY2389575 independently amplifies Aβ toxicity; LY379268, which is otherwise highly protective, becomes neurotoxic in the presence of LY2389575. These findings suggest that mGluR3 is protective against Aβ-induced toxicity, while mGluR2 promotes Aβ toxicity. Hence, the ratio of mGluR3:mGluR2 activity modulates AD risk.
Group II mGluRs have emerged as a possible target for AD therapeutic intervention to reduce Aβ burden at the synapse. This thought is based on the hypothesis that activation of group II mGluRs may trigger synaptic activation of all three secretases, as well as preferential production and release of Aβ42 peptides from nerve terminals [138]. Group II mGluR agonist DCG-IV administration leads to initially increased levels of AβPP CTFs of isolated neuronal synaptosomes taken from TgCRND8 mice. This phase was shortly followed by a CTF cleavage characterized by sustained accumulation of Aβ42 peptides, but surprisingly little effect on the release of Aβ40 peptides [138]. Consequently, the Aβ42:Aβ40 ratio is increased, mimicking a common feature of hereditary AD and animal models of AD [176], which suggests a role of group II mGluRs in amyloidogenic synaptic events. Indeed, when Dutch Aβ-oligomer-forming APP transgenic mice (APP E693Q) were chronically administered the group II mGluR antagonist BCI-838, these mice exhibited a reversal of transgene-related learning and memory impairment, increased hippocampal neurogenesis, reduced anxiety, and reduced Aβ monomers and oligomers in the hippocampus and cortex [177].
Group III mGluRs in AD
The role of group III receptors in AD pathogenesis is still being explored, though it is believed that their activation has neuroprotective effects in the case of AD through regulation of the NMDAR and PI3K pathway [166]. Although less studied than other groups, group III mGluRs play an important role in nervous system disorders and their activation has been found to be beneficial against neurodegeneration [178]. Identification of selective compounds for its subtypes has catapulted studies focusing on the physiological roles and therapeutic potential of group III mGluRs [179]. Group III mGluRs are believed to exert neuroprotective effects by inhibiting glutamate release and dampening its excitotoxicity [180]. In case of AD, they reportedly confer neuroprotection by regulating neurotransmitter release and improving AD symptoms while possessing an improved safety profile. The agonists of group III also reduce the levels of pro-inflammatory cytokines from activated astrocytes, thus preventing cellular demise due to neuroinflammation, a common hallmark of AD [181]. Disruption of calcium homeostasis is one of the primary outcomes of Aβ neurotoxicity. In vitro studies show that a pretreatment of neuronal cells with group III mGluR agonists rescued the cells from Aβ-induced neurotoxicity by reducing the elevated intracellular calcium levels that would otherwise be damaging [182].
mGluR4 agonists have been found to protect degenerating neurons in the caudate nucleus of wildtype mice but not in the mGluR4 knockout mice, suggesting its ability to induce neuroprotection [54]. Its activation also promoted a decline in neurotoxicity associated with excessive activation of microglia by Aβ, a hallmark of AD pathology, suggesting a potential use of mGluR4 agonists in AD therapy [183, 184]. Besides being neuroprotective, mGluR4 also influences LTP at CA1 synapses. The mGluR4–/– mice exhibit improved LTP in the CA1 region of hippocampus, but no such effect has been observed in the prefrontal cortex or dentate gyrus, likely due to the varied expression profile of mGluR4 in these brain regions. Again, the critical involvement of synaptic plasticity at CA1 synapses in AD, mGluR4 inhibition is a potential therapeutic target [185].
Despite a paucity of suitable pharmacological compounds and ligands against mGluR7, studies conducted with knockout mice at the cellular and molecular levels reveal that this particular receptor holds a lot of potential in therapeutic implications against neurodegenerative disorders like AD [186]. Unlike other group III members, mGluR7 is highly abundant in basal forebrain neurons. When this receptor is activated, it suppresses NMDAR currents thus protecting neurons against NMDAR-mediated excitotoxicity. However, Aβ inhibits this protective action of mGluR7, specifically in cholinergic neurons in the basal forebrain area [187]. mGluR7 is also known as the emergency brake system of the cell in the case of abnormal elevation in glutamate levels due to its minimal affinity for glutamate. This was further corroborated by a study involving mGluR7 KO mice that exhibited spontaneous seizures in absence of mGluR7 [15, 188]. Another study reported a reduction in mGluR7 levels in response to elevated miRNA-34a levels in 3×Tg-AD mice models. GRM7, the gene encoding mGluR7, is a target of miR-34a associated with anxiety [189]. The finding corroborated an earlier study involving mGluR7 KO mice exhibiting anxiety-like behavior [190], confirming its role in cognition and behavior.
Mice with targeted mGluR8 gene deletion exhibit subtle behavioral changes, mainly related to a diminished acclimatization to novel surroundings, interrupted context-based freezing response against fear, as well as hyperactivity and anxiety [191]. These changes are likely due to reduced glutamate release along with an abnormal GABA release and lead to the dysfunctional hippocampus. Since hippocampal neurotransmission anomalies are some of the pronounced effects of AD, activation of mGluR8 should be explored as an AD therapeutic target [192].
SUMMARY
Glutamate excitotoxicity occurs when the glutamate receptors are activated beyond a normal threshold, resulting in the loss of dendrites, cell bodies, and other postsynaptic structures. This is one of the major underlying reasons for synaptic failures and neurodegeneration in AD [193]. The ability of mGluRs to modulate glutamate release bodes very well with their contributions to the synaptic failure of AD. Perplexingly, modulation of mGluR activity for AD therapeutic intervention remains underexplored. If research efforts will be growing in this area, including more functional and pharmacological studies as well as pre-clinical explorations, growing knowledge will eventually shed light on novel discoveries of mGluR modulators in AD treatment.