
Abstract
Microglia constitute the brain’s immune system and their involvement in Alzheimer’s disease has been discussed. Commonly, and in line with the amyloid/neuroinflammation cascade hypothesis, microglia have been portrayed as potentially dangerous immune effector cells thought to be overactivated by amyloid and producing neurotoxic inflammatory mediators that lead to neurofibrillary degeneration. We disagree with this theory and offer as an alternative the microglial dysfunction theory stating that microglia become impaired in their normally neuroprotective roles because of aging, i.e., they become senescent and aging neurons degenerate because they lack the needed microglial support for their survival. Thus, while the amyloid cascade theory relies primarily on genetic data, the dysfunction theory incorporates aging as a critical etiological factor. Aging is the greatest risk factor for the sporadic (late-onset) and most common form of Alzheimer’s disease, where fully penetrant genetic mutations are absent. In this review, we lay out and discuss the human evidence that supports senescent microglial dysfunction and conflicts with the amyloid/neuroinflammation idea.
INTRODUCTION
The brain’s immune system is comprised primarily of microglia and other myeloid cells [1], and there is widespread consensus that these cells are important components of Alzheimer’s disease (AD) pathogenesis. However, opinions and hypotheses about the nature of microglial involvement are divergent, and these are summed up by two theories: amyloid/neuroinflammation and microglial dysfunction (Fig. 1). In this review, we are focused on discussing the differences in opinions and why we favor one theory over the other. It is important to note that the dysfunction theory is based mainly on neuropathological examination of human tissues from many nonselected (random) individuals in the general population, whereas the amyloid theory is based on the genetics of rare familial forms of AD affecting few, highly selected individuals in the human population.
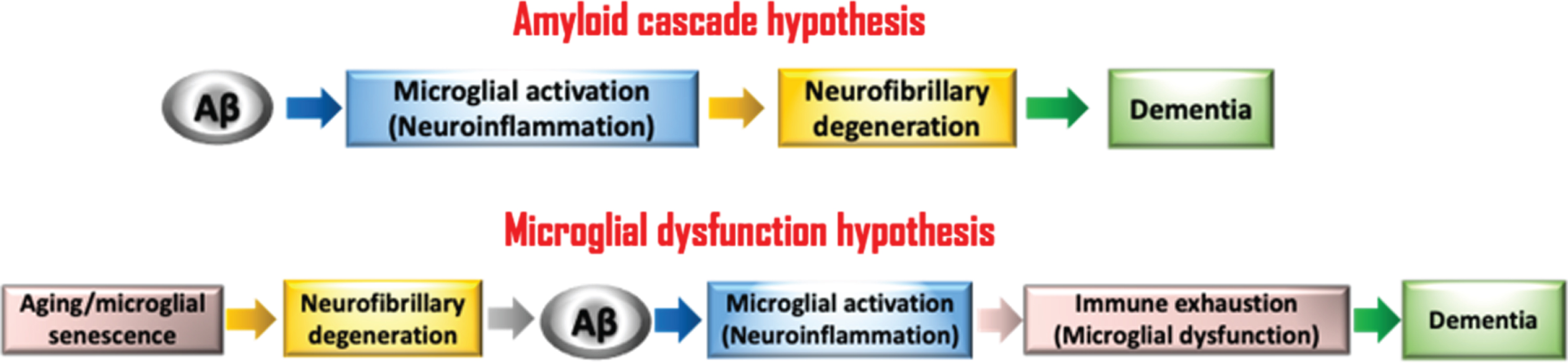
Schematic shows the perceived chains of events underlying the two different theories concerned with microglial involvement in AD pathogenesis. The fundamental difference between the two is that one sees microglia as potentially neurotoxic aggressors while the other sees them as weakened neuroprotectors. Aging, the greatest risk factor for LOAD, is considered in the microglial dysfunction theory to account for microglial senescence.
ARE MICROGLIA HEROS OR VILLAINS?
Our view of microglia as neuroprotective heroes is the result of numerous experiments in various animal models of neuronal injury focused on detailing histopathological changes associated with microglial activation post-injury. The essential conclusion from years of in vivo studies in rodents was that microglial activation is beneficial and neuroprotective, i.e., that microglia activated by acute neuronal injury function as first responders attempting to repair injured neural tissue and to restore homeostasis. We have previously reviewed in detail this body of experimental in vivo evidence favoring microglial neuroprotection because it led us to hypothesize that chronic malfunctioning microglial neuroprotection accounts for aging-associated neurodegeneration in AD [2, 3]. Specifically, it caused us to claim that senescence-related microglial dysfunction could lead to weakened neuroprotection and thus to neurodegeneration in AD, and possibly other aging-related neurodegenerative diseases. The evidence for microglial senescence was generated initially by studying microglial morphology in aged, non-demented humans, showing that with aging there is an increase in numbers of dystrophic microglia, cells that display uniquely abnormal morphological features indicative of senescence, such as atrophic or fragmented cytoplasmic processes [4]. Later on, we showed that dystrophic microglia are abundant in areas of the AD brain undergoing neurofibrillary degeneration, supporting our belief that microglial dystrophy and neurodegeneration were linked [5]. A few years after that, genetic evidence in favor of microglial dysfunction emerged showing that a rare coding variant in the TREM2 gene is present in approximatively 0.1% of the general population found to be associated with increased AD risk [6, 7], consistent with prior work showing that TREM2 is among the highest upregulated mRNAs and the protein colocalizes with microglia at plaques in APP23 overexpressing mice [8]. Since then, other gene variants associated with increased AD risk have been linked to microglia, and this has helped move the idea of microglial dysfunction to the forefront of current thinking about AD etiology [9].
At the time when we first posited the micro-glial dysfunction theory [3], most neuroscientists considering the role of microglia in AD were investigating the neuroinflammation theory of AD stating that neurodegeneration results from chronic inflammation involving activated microglial cells. In contrast to the dysfunction hypothesis, the neuroinflammation theory views activated microglia as dangerous immune effector cells that produce neurotoxic, or autotoxic substances [10 –14]. We have discussed and critically evaluated the evidence that gave rise to the neuroinflammation theory in a previous paper [15]; suffice it to say here that in light of failed clinical trials with anti-inflammatory drugs, it is all but impossible to adhere to this theory any longer. The neuroinflammation idea represents an essential link in the amyloid cascade hypothesis, which has (mis)guided AD research for the last three decades [16, 17]. To date, every clinical trial aimed at amyloid reduction or removal has failed. One major, basic problem with the amyloid theory in our view has been the singular focus on one molecule, amyloid-beta protein (Aβ), which is seen as the cause of a rare familial form of AD (FAD), but these fully penetrant disease-causing mutations in amyloid precursor protein (APP) and presenilins account for less than 5% of all AD cases. Unknown reasoning seems to support a belief that therapies developed in transgenic mice overexpressing mutated APP and ostensibly mimicking FAD apply to the common sporadic or late-onset form of AD (LOAD). This is suggested by the fact that anti-amyloid therapies developed in FAD mouse models are tested in patients with LOAD in clinical trials and consequently fail. Not only does the amyloid theory appear to be wrong, the implication that FAD pathogenesis is the same as LOAD pathogenesis is not justifiable scientifically, because even if Aβ deposits are the initial lesions to develop in FAD, the situation is reversed in LOAD, where neurofibrillary degeneration precedes amyloid deposition by many years, even decades [18]. In the paragraphs following, we discuss the chain of events that characterize pathological lesion development in preclinical and early LOAD and the compelling evidence showing that LOAD does not begin with amyloid deposition but with neurofibrillary degeneration (tau pathology).
THE NEUROPATHOLOGY OF SPORADIC AD (LOAD)
The principal neuropathological lesions, plaques and tangles, have been inextricably linked to AD for about a century (Alois Alzheimer died in 1915); they refer to extracellular deposits of Aβ (plaques) and intracellular neurofibrillary tangles (NFTs). A key question is how these two pathognomonic lesions are connected to one another, and this issue drives most hypotheses regarding AD pathogenesis. Aβ deposited in extracellular plaques can become aggregated and stain positive with Congo red, at which time it is referred to as amyloid. Not all Aβ deposits are created equal and they present in different shapes and sizes under the microscope, thus many descriptors have been used to refer to different plaque morphologies. Perhaps the most useful terms are cored plaques, diffuse plaques, burnt-out plaques, and neuritic plaques; they are described in detail elsewhere [19]. In contrast to extracellular Aβ plaques, NFTs are intracellular neurodegenerative lesions consisting of paired helical neurofilaments rich in aggregates of hyperphosphorylated tau protein (phospho-tau; p-tau). p-tau is reliably visualized in immunohistochemical preparations which facilitates identification of other neurodegenerative structures, such as neuropil threads (NPTs), neuronal pretangles, and neuritic plaques. Collectively, NFTs, NPTs, pretangles, and neuritic plaques may be referred to as neurofibrillary degeneration (NFD) or tau pathology. Aside from NFD, another neurodegenerative change associated with AD is granulovacuolar degeneration (GVD). GVD affects primarily hippocampal pyramidal neurons and, like NFD, it increases with aging and in dementia [20, 21]. However, GVD, presents differently from NFD in terms of morphology, i.e., it is characterized by presence of numerous small, intracytoplasmic vacuoles a few micrometers in diameter. Although these vacuoles are positive for p-tau, GVD is generally discussed as a separate neurodegenerative change that may be linked to the formation of autophagosomes [21]. Like NFD, it is commonly mentioned in the context of AD but is not specific for it.
A third component of AD pathology, neuroinflammation, was recognized much later than plaques and tangles. Neuroinflammation in an AD context refers to accumulations (clusters) of reactive (activated) microglial cells in and around extracellular amyloid deposits. Such localized microglial clusters were first described in human AD brain by McGeer et al. [22]. The observation that activated microglia were present in AD was rapidly incorporated into the amyloid theory by claiming existence of a chronic inflammatory response where amyloid-activated microglia produce various neurotoxins, as well as proinflammatory cytokines, such as Il-6, Il-1β, and TNF-α, and others that allegedly trigger NFD. Since then, clinical trials with nonsteroidal anti-inflammatory drugs have shown that this form of treatment is ineffective in terms of slowing development of NFD or AD progression [23 –25]. In our view, the idea that neuroinflammation directly causes NFD is unsubstantiated and was introduced prematurely to establish a link between allegedly dangerous, activated microglia and NFD. Regrettably, some investigators have further perpetuated the neuroinflammation idea by claiming overactivation of microglia to tout the perceived detrimental nature of activated microglia not only in AD but also other neurodegenerative and/or neuropsychiatric disorders where presence of activated microglia cannot always be demonstrated in humans [26]. The term “overactivation” is subjective and not defined, and to date there are no data that document how molecular/cellular characteristics of activated microglia differ from so-called overactivated microglia. Overactivation was introduced in the context of in vitro studies where primary cultures of microglia were stimulated excessively with bacterial endotoxin, lipopolysaccharide (LPS) [27]. The LPS in vitro model mimics infectious etiology and bears little, if any, resemblance to what occurs in AD. That said, an inflammatory reaction involving activated microglia does indeed occur at some point during AD pathogenesis but is likely of limited duration. Our recent work, discussed in detail below, has identified the point when neuroinflammation sets in during AD pathogenesis, which is when preclinical (non-symptomatic) LOAD transitions to clinical (symptomatic) LOAD [28].
MICROGLIAL INVOLVEMENT IN THE DEVELOPMENT OF AD LESIONS
LOAD takes many decades to advance to a point when clinical symptoms appear because there is a prolonged preclinical, asymptomatic period during which microscopic lesions develop gradually over time without causing neurological deficits. So, what is the nature of this preclinical neuropathology? The issue has been addressed in great detail by the extensive work of Braak and colleagues, as well as others, and is very well documented [18 , 29–32]. The data compiled from thousands of nonselected (random) humans of all ages shows unequivocally that early stages of tau pathology can be detected in virtually every adult human being, and even in some adolescents [33], and that in the vast majority of humans NFD initially develops in the absence of Aβ deposition. Clearly, this does not align with the amyloid cascade hypothesis, because how could Aβ be a cause of NFD if it is not present when NFD occurs. This observation goes back all the way to Alzheimer’s original work from more than a hundred years ago, where he described both neurofibrillary changes as well as amyloid plaques, called “Drusen” at the time, commenting that plaques are merely a “Begleiterscheinung” (German for “side effect”) of the disease [34]. His statement was based on the fact that in some cases of dementia and in some brain regions showing NFD plaques were not very numerous, and so he concluded that plaques could not be the cause of neurodegenerative changes. Later, Braak & Braak noted some cases with moderate neurofibrillary changes (up to Braak stage III) did not reveal amyloid plaques at all [35]. Despite, or maybe because of the neuropathological findings, proponents of the amyloid theory have revised the theory and identified invisible Aβ oligomers as toxic inducers of NFDs rendering moot the issue of poor correlation between Aβ deposits and disease, but this has created additional questions and uncertainties [17].
Our recent work conducted in a cohort of 66 nonselected, nondemented individuals has not only confirmed Braak’s findings but has also extended them by identifying the place of microglial activation (neuroinflammation) in the chain of pathological events [28]. Our postmortem histopathological evaluations of the inferior temporal lobe (including entorhinal cortex and hippocampus) from subjects aged 42 to 93 found that all 66 human brains showed presence of NFD, which increased with aging and reached a maximum of Braak stage III-IV in a few individuals [35]. About one third (23/66) of individuals showed presence of mostly diffuse Aβ deposits, the youngest of which was 61 years of age. Neuroinflammation consisting of clusters of activated microglia was present in 5 individuals with Aβ deposits, but only in those where the Aβ deposits stained positive with Congo red indicating that aggregation of Aβ into insoluble amyloid had occurred. Based on these numbers from preclinical subjects, i.e., 100% NFD, 35% Aβ, and 8% neuroinflammation, we concluded that the sequence of pathological lesion development begins with NFD, followed by Aβ deposition, followed by neuroinflammation. This sequence of events, which is based exclusively on human neuropathology, is opposite to a hypothetical model of the pathological cascade based on cerebrospinal fluid biomarkers and neuroimaging data from AD patients, where amyloid is seen as the initial event followed by NFD [36]. Thus, the development of AD neuropathology does not corroborate the model by Jack et al. [36], which also lacks information on neuroinflammation.
Our work shows that microglia become activated only after Aβ has aggregated [28]; prior to that microglia are non-reactive to deposits of diffuse Aβ, as well as any tau pathology already present, which consists primarily of NPTs, pretangles, and NFTs. Neuritic plaques were not prevalent in our cohort of preclinical subjects and found in a few individuals only. One obvious corollary from these observations is that neurofibrillary tangles occur in the absence of neuroinflammation and Aβ deposits, contradicting the amyloid hypothesis. Tangentially, it is appropriate here to mention briefly that transgenic mouse models overexpressing APP and forming widespread amyloid deposits triggering microglial activation more robust than in humans also do not develop NFD [37 –39]. The conclusion that neither amyloid nor activated microglia trigger NFD is inescapable. What then accounts for the development of tau pathology? Our conjecture is that tau pathology results from oxidative stress, which we have recently discussed in greater detail [40].
Cumulative oxidative stress associated with aging affects all cells and is important regarding microglial involvement in LOAD. Sixteen years ago, we introduced the notion of microglial senescence reporting that numbers of dystrophic cells increase in the aging human brain, and we therefore believe that microglial dystrophy is the morphological reflection of senescence [4]. We have shown widespread presence of dystrophic microglia in aging and in neurodegenerative diseases [5 , 41], findings that have been corroborated subsequently by other investigators in humans [42 –46], and recently in tree shrews [47]. We and others have also shown that dystrophic microglia often stain positive for the iron storage protein, ferritin [48, 49], raising the possibility that microglial dystrophy may be the result of iron-mediated oxidative stress, and/or of low brain pH [44]. It has long been known that levels of brain iron increase with aging and are involved in AD pathogenesis, yet exact mechanisms of aging-related iron accumulation in the CNS and its downstream consequences remain largely speculative and unknown [50 –52]. For now, we know that microglial dystrophy, NFTs, and Aβ plaques can all be linked to oxidative stress and iron [28 , 52–55].
Our view of the significance of microglial ferritin expression and concomitant iron sequestration is that it represents a most important neuroprotective function of microglia that increasingly comes to bear with aging and especially with AD when ferritin expression increases [28, 48]. Presence of free brain iron is known to generate free radicals via Fenton chemistry that can cause neuronal damage, and thus one function of microglia is to sequester free iron in order to prevent or minimize such damage. Given the notable increase in microglial ferritin expression that occurs with aging and AD, one is tempted to conclude that this neuroprotective function is being stretched to its limits and may therefore contribute to microglial exhaustion.
Microglia and amyloid plaques
There is a close relationship between extracellular amyloid deposits and microglial activation, which has been known ever since McGeer and colleagues reported presence of reactive microglia in association with amyloid plaques in AD brain [22]. Specifically, aggregated Aβ (i.e., amyloid) will cause microglia to become activated because it forms a foreign body that attracts these endogenous brain macrophages, which move towards it and then attempt to phagocytize and remove the insoluble material [28]. Such activation of microglia is accompanied by numerous molecular changes, including the increased production and release of various cytokines. This relationship between amyloid and microglial activation is the only sequence of events that both the amyloid cascade hypothesis and the microglial dysfunction hypothesis have in common (Fig. 1). The microglial response to amyloid is highly specific, meaning that activated cells are present only in the immediate vicinity of amyloid deposits and not in the neuropil between amyloid plaques, e.g., see Fig. 1 in Prokop et al. [56]. This is important because it shows the extraordinary specificity of microglial activation towards amyloid only. For that reason, it is difficult to understand the reasoning of the amyloid theory that chronic microglial activation leads to NFD, which implies the targeting of neurons rather than of amyloid by activated microglia. As pointed out already, NFD occurs without amyloid or activated microglia being present, not only in preclinical LOAD but also in other tauopathies, such as Down syndrome and diffuse Lewy body disease [26, 41]. Upon clustering around Congo red positive amyloid deposits microglia struggle to internalize and digest the insoluble amyloid, and they ultimately fail at degradation [57, 58]. This is also supported by the fact that internalization of amyloid inside phagocytic microglia is rarely observed during neuropathological examination of human brain. Instead, what is seen much more frequently in microglial clusters associated with amyloid plaques is dystrophic fragmentation of microglial processes [5, 40]. This has led us to conclude that indigestible amyloid contributes towards microglial immune exhaustion and senescence [28, 59] (Fig. 1). The idea of senescence-associated immune exhaustion is consistent with findings showing that neuroinflammation in AD wanes with aging [60].
Microglia and NFD
The relationship of activated microglia to tau pathology is more enigmatic than their relationship to amyloid plaques because there is no histologically detectable activation of microglia to neuropil threads, pretangles, or NFTs, which is puzzling because microglial macrophages typically do engulf and phagocytize degenerating neurons and/or synapses. Considering that the p-tau aggregates contained in NPTs, NFTs, and pretangles remain intracellular and surrounded by the neuronal plasma membrane suggests that microglia do not recognize the intracellular tau aggregates as degenerative changes and therefore do not remove them. The only form of tau pathology associated with reactive microglia is the neuritic plaque, which is composed of both amyloid and p-tau+neuronal structures, and it is very likely that microglial activation associated with neuritic plaques is triggered by the amyloid. In fact, this colocalization of amyloid, activated microglia, and tau pathology in neuritic plaques has been a critical, albeit the only neuropathological lesion to support the amyloid cascade theory.
The relationship between tau pathology and micro-glia is different in the sense that it is not activated microglia that are associated with tau pathology but instead dystrophic microglia. While there are instances where dystrophic microglia are immediately adjacent to NFTs, pretangles, or NPTs, dystrophic cells are also found in association with neuritic plaques, and thus the entire spectrum of lesions grouped under tau pathology can be observed in association with microglial dystrophy. This suggests that both neuronal and microglial degeneration occur largely side-by-side and that they may have a common cause, such as oxidative stress [40]. The parallel development of microglial dystrophy and NFD might also be linked in that aging neurons depending on microglial support and protection do not receive their needed support any longer because microglia are aging as well, setting up a vicious cycle that only becomes worse with continued aging. This could of course be the basis for the rapid progression of LOAD once it reaches the symptomatic stage when NFD has become widespread.
A COMPREHENSIVE MODEL OF LOAD PATHOGENESIS
The one fact about LOAD that is beyond discussion is that aging is the greatest risk factor; our view of LOAD pathogenesis therefore begins with aging. Mechanistically, oxidative stress is very likely to be involved in the generation of AD pathology because it is inextricably linked to aging [51]. We hypothesize that both tau pathology and microglial dystrophy are related to aging/oxidative stress because these neuronal and microglial pathologies increase with aging; they are detectable at low levels even in relatively young individuals and gradually worsen over time (Fig. 1 in Streit et al. 2020 [40]). At some point later in life when NFD has become more widespread Aβ deposition sets in and plaques appear. The fact that Aβ deposition ensues after NFD is already underway has prompted us to hypothesize that Aβ deposition occurs in response to neurodegeneration, and that it may reflect an endogenous compensatory response to counter neurodegeneration, i.e., we believe Aβ may be produced to protect against advancing neurodegeneration. However, such presumed protective ability of Aβ may be of limited duration because the protein eventually aggregates into amyloid rendering it insoluble and inert and, presumably, biologically inactive. Once amyloid has formed, it essentially represents a foreign body that attracts the attention of microglia-derived macrophages, which will attempt to remove this undesirable and potentially toxic material through phagocytosis. Unfortunately, amyloid is quite resistant to degradation and removal [57], and this may result in so-called “frustrated phagocytosis” [61], an anthropomorphic term that is a suitable metaphor for describing inability of microglia to remove amyloid. Eventually microglia become exhausted by futile efforts to remove amyloid and become dystrophic thus exacerbating already ongoing, aging-related microglial dystrophy, which in turn will promote the progression of NFD to an advanced state that coincides with onset of dementia.
SUMMARY OF ARGUMENTS
Throughout this paper we have discussed why the amyloid/neuroinflammation theory of AD pathogenesis does not apply to LOAD, and why a microglial dysfunction theory makes sense. In this final paragraph we are summarizing the key points of this discussion, which are as follows: first, there are no fully penetrant genetic mutations associated with LOAD that would provide a genetic basis for a causative role of amyloid in LOAD neurodegeneration. Second, aging is the greatest risk factor for LOAD and must therefore be incorporated into any theory about its pathogenesis. We have done so by incorporating microglial senescence into the dysfunction theory. Third, the temporal sequence of lesion development during preclinical LOAD begins with NFD, which is followed by Aβ deposition and microglial activation, i.e., NFD occurs in the absence of Aβ deposits and neuroinflammation. Fourth, transgenic rodents overexpressing APP develop amyloid plaques and neuroinflammation but do not develop NFD. Fifth, activated and allegedly neurotoxic microglia producing cytokines and inflammatory mediators are found accumulated around amyloid and not around p-tau positive neurons. Sixth, recent genome-wide association studies have identified several microglia-related gene variants associated with higher AD risk supporting microglial malfunctioning in LOAD. Seventh, all clinical trials employing anti-inflammatory and anti-amyloid strategies have failed.
Collectively, these points provide strong evidence against the amyloid/neuroinflammation theory that has dominated and potentially misled AD research for about 30 years producing no effective therapies. It is a “Me TOO” moment for AD research in that this theory must be critically reevaluated if progress for AD prevention and treatment is to be made.
CONCLUSIONS
It is time to seriously consider moving away from thinking that LOAD is caused by one molecule, Aβ, and ensuing neuroinflammation. Decades and large amounts of money have been spent in efforts to support a theory of pathogenesis based mostly on genetics for a disease that is largely related to aging and not as much to genetic mutations. We propose to refocus thinking on undisputable facts known about LOAD, which is that aging is the greatest risk factor. Aging and LOAD are inseparable and, while it may not be possible to model human aging in mice, careful analyses of human brains is likely to shed light on cellular/molecular mechanisms that underlie the sequential development of AD lesions and therefore elucidate pathogenesis. Developing strategies for treatment and/or prevention is only meaningful if LOAD pathogenesis is fully understood.
Footnotes
ACKNOWLEDGMENTS
This study was supported by the Deutsche Forsch-ungsgemeinschaft (DFG) Collaborative Research Center 1052-A9, by NIH grants R01NS071122 and R21 NS103108, and by a Florida Department of Health Grant 8AZ19. We also thank the Cooper family of Indialantic, FL for their support of dementia research. The authors wish to thank Heidrun Kuhrt and Angela Ehrlich for expert technical assistance.