
Abstract
Background:
Iron plays an important role in maintaining cell survival, with normal iron trafficking known to be regulated by the ceruloplasmin-transferrin (Cp-Tf) antioxidant system. Disruption to this system is thought to be detrimental to normal brain function.
Objective:
To determine whether an imbalance of iron and the proteins involved in its metabolism (ceruloplasmin and transferrin) are linked to Alzheimer’s disease (AD) and to the expression of amyloid-beta (Aβ) peptide 1–42 (Aβ1–42), which is a major species of Aβ, and the most toxic.
Methods:
We evaluated the concentrations of iron, calcium, magnesium, and Aβ1–42 in the cerebrospinal fluid (CSF) of patients with AD and cognitively normal controls. Correlations between the components of the Cp-Tf antioxidant system in plasma were studied to determine the role of peripheral blood in the onset and/or development of AD. We used commercial ELISA immunoassays to measure Aβ1–42, immunoturbidimetry to quantify ceruloplasmin and transferrin, and colorimetry to quantify iron, calcium, and magnesium.
Results:
We found that the AD group had lower CSF concentrations of Aβ1–42 (p < 0.001) and calcium (p < 0.001), but a higher CSF concentration of iron (p < 0.001). Significantly lower plasma concentrations of ceruloplasmin (p = 0.003), transferrin (mean, p < 0.001), and iron (p < 0.001) were observed in the AD group than in cognitively normal adults. Moreover, we found a strong interdependence between most of these components.
Conclusion:
Iron dyshomeostasis has a crucial role in the onset of AD and/or its development. Correcting metal misdistribution is an appealing therapeutic strategy for AD.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is the most common type of dementia worldwide, contributing to around 80% of total dementia cases, and one of the greatest global healthcare challenges [1, 2]. The World Health Organization estimates that by 2050, more than 135 million people worldwide will have dementia, which would triple the current number.
AD is a progressive neurodegenerative disorder initiated by modifications in the brain that occur years before the appearance of clinical symptoms. There are two forms: early-onset AD and late-onset (or sporadic) AD. Early-onset AD affects patients <65 years of age, most often those aged 30–50 years [3, 4]. It is a familial form of the disease caused by mutations occurring in one of many genes, including amyloid-β protein precursor (AβPP), presenilin 1 (PSEN1), or presenilin 2 (PSEN2), which together account for only 11% of cases of early-onset AD [5]. A strong association between the APOE ɛ4 allele and early-onset AD has been reported [6, 7]. The origin and cause of late-onset (sporadic) AD, which generally occurs in patients ≥65 years old [8], remain controversial, but the risk of developing this form is also increased by the APOE ɛ4 allele [9–12]. Both forms are characterized by amyloid protein deposits and neurofibrillary tangles consisting of hyperphosphorylated tau protein [13]. These two types of histopathological lesions have become the fundamental markers of cases of AD that lead to neurological dysfunction [14]. Furthermore, both forms of AD share many similarities at the molecular level, such as the accumulation of amyloid-β (Aβ) and hyperphosphorylated tau protein; oxidative stress; dyshomeostasis of metal ions; and the disturbance of N-methyl-D-aspartate (NMDA) receptor (NMDAR) signaling. All of these changes occur during the spread of the disease from the hippocampus to other areas of the brain. This leads to the clinical symptoms of AD, which start with memory problems, the gradual disappearance of notions of space and time, impaired recognition of objects and people, and the disruption of language mechanisms [15].
To date, the most promising biomarkers for AD have been Aβ peptide and tau protein; however, the role of biometal-induced oxidative stress has also been investigated. It has been shown that intracellular iron accumulation induces the production of reactive oxygen species (ROS), which cause oxidative damage to proteins, lipids, and DNA [16]. The detrimental roles of oxidative stress and iron dyshomeostasis in the brain have been described in several outstanding reviews [17–19].
In this context, investigating the components of cerebrospinal fluid (CSF) and plasma represents an effective way of understanding the mechanisms underlying the disease. Using this approach, it may be possible to discover a novel biochemical biomarker and validate it as a non-invasive way of diagnosing the disease.
Given that iron dyshomeostasis is pathologically linked to Aβ aggregation [20, 21], which is considered an early biomarker for AD, we hypothesized that iron dysmetabolism would be associated with disturbances in Aβ levels in the CSF. To test this, we sought to determine whether the concentration of Aβ in the CSF correlates with metal ions (iron, calcium and magnesium) in the plasma and/or CSF and examined systemic variations in the markers of iron metabolism (ceruloplasmin and transferrin). Our aims were to identify new indicators of disease development and to observe variations in these indicators due to the disease.
MATERIAL AND METHODS
Patients
For this study, 106 patients with AD and 61 age- and gender-matched cognitively normal (CN) participants (control group) were recruited from the Neurological Institute, Mongi Ben Hamida, La Ra-bta, Tunis, Tunisia. Our clinical assessment confirmed that 106 participants from the whole cohort had Alzheimer’s disease (Mini-Mental State Examination [MMSE] score ≤25) and 25 had mild cognitive impairment (MMSE scores of 25–28) or a questionable diagnosis. Those with a questionable diagnosis, cancer, or a history of alcohol abuse were excluded from the study. Patients with mild cognitive impairment will be asked to participate in a separate study. The control group comprised a group of CN participants who were neurologically and psychologically healthy. The same clinical examination was performed in the control group (with an MMSE score of ≥28 considered to indicate CN status).
All participants in the study gave informed consent for a blood and/or CSF sample to be taken for analysis. The study protocol was approved by the Ethics Committee of the institute and the study was performed in accordance with the ethical standards laid down in the 1975 Declaration of Helsinki and its later amendments.
Clinical examination
A complete neurological and somatic examination was performed by a physician using the MMSE. We also assessed the general and cardiovascular condition of the patients, their vigilance (mental confusion), and any sensory (visual or auditory) or motor deficits that may interfere with taking neuropsychological tests.
Neuropsychological assessment
We performed an in-depth neuropsychological ass-essment using validated and standardized neuropsychological tests adapted to the Tunisian population by the Clinical Neuropsychology and Language Research Unit of the Neurology Department of La Rabta Hospital in Tunis. This exhaustive assessment examined each of the cognitive functions, in particular episodic memory, semantic memory, executive function, attention, and instrumental functions (language, praxis, visuo-constructive functions).
Brain imaging
All patients underwent brain imaging. Magnetic resonance imaging was preferred to computed tomo-graphy and performed routinely. Imaging enabled us to exclude the existence of a neurosurgical etiology of dementia (expansive process, chronic subdural hematoma, normal pressure hydrocephalus, etc.) and to search for degenerative or vascular lesions. It also allowed us to assess hippocampal atrophy and corticosteroid-induced subcortical atrophy by qualitative analysis.
Etiologic diagnosis of AD
Our patients were diagnosed with AD according to two sets of diagnostic criteria: the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM IV) and the NINCDS-ADRDA criteria [22, 23].
Sample preparation and analysis
Blood samples were collected from patients in the morning without overnight fasting, and plasma was separated by centrifugation at 3000×g for 10 min at 4°C, in tubes containing anticoagulant (lithium heparin). The supernatant was then aliquoted into 500μl samples in polypropylene tubes and stored at –80°C until use. Prior to assay, the samples were thawed for 15 min and centrifuged for 5 min at 1000×g.
CSF samples were collected from all participants in the patient and control groups by a single health professional, using a lumbar puncture. Lumbar puncture was performed using the syringe method, which consists of inserting a thin needle in the widest space between two vertebrae of the lower back, generally at the level of the lumbar vertebra L3–L4 or L4–L5. The CSF was collected using a needle and mandrel for lumbar puncture and a sterile (polypropylene) analysis vial. CSF was centrifuged at 2000×g for 10 min at 4°C. The CSF supernatant was taken in volumes of 1000μL at a time and transferred to a 1.5 ml polypropylene Eppendorf microtube; we repeated this as many times as was necessary to use up all the supernatant. Hemorrhagic or purulent samples were excluded.
All plasma and CSF measurements were performed in Bechir Hamza Children’s Hospital, Bab Saadoun, Tunisia, using immunoturbidimetry to measure plasma concentrations of ceruloplasmin and transferrin [24], and colorimetry [25] to measure CSF concentrations of iron, calcium, and magnesium (COBAS INTEGRA analyzer, Roche Diagnostics). The concentration of Aβ1–42 in CSF was measured by the enzyme-linked immunosorbent assay (ELISA) using the Amyloid Beta 42 Human ELISA Kit (#KHB3441; Thermo Fisher Scientific, Charguia, Tunisia).
Statistical analysis
Statistical analysis was performed using SPSS 20.0 (IBM, Armonk, NY USA). For each quantitative parameter, we calculated the mean and the standard deviation in each of the population groups. ANOVA and Chi-squared tests were used to assess differences in demographic, biochemical, and Aβ1–42 variables between patients in the AD and CN groups, adjusting for sex and age. Pearson’s rank correlation test was used for correlation analyses of all parameters. Differences with p < 0.05 were considered significant.
RESULTS
The demographic data of the AD and CN groups are shown in Table 1, while the results of the CSF assays of calcium, iron, magnesium, and Aβ1–42 are shown in Table 2. The average iron concentration in the CSF of the AD group was significantly higher than that in the controls (p < 0.001). There was also a significant difference between the mean calcium concentration in the CSF of participants in the AD group and those in the CN group. The mean concentrations of transferrin, ceruloplasmin, and iron in the plasma were also significantly lower in the AD group than in CN controls (p < 0.001, p = 0.03, and p < 0.001 respectively). We found no significant difference between the groups in the CSF concentration of magnesium. Participants in the AD group had lower MMSE scores than those in the CN group (p < 0.001). In addition, we found strong associations between these components (ceruloplasmin, transferrin) at the plasma level and calcium, iron and Aβ1–42 in CSF concentrations and both reported to be associated with cognitive decline, as noted by the MMSE (Table 3).
Demographics, MMSE scores, and comorbidity of participants in the AD and CN groups
MMSE, Mini-Mental State Examination; HBP, high blood pre-ssure (≥140/90 mmHg for patients≤80 years old;≥150/90 mmHg for patients > 80 years old); DM, diabetes mellitus (defined as abnormally high levels of glucose (> 200 mg/dL)); NA, not applicable; AD, Alzheimer’s disease; CN, cognitively normal. Differences with p < 0.05 were considered significant.
Changes in concentration of biochemical components of CSF and plasma in the AD and CN groups
AD, Alzheimer’s disease; CN, cognitively normal; CSF, cerebrospinal fluid; Aβ1–42, amyloid beta peptide 1–42; Fe, iron; Ca, calcium; Mg, magnesium; Cp, ceruloplasmin; Tf, transferring.
Matrix of correlations
Aβ1–42, amyloid beta peptide 1–42; Fe, iron; Ca, calcium; Mg, magnesium; Cp(pl), plasma ceruloplasmin; Fe(pl), plasma iron; tf(pl), plasma transferrin; MMSE, Mini-Mental State Examination. *p < 0.05 (2-tailed); **p < 0.01 (2-tailed).
DISCUSSION
Under normal conditions, ceruloplasmin catalyzes and oxidizes the conversion of ferrous iron (Fe2 +) into ferric iron (Fe3 +) (Eq. 1). This reaction helps to eliminate iron from the periphery by the binding of ferric iron (Fe3 +) to transferrin, forming holo-transferrin (the iron-saturated form of transferrin), which reaches cells and organs later [26]:.
Under pathological conditions (as in our study), Fe2 + is not oxidized sufficiently by ceruloplasmin (which we found to be low in plasma, p = 0.03; Table 2) and is engaged in other reactions consi-dered harmful to the organism (i.e., the Fenton reaction, which generates toxic free radicals and ROS) [27–29]:
We found low levels of ceruloplasmin and transferrin, which are considered to be important for iron metabolism, in our AD group compared to the CN group (0.229 mg/l versus 0.261 mg/l [p = 0.03] and 2.657 g/l versus 3.259 g/l p < 0.001], respectively). The same observation for transferrin was reported by Ashraf et al. [30]. These changes may have caused oxidative damage arising from the imbalance between free radicals and the antioxidant effect of Cp-Tf during iron metabolism, a result reported previously by Patel et al. [31].
Holo-transferrin formation helps to reduce levels of ferrous iron, which is dangerous for the organism because this form of iron can produce highly reactive radicals (Eq. 2 and 3) [32, 33]. Therefore, a change in the regulation of ceruloplasmin and transferrin (as in our AD cohort) may elevate the amount of Fe2 +, which the organism will then redistribute to other organs. Fe2 + may end up in the brain, thereby potentially explaining our results.
Hare et al. [34] reported the same result, namely a decreased level of iron in plasma, in line with that shown by Vural et al. [35] and with our finding of an increased level of iron in CSF (Table 2). Ferric ion cannot be absorbed in its oxidized state but has to be reduced to ferrous iron or bound to a heme group to facilitate its absorption. Hare et al. [34] demonstrated a decrease in iron-bound Tf, but also found that this decrease was not due to a decrease in total plasma Tf levels, in contrast to our observation of a decreased level of transferrin in plasma (Table 2).
Studies in the literature have described an increase of ferritin and a decrease of transferrin in plasma [21, 36–39], in line with our transferrin results. These findings can be explained in two ways: 1) as the establishment of a protective process in response to the appearance of AD; or 2) as a result of part of the pathogenic process of AD. However, at this point, it is difficult to distinguish between these two ideas.
The observations made above all highlight the crucial role of iron metabolism in the bloodstream and the role of its regulation by the Cp-Tf system in the onset and/or development of AD. Furthermore, they point to the potential efficacy of targeting plasma iron metabolism as an eventual therapeutic strategy for AD.
How this excess of Fe2 + enters the brain through the blood-brain barrier (BBB) remains an open question. The BBB is composed of endothelial cells characterized by a sophisticated mechanism for ion trafficking, which is heavily regulated [40, 41]. To date, the mechanism by which the iron reaches the brain remains unclear; however, it has been demonstrated that plasma iron deficiency, as observed in our study (p < 0.001; Table 1), induces iron and transferrin uptake through the BBB [42, 43].
With respect to this process, the divalent metal tra-nsporter 1 (DMT-1) plays a key role through its involvement in non-vesicular iron uptake. Voltage-gated calcium channels (VGCCs), neuronal NMDARs, Zip8 and Zip14 may also participate in this process [44, 45]. DMT-1, VGCCs, and the NMDAR are expressed in brain endothelial cells and in the choroid plexus cells that form the BBB [46–51]. Endothelial cells contributing to the function of the BBB also express the transferrin receptor, ferroportin, and ceruloplasmin (forming the system regulating iron distribution).
Moreover, in ceruloplasmin-knockout mice there is an upregulation of divalent metal transporter 1 (DMT-1) mRNA expression in the cerebellum [52]. A significant elevation in this mRNA was also found in the APPswe/PS1E9 AD mouse model [53]. This strongly suggests a pivotal role for ceruloplasmin in the etiology of AD, given that ceruloplasmin catalyzes the oxidation of ferrous iron to ferric iron, as mentioned above.
Furthermore, mice with DMT-1 knockdown show a reduced level of IL-1β-induced oxidative stress resulting from lowered iron levels [54]. This encouraging result from beta cells can be generalized and verified in neuronal cells, especially at the BBB le-vel, assuming that the same model of iron trafficking occurring via DMT-1 in beta cells also takes place in the BBB. This suggests that iron dysmetabolism might also trigger an oxidative stress-induced inflammatory response, in which astrocytes and microglia release inflammatory cytokines such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and IL-6 [55] in response to Aβ1–42 accumulation. This response is characterized by Fenton-reaction-dependent ROS generation and may explain the localization of astrocytes and microglia around neurons [56, 57]. Understanding the mechanism behind this could lead to a promising therapeutic strategy for AD [58].
Together, these observations strongly indicate that there is ultimately an interaction between the mechanisms underlying AD and markers of iron metabolism (ceruloplasmin and transferrin). This may lead to the BBB taking up ferrous iron as result of a defective Cp-Tf system, leading to its eventual release in the brain through specialized transporters such as DMT-1, VGCCs, and the NMDAR [59] (Fig. 1). The resulting iron overload in the brain induces neuroinflammation, which is recognized as an important feature of AD in addition to Aβ.
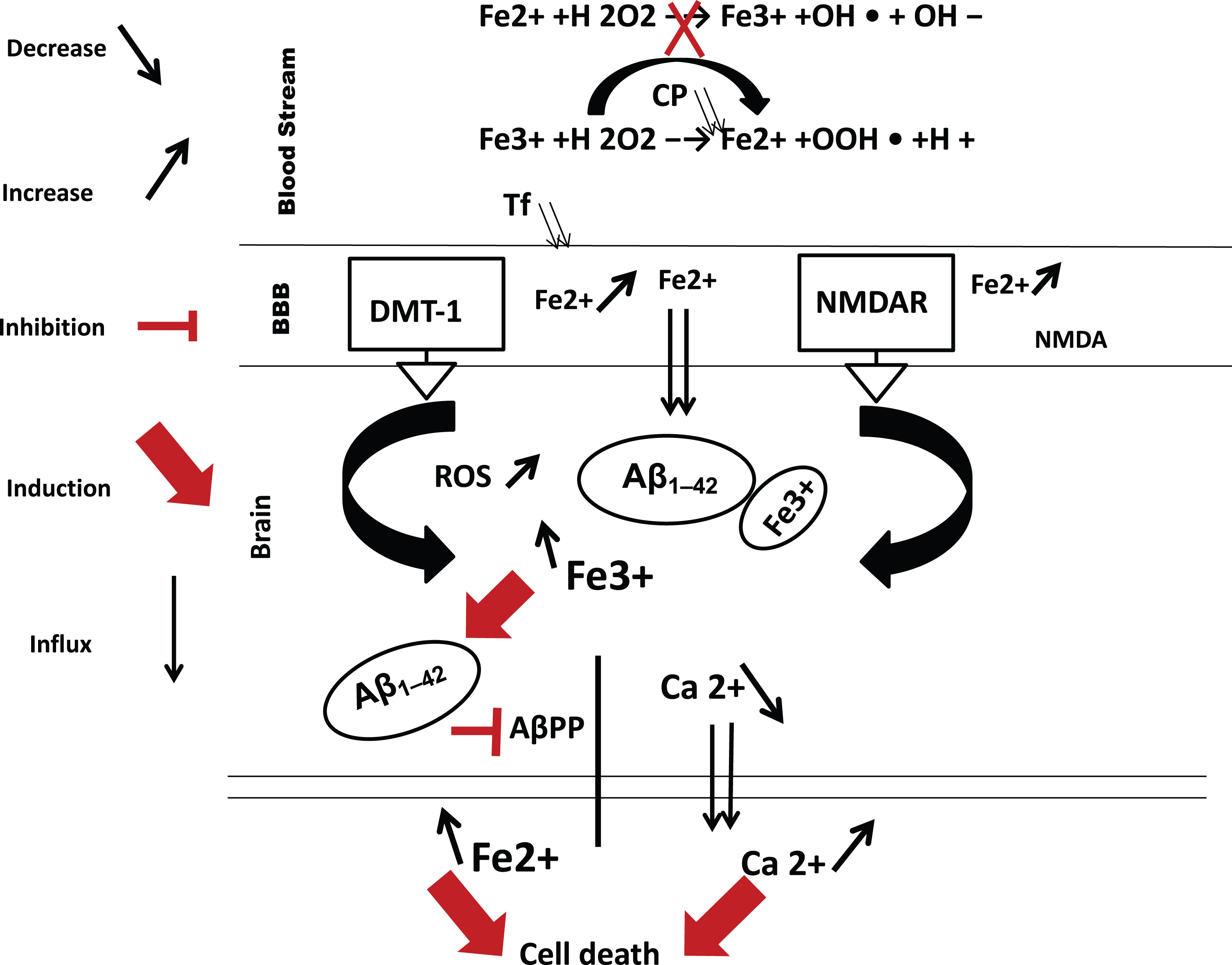
Hypothesis for the interaction of iron, calcium, and Aβ1–42 in AD. AβPP regulates the level of ferrous iron in cells by facilitating its export via ferroportin [74]. An increased level of the insoluble form of Fe3 + accelerates the production of free radicals, causing oxidative stress [78]. This process may grow out of control as Fe3 + continues to accumulate from the periphery in the brain, and plaque formation becomes excessive. Finally, an excessive influx of Ca2 + induced by NMDAR overstimulation [65] and Aβ1–42 can generate ROS formation and induce cell death, favoring the development and worsening of AD pathology. ROS, reactive oxygen species; Cp, ceruloplasmin; Fe2 +, ferrous iron; Fe3 +, ferric iron; Ca2 +, calcium ion; Aβ1–42, amyloid beta peptide 1–42; NMDAR, N-methyl-D-aspartate; DMT-1, divalent metal transporter 1; AβPP, amyloid-β precursor protein; BBB, blood-brain barrier.
This conclusion is strengthened by the study of Xu et al. [60], which reported increased iron deposition in the brains of patients with aceruloplasminemia, a disease characterized by the absence or dysfunction of ceruloplasmin. In other words, once Fe3 + has been reduced to Fe2 + under pathological conditions, these Fe2 + ions may be transported into the brain by the DMT-1 transporter protein, VGCCs, and/or the NMDAR (serving as a direct importer of non-transferrin-bound iron). Then they can be either stored in ferritin or released from endothelial cells in the brain via ferroportin 1 [59, 61]. This may explain the report that ferritin and iron are enhanced in the CSF of participants with AD compared to the CN group [62].
Interestingly, in our AD cohort we found an increased level of iron and a decreased level of calcium in the CSF [62, 63]. We also noted a significant negative correlation between calcium and iron levels in the CSF (r(161) = –0.25, p = 0.001) (Fig. 2). This leads us to consider calcium signaling and its eventual harmful interaction with iron metabolism, especially given our finding that the level of calcium correlates negatively with that of iron in CSF and positively with the level of iron in plasma (Table 3).
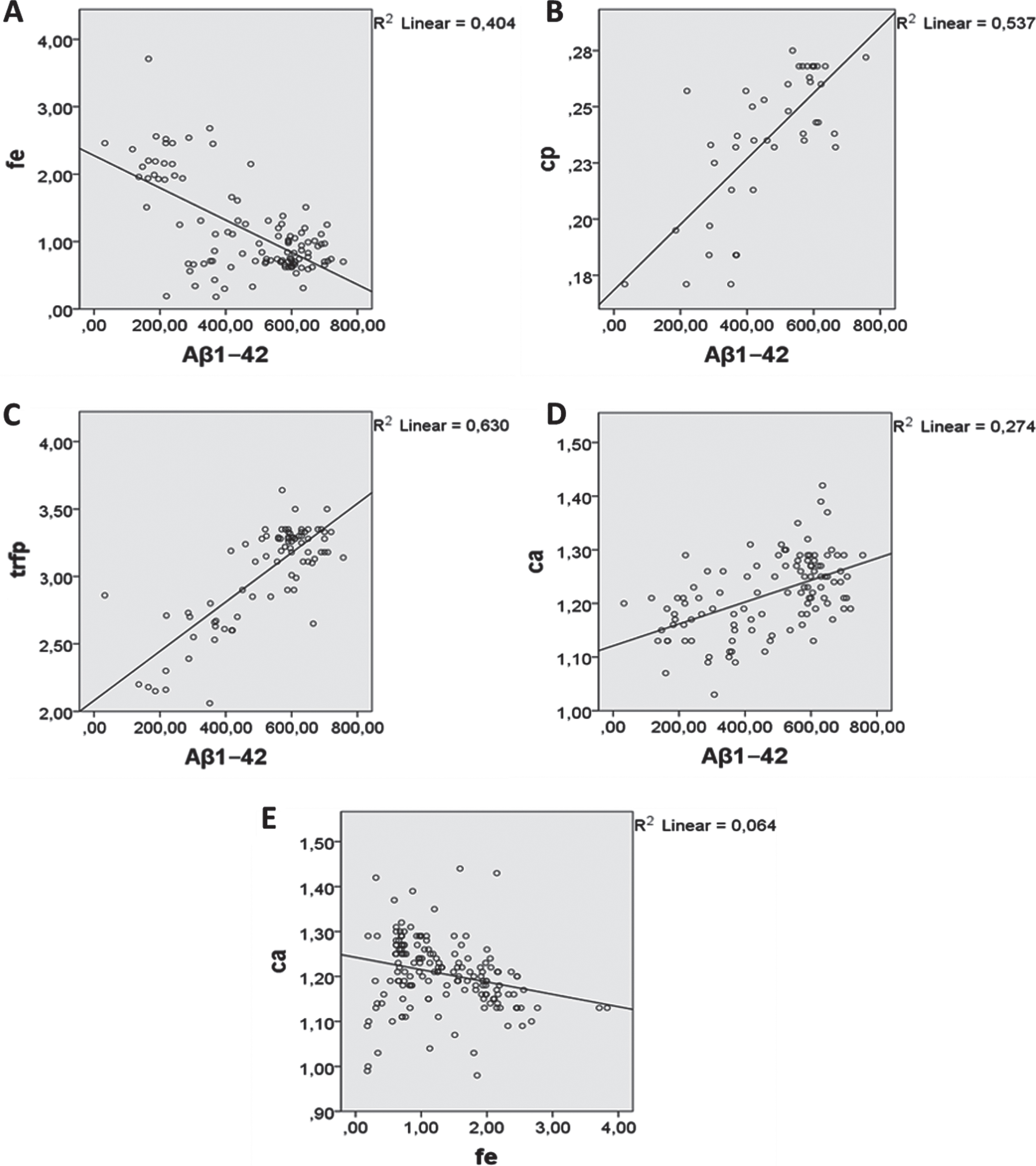
Levels of Aβ1–42 correlate with levels of: A) iron in CSF (Pearson’s r = –0.635, p < 0.001, n = 109); B) ceruloplasmin in CSF (Pearson’s r = 0.733, p < 0.001); C) transferrin (Trf) in plasma (Pearson’s r = 0.794, p < 0.001, n = 62); and D) calcium in CSF (Pearson’s r = 0.524, p < 0.001, n = 111). E) Levels of calcium in CSF correlate negatively with those of iron in CSF (Pearson’s r = –0.253, p < 0.001, n = 163). Aβ1–42, amyloid beta peptide 1–42; fe, iron; cp, ceruloplasmin; trfp, plasma transferrin; ca, calcium.
Recent evidence indicates an interaction between DMT-1 and calcium channels in relation to neurodegeneration. In cases of excess iron, DMT-1 is responsible for facilitating iron uptake [64]. The same study also demonstrated that an overactivation of the NMDAR gives rise to a state of neurotoxicity which can be avoided by iron chelation.
Intriguingly, we now know that DMT-1 is activated by the NMDAR (a calcium channel) via a series of specific steps, starting with calcium entry into cells; this may explain the significant decrease of calcium we observed in the CSF of participants in the AD group. These steps eventually lead to the activation of DMT-1 (Fig. 1) [64]. Albensi [65] reported increased entry of calcium immediately following NMDA exposure, with the addition of NMDA antagonists such as MK801 or D-APV reported to suppress this effect [66]. However, NMDA agonists were reported to induce neuronal death in a manner proportional to the agonist-induced increase in calcium entry [67].
Furthermore, the addition of iron or NMDA to hip-pocampal neurons in the rat activates calcium signaling and stimulates the extracellular signal-regulated kinase (ERK) signaling pathway. This effect was suppressed by the addition of the scavenger deferoxamine, an iron chelator. Thus, we can conclude that an excess of iron can play an NMDA-like role with respect to calcium entry into a cell [68, 69].
All of these observations point to a high level of iron activating the NMDAR and indicate that hyperactivation of the NMDAR by excess iron affects normal calcium trafficking, generating ROS formation [17], especially at the synaptic and BBB levels. This can subsequently lead to the synaptic failure and cell death seen in AD (Fig. 1).
Our results revealed a significant decrease in Aβ1–42 in the CSF of participants in the AD group compared to those in the CN group This finding is in line with previous reports that showed a low level of Aβ1–42 in CSF [70, 71]. In addition, our results showed a significant elevation of iron in CSF, positive correlations between Aβ1–42 and calcium levels in CSF, and between Aβ1–42, transferrin and ceruloplasmin in plasma (calcium: r(108) = 0.52, p < 0.001; transferrin: r(90) = 0.56, p < 0.001; ceruloplasmin: r(46) = 0.49, p < 0.001), and a negative correlation between the levels of Aβ1–42 and iron in CSF (r(107) = –0.63, p < 0.001) (Tables 2 3, Fig. 2).
Based on these observations, an important question arises: what is the relationship between all these components of CSF and plasma? The answer to this question will help establish a full picture of the etiology of AD.
Aβ1–42 and iron are located in the same regions of the brain [72], which can be explained by the fact that Aβ1–42 can bind iron. This might indicate a mechanism of clearing excess iron in the brain via plaque formation, which grows out of control as pla-que formation becomes excessive [73]. This proposed mechanism could change our understanding of the role of Aβ in the etiology of AD.
Duce et al. [74] demonstrated that AβPP does bind ferroportin and may help promote cellular iron export via stabilizing ferroportin. In the same study, the authors also reported that Aβ1–42 can inhibit this AβPP activity. Moreover, Abramov et al. [75] rep-orted that Aβ peptide causes calcium-dependent oxidative stress, and that the addition of Aβ1–42 cau-ses dramatic changes in calcium signaling.
All these observations suggest that iron meta-bolism, calcium metabolism, amyloidogenesis, and senile plaque formation are strongly linked, and that Aβ1–42 might be involved in a process that protects the brain against iron overload under physiological conditions.
In summary, our study revealed dysfunction of the iron antioxidant metabolism system in the bloodstream (as shown by low levels of Cp, Tf, and iron in plasma), dysregulation of calcium metabolism in CSF (as shown by a low level of calcium in CSF), and a low level of Aβ1–42 in CSF (Table 3).
A state of iron overload in the brain and the resulting accumulation of excess ROS may induce the inhibition of AβPP by Aβ1–42, which binds Fe3 + in the extracellular space in order to clear it. In addition, it may inhibit AβPP activity, reducing the ability of cells to export iron [49], and this may serve to correct the excess level of iron that has been created in the extracellular space. Under pathological conditions, a continuous supply of iron from the periphery to the brain causes the situation to become out of control, while intracellular calcium, which is increased by the iron overload, worsens the situation by inducing cell death (Fig. 1) [76]. This conclusion is also supported by the study of Crapper McLachan et al. [77], who reported that the administration of the chelator deferoxamine may slow down the progression of AD.
One particularly interesting observation from our results is that levels of iron and Aβ1–42 in CSF were found to be age-related (iron: r = 0.21, p = 0.008; Aβ1–42: r = –0.31, p = 0.001). However, levels of calcium in CSF, and transferrin and ceruloplasmin in plasma, seemed to be closely related to AD rather than age. This suggests that changes in iron metabolism occurring in the bloodstream play a role in the onset of AD, and merits further investigation.
These results also confirm that iron levels are age-related and that when this age-related effect is combined with defective iron metabolism in the blo-odstream, this may lead to the development of AD. Furthermore, our results show a direct link between cognitive deficits (MMSE) and the imbalance obs-erved in the bloodstream (transferrin r(90) = 0.81, p < 0.001; ceruloplasmin, r(46) = 0.37, p = 0.01), in addition to a link between the levels of calcium, iron, and Aβ1–42 in CSF. Similar findings were recently reported in a study by Ashraf et al. [30].
The greatest challenges and limitations of this study were the difficulty in recruiting the control group and the unexpected losses (participants withdrew from the study, resulting in a small sample size) that occurred during the study. A strength of the present study was that it considered AD as a multifactorial disease with several mechanisms contributing to its multiple etiology; this has often not been considered in previous research. This study aimed to fill the missing pieces of this puzzle by studying the most important components involved in iron metabolism and their interactions with Aβ. To our knowledge, this is the first time that these interactions have been investigated in CSF.
CONCLUSIONS
Studying the causative role of metal ions in neu-rodegenerative disease, especially in AD, is becoming increasingly attractive to neurobiologists. The main aims of this study were to investigate changes in iron metabolism and their role in the mechanisms underlying AD, and to determine the interaction of such changes with the primary marker protein of the disease, Aβ1–42.
Our results suggest that decreased activity in the plasma Cp-Tf system may contribute to the neurodegenerative process in AD by increasing the level of iron in CSF, leading to the production of ROS that cause oxidative damage to proteins, lipids, and DNA [16]. Furthermore, they suggest that the elevation of iron-induced ROS in CSF induces calcium uptake into cells, explaining the mechanism by which cell death occurs.
The resulting deleterious link between the dysfunction of iron, Aβ1–42, and calcium metabolism in plasma needs to be further investigated in order to understand exactly which dysfunction occurs first: iron, calcium, or Aβ accumulation. Although the kno-wledge gathered to date by studies at the systemic level is clearly insufficient, disruption at this level se-ems to be directly associated with amyloid pathology.
In summary, these findings indicate that the components of iron metabolism in the bloodstream influence the pathophysiological processes of AD and are potential therapeutic targets. In addition, the measurement of iron’s metabolism, regulation, and transporters in the bloodstream could be useful additions to existing biomarker panels in disease diagnosis.