
Abstract
Background:
Toxic amyloid-β protein (Aβ) conformers play an important role in the progression of Alzheimer’s disease (AD). The ratio of toxic conformer to total Aβ42 in cerebrospinal fluid (CSF) was significantly high in AD and mild cognitive impairment (MCI) due to AD using an enzyme-linked immunosorbent assay kit with a 24B3 antibody.
Objective:
We compared the toxic Aβ42, conformer at different stages of AD to identify its contribution to AD pathogenesis.
Methods:
We compared 5 patients with preclinical AD, 11 patients with MCI due to AD, 21 patients with AD, and 5 healthy controls to measure CSF levels of total Aβ42, total tau, tau phosphorylated at threonine 181 (p-tau), and toxic Aβ conformers. All were classified using the Clinical Dementia Rating. Cognitive function was assessed using the Japanese version of the Mini-Mental State Examination (MMSE-J).
Results:
Toxic Aβ conformer level was insignificant between groups, but its ratio to Aβ42 was significantly higher in AD than in preclinical AD (p < 0.05). Toxic Aβ42 conformer correlated positively with p-tau (r = 0.67, p < 0.01) and p-tau correlated negatively with MMSE-J (r = –0.38, p < 0.05).
Conclusion:
Toxic Aβ conformer triggers tau accumulation leading to neuronal impairment in AD pathogenesis.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is the most common age-related neurodegenerative form of dementia. It is characterized by pathological aggregates of amyloid-β protein (Aβ) and tau protein, senile plaques, and neurofibrillary tangles. Based on genetic evidence, biochemical data, and animal models, Aβ is thought to be responsible for the pathogenesis of AD (the amyloid hypothesis) [1–3]. Aβ is cleaved from amy-loid-β protein precursor (AβPP) by β- and γ-secre-tases and aggregates into the intermolecular β-sheet formation via a change from Aβ monomers into amyloid fibrils via oligomers or protofibrils. Aβ oligomer accumulation may be among the earliest signs of AD progression compared to other AD-re-lated events such as hyperphosphorylation of tau protein, decreased hippocampal volume, and lowered glucose metabolism [4].
24B3 is an antibody highly specific to the toxic conformer with a turn at 22 and 23 of Aβ42 (toxic turn) and recognizes not only the monomer with the toxic turn but also a potential dimer of Aβ42. Irie et al. previously reported that the ratio of toxic conformers to total Aβ42 in the cerebrospinal fluid (CSF) of patients with AD and mild cognitive impairment (MCI) due to AD was significantly higher than that of age-matched non-AD controls using an enzyme-linked immunosorbent assay (ELISA) kit with a 24B3 antibody [5]. Akiba et al. also found that the ratio of toxic Aβ42 conformer to total Aβ42 in patients with idiopathic normal pressure hydrocephalus (iNPH) was significantly higher than that in control groups, and the toxic conformer ratio was high in iNPH progressing to AD [6].
In this report, we compared the toxic Aβ42 conformer at different stages of AD and Parkinson’s disease (PD) using a 24B3 antibody ELISA and ide-ntified its contribution to pathogenesis and its usefulness for the diagnosis of AD.
MATERIALS AND METHODS
Participants
This study was approved by the Ethics Committees of Showa University Hospital (clinical trial identifier number: 1997) and conducted according to the principles of the Declaration of Helsinki. All participants and their proxies provided written informed consent before participation, with the possibility of opting out later.
All patients were registered in the memory clinic of Showa University Hospital between 2015 and 2019. All AD participants underwent brain MRI 99mTc, ECD-SPECT, and CSF analysis. All patients with PD underwent brain MRI, CSF analysis, dopamine transporter imaging using 123I-ioflupane SPECT, and car-diac 123I-metaiodobenzylguanidine scintigraphy to confirm PD diagnosis. We revised the diagnosis of each disease, and standardized neuropathological assessment was made according to internationally accepted criteria [7–10]. We included probable AD patients, a high likelihood of MCI due to AD, and preclinical AD (PC) patients with decreased evidence of CSF Aβ42 without dementia, based on the National Institute on Aging-Alzheimer’s Association criteria with high-level evidence of AD pathophysiological process. All PC, MCI, and AD patients were cla-ssified by clinical demented rating (CDR) and underwent the Japanese version of the Mini-Mental State Examination (MMSE-J) for cognitive assessment [11–13]. None had a history of cerebrovascular problems. We identified 23 age-matched AD, 13 MCI, and 23 PD patients. We also identified five patients with PC and prepared five age-matched normal CSF subjects from the National Center of Neurology and Psychiatry as a healthy control group (HC). For PC, two of five patients were preclinical stage 1 (asy-mptomatic amyloidosis), two other patients were preclinical stage 2 (amyloidosis with neurodegeneration) due to increased CSF p-tau, and the last patient was stage 3 (amyloidosis with neurodegeneration with subtle cognitive decline) due to high t-tau with subtle cognitive decline in preclinical AD [9].
We measured Aβ42, p-tau, t-tau, and toxic Aβ42 conformer in CSF and compared HC, PC, MCI, and AD. We correlated toxic Aβ42 conformer with p-tau and MMSE-J for AD (PC, MCI, and AD). We also compared 23 PD patients with MCI and AD (MCI/AD) for toxic Aβ42, conformer and its relationship to other CSF biomarkers.
CSF analysis
All CSF samples were centrifuged at 4°C and 1690×g for 10 min to remove cells and debris, ali-quoted, and stored in polypropylene tubes at –80°C until biochemical analysis. CSF concentrations of Aβ42 were analyzed using V-PLEX Aβ Peptide Panel 1 (6E10) (Meso Scale Discovery, Rockville, MD) with MESO QuickPlex SQ 120 (Meso Scale Diagnostics, LLC, Rockville, MD). CSF concentrations of total tau (t-tau) and tau phosphorylated at threonine 181 (p-tau) were measured using commercially available enzyme-linked immunosorbent assay kits, INNOTEST hTAU Ag and PHOSPHO-TAU (181P) (Fujirebio Europe, Inc., Ghent, Belgium) according to the manufacturer’s instructions. The inter- and in-tra-assay coefficients of variation were less than 20% for all assays. We should also note that the laboratory at Niigata University participates in the Alzheimer’s Association’s external quality control program for CSF biomarkers.
The toxic Aβ42 conformer was analyzed using the Human Amyloid β Toxic Oligomer (No. 27709, IBL, Japan) [5]. The bicinchoninic acid method was used for total protein measurement.
Statistical analysis
Demographic comparisons across the study groups were analyzed using ANOVA followed by the Tukey-Kramer test for multiple comparisons with R (ver. 3.5.0) software. Comparative analyses between the two groups were performed using t-tests. Correlation coefficients between values were calculated using Spearman’s rank correlation coefficient. A p-value less than 0.05 was considered statistically significant.
RESULTS
Demographic background of the groups
The demographic data between the groups are shown in Table 1. The mean age was 64.8 years for HC and 66.4 years for PC. They were significantly younger than the patients with MCI (mean age, 78.3 years) and AD (mean age, 78.6 years) (F (3,31) = 7.34, p < 0.01; NC versus AD; p < 0.01; NC versus MCI, p < 0.01; PC versus AD, p < 0.05; PC versus MCI, p < 0.05; PC versus NC, p = 0.98; MCI versus AD, p = 0.91)). The ɛ4 allele of the apolipoprotein E gene carrier ratio was 25% for MCI and 14.2% for AD. CDRs were 0 for HC and PC, 0.5 for MCI, and 1 to 3 for AD. The MMSE-J score was 16.7 for AD, which was significantly lower than those for PC (29.2) and MCI (24.9) (F(2, 27) = 25.4, p < 0.01; AD versus MCI, p < 0.01; AD versus PC, p < 0.01; MCI versus PC, p = 0.14). Aβ42 was significantly lower for PC (316 pg/mL), MCI (209 pg/mL), and AD (218 pg/ml) than for HC (1089.2 pg/mL) (F (3,31) = 129.36, p < 0.01; NC versus AD, p < 0.01; NC versus MCI, p < 0.01; NC versus PC, p < 0.01; PC versus MCI, p = 0.19; PC versus AD, p = 0.18; MCI versus AD, p = 1.00) (Fig. 1B). P-tau in AD was 84.3 pg/ml and it was significantly higher than that in PC (47.6 pg/mL) but insignificant compared to that in MCI (60.4 pg/mL) (F (2, 27) = 5.44, p = 0.01; PC versus AD, p < 0.05; PC versus MCI, p = 0.64, MCI versus AD, p = 0.08). T-tau in AD was 551 pg/mL, which was significantly higher than that in PC (224 pg/-mL), but was insignificant compared to that in MCI (349 pg/mL) (F (2,27) = 7.01, p < 0.01; MCI versus AD, p = 0.05; PC versus AD, p < 0.01; PC versus MCI, p = 0.49). For PD and MCI/AD, age was in-significant between the groups (p = 0.47). The mean Aβ42 in PD was 383.8 pg/mL and was significantly higher than that in MCI/AD (215.6 pg/mL) (p < 0.01). The mean p-tau in PD was 41.4 pg/mL and significantly lower than that in MCI/AD (76.7 pg/mL) (p < 0.01). The mean t-tau in PD was 201.7 pg/mL and significantly lower than that in MCI/AD (486 pg/mL) (p < 0.01).
The demographic data of HC, PC, MCI, AD, and PD groups
Results are presented as the median (minimum and maximum) or mean±SD and conducted by ANOVA followed by the Tukey-Kramer test formultiple comparisons. p values are shown as follows: p1 = HC versus AD, p2 = HC versus MCI, p3 = PC versus AD, p4 = PC versus MCI, p5 = PC versus HC, p6 = MCI versus AD, and p7 = MCI/AD versus PD. *p < 0.05, **p < 0.01 were considered to be statistically significant. HC, healthy control; PC, preclinical Alzheimer’s disease; MCI, mild cognitive impairment due to Alzheimer’s disease; AD, Alzheimer’s disease; PD, Parkinson’s disease; APOE, apolipoprotein; CDR, Clinical Dementia Rating; MMSE-J, Japanese version of the Mini-Mental State Examination.

Comparison of Aβ42 and toxic conformer between AD groups. Panel A shows that the toxic conformer was insignificant between groups (F (2, 32) = 0.37, n. s.). Panel B compares Aβ42 between the groups. PC, MCI, and AD were significantly lower than HC (p < 0.01; p < 0.01; p < 0.01). p < 0.05, based on variance (ANOVA) followed by the Tukey-Kramer test for multiple comparisons. Panel C compares the toxic conformer ratio to Aβ42. HC was significantly lower compared to PC, MCI, and AD (p < 0.01; p < 0.01; p < 0.01). AD was significantly higher than PC (p < 0.05). Panel D shows that toxic Aβ42 conformer and p-tau were positively correlated (r = 0.69, p < 0.01). p < 0.05, based on Spearman’s rank correlation coefficient. Panel E shows that p-tau was negatively correlated with MMSE-J (r = –0.35, p < 0.05).
Toxic Aβ42 conformer ratio differentiates between HC, PC, MCI, and AD
The level of toxic Aβ42 conformer was insignificant between HC, PC, MCI, and AD (F (3, 31) p = 0.09, n.s.) (Fig. 1A). The toxic Aβ42 conformer ratio to Aβ42 was significantly lower in HC (HC versus PC, p < 0.01; HC versus MCI, p < 0.01; HC versus AD, p < 0.01) and higher in AD than in HC and PC (AD versus HC, p < 0.01; AD versus PC, p < 0.05) (Fig. 1C). Toxic Aβ42 conformer correlated positively with p-tau (r = 0.67, p < 0.01) (Fig. 1D), and p-tau was negatively correlated with MMSE-J (r = –0.38, p < 0.05) (Fig. 1E).
Toxic Aβ42 conformer between MCI/AD and PD
For PD and MCI/AD, the toxic Aβ42 conformer was insignificant between groups (Fig. 2A), and Aβ42 was higher in PD (p < 0.01) (Table 1 and Fig. 2B). The toxic conformer ratio to Aβ42 was significantly lower in PD than in MCI/AD (p < 0.01) (Fig. 2C). The toxic conformer and Aβ42 positively correlated with p-tau (r = 0.80, p < 0.01; r = 0.62, p < 0.01). p < 0.05, based on Spearman’s rank correlation coefficient (Fig. 2D, E).
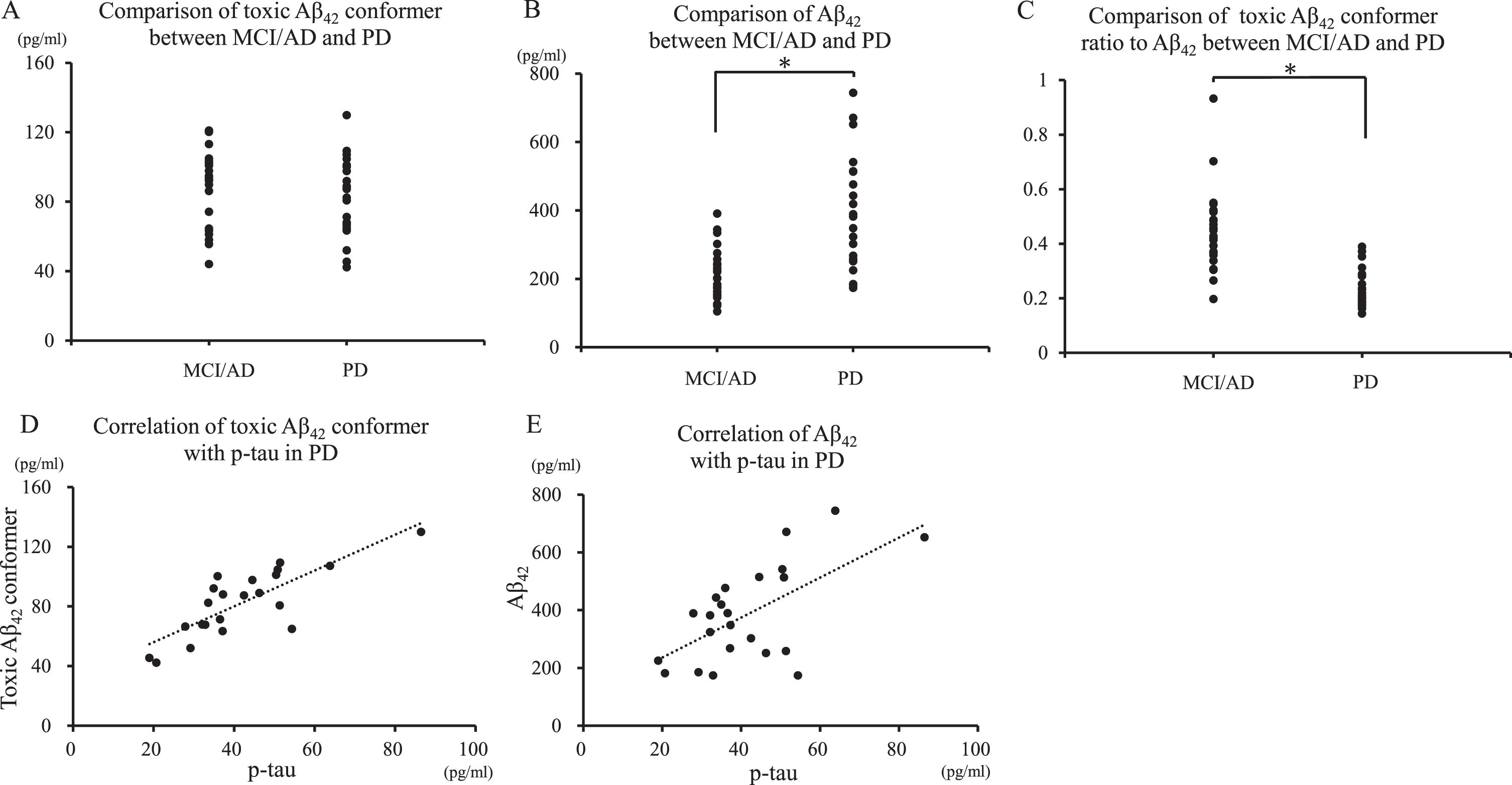
Comparison of Aβ42 and toxic conformer between MCI/AD and PD. Panel A shows that the toxic conformer was insignificant between MCI/AD and PD. Panel B shows that MCI/AD was significantly lower than PD (p < 0.01). p < 0.05, based on variance (ANOVA) followed by the Tukey-Kramer test for multiple comparisons. Panel C shows that the toxic conformer ratio to Aβ42 was higher in MCI/AD than in PD (p < 0.01). Panels D and E shows that toxic conformer and Aβ42 positively correlated with p-tau (r = 0.80, p < 0.01; r = 0.62, p < 0.01). p < 0.05, based on Spearman’s rank correlation coefficient.
DISCUSSION
The main finding of this study was that the toxic conformer ratio to Aβ42 increased exponentially between PC and AD groups, and toxic Aβ42 conformer was independently related to cognitive decline via positive correlation with tau pathology. Aβ and human tau accelerate each other’s pathology.
The mutation sites related to familial AD are turns in the central region of Aβ42. A deletion at Glu22 (E22Δ) of Aβ, known as an Osaka mutation, induces the formation of putative Aβ42 dimers and trimers and accelerates low n-order Aβ oligomerization without amyloid plaque formation [14–16]. The APP transgenic mice harboring this mutation showed intraneuronal accumulation of Aβ oligomers, hyperphosphorylation of tau, synapse loss, and memory impairment [17].
24B3 is an antibody highly specific to the conformer with a turn at 22 and 23 of Aβ42 (toxic turn) and recognizes not only the monomer with the toxic turn but also a potential oligomeric Aβ42 mainly dimer. The toxic turn enhanced aggregative ability, neurotoxicity, radical generation, and oligomeriza-tion in systematic proline scanning, solid-state nuc-lear magnetic resonance studies, and electron spin resonance [18, 19]. The toxic Aβ42 conformer ind-uced cognitive dysfunction without neuronal loss, indicating that Aβ oligomers induced only synapse toxicity in an APP transgenic mouse study [20]. Administration of 24B3 in AD model mice, double transgenic mice (PS2Tg2576), relieved executive dysfunction and spatial cognitive impairment and significantly reduced the toxic Aβ42 conformer levels in the soluble fraction without affecting amyloid plaque pathology [21].
Antibody therapy for AD targeting Aβ aggregates, including toxic oligomers, is now attracting attention. It is important for future research to clarify the elevation of toxic Aβ oligomers at different stages of AD. A previous study showed that the ratio of toxic to total Aβ42 in the CSF of AD patients is significantly higher than that of control subjects, as measured by sandwich ELISA using antibodies 24B3, suggesting that the proportion of toxic Aβ42 conformer may contribute to AD pathologies [5]. In this study, using more samples, the analysis was performed separately for preclinical AD, MCI due to AD, and AD, and revealed that the toxic Aβ42 conformer ratio to Aβ42 was elevated in the preclinical stage of AD.
In this study, the toxic conformer ratio to total Aβ42 was positively correlated with CSF p-tau levels. As reported by Bateman et al. [22], a decrease in Aβ42 concentration in CSF precedes Aβ deposition and increased tau protein levels in the CSF. It has also been reported that exogenous Aβ42 oligomers induce hyperphosphorylation and neurodegeneration of tau in cultured primary rats [23, 24]. Furthermore, it is known that serum insulin secretory deficiency promotes toxic Aβ42 conformer and co-aggregation of phosphorylated tau in the hippocampus in animal experiments [25]. The toxic Aβ42 conformer triggers tau accumulation, leading to neuronal impairment in AD pathogeneses and can cause cognitive decline.
Our study clearly showed that there was a significant amount of toxic Aβ42 conformer in HC. This may be because the anti-toxic turn antibody 24B3 may also detect the toxic conformer as a monomeric form of Aβ42 as well as its toxic oligomers in HC. Furthermore, we cannot exclude the possibility that elevation of toxic Aβ42 conformer might begin at an earlier stage of AD pathogenesis and that the HC group of around 65 years included subjects with an earlier stage of AD. The monomeric form of toxic Aβ42 conformer might be less toxic to cognition in the presence of significant amount of Aβ42 . Toxic Aβ42 conformer forms dimers and trimers and high molecular weight oligomers throughout the progression of AD. The elevation of its ratio to Aβ42 may be important for the pathogenesis and diagnosis of AD.
Hypothetical models of dynamic biomarkers of the AD pathological cascade have been proposed [9, 22]. AD initiates from abnormal processing of Aβ peptides, leading to the formation of Aβ plaques in the brain and reduction of Aβ42 in CSF, while individuals are still cognitively normal. After a lag period, neuronal dysfunction and neurodegeneration become the dominant pathological processes. Biomarkers of neuronal injury and neurodegeneration are increased CSF tau and structural MRI measures of cerebral atrophy. Referring to these models with a recent longitudinal study [26], we present an estimated timeline of dynamic biomarkers and the CSF toxic Aβ42 conformer in the AD pathological cascade (Fig. 3). The CSF toxic conformer increased its ratio to CSF Aβ42 in the early preclinical stage. It accelerates tau accumulation in the late PC stage, leading to neuronal impairment in the MCI stage and causes cognitive decline throughout the MCI and AD stages. Its ratio to CSF Aβ42 is shown as a dotted line in the late AD stage because further research is needed.
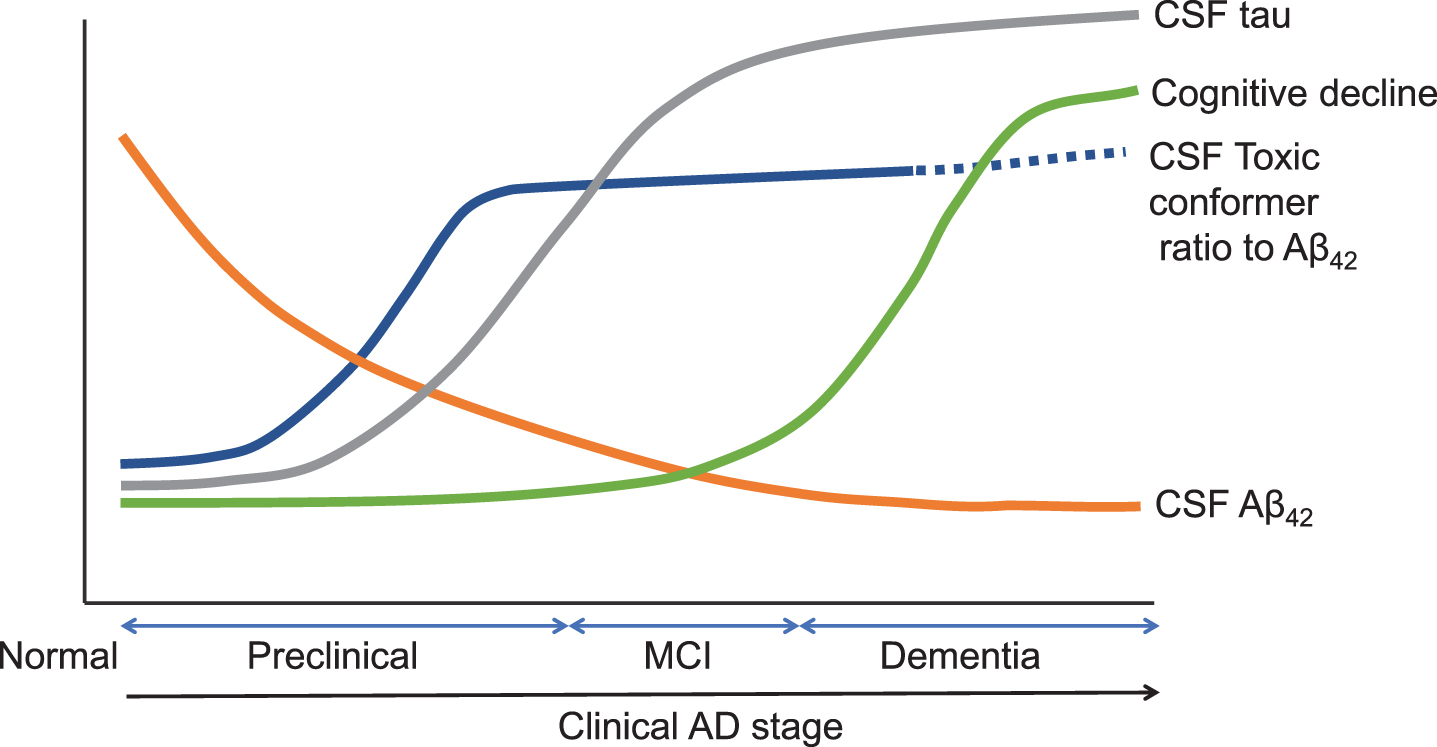
Estimated timeline of dynamic biomarkers and the CSF toxic Aβ42 conformer in AD pathological cascade. Estimated timeline of AD and relations with CSF Aβ42, CSF tau, CSF toxic Aβ42 conformer ratio to Aβ42, and cognitive decline. CSF biomarker changes and cognitive decline (y-axis) as a clinical AD stage (x-axis). The figure illustrated referring to Sperling et al. [9] and Bateman et al. [22].
We also compared the toxic Aβ42 conformer between PD and AD. Its ratio to Aβ42 in PD was significantly lower than that of MCI/AD but correlated positively with p-tau in CSF. This indicates that it may also be involved in the pathological changes in PD. α-synuclein (αS) is a neuropathological hallmark of PD. Because fibrils and oligomers of Aβ42 and αS act as seeds and affect each other’s aggregation pathways in vitro, interactions between Aβ and αS are thought to be involved in the pathogenesis of AD and Lewy body diseases [27]. In PD with dementia and dementia with Lewy body patients with AD co-pathology, regional tau pathology relates to cognitive performance [28]. Cerebral tau pathology, in addition to αS and Aβ pathology, are the strongest pathological predictors of a shorter interval between onset of motor and dementia symptoms and survival in Lewy body disorder (PD and dementia with Lewy bodies) [29]. Recently, αS has also been reported as a modulator of tau pathology and spreading in the brains of patients with AD, PD with dementia, and dementia with Lewy bodies. Both in vitro and in vivo, accumulation of tau aggregates increased by simultaneous introduction of αS mouse preformed fibrils and AD lysate–derived tau seeds (AD-tau) [30]. Taken together with our results, toxic Aβ42 conformer is involved not only in AD pathogenesis but also in PD pathogenesis via accelerating the interactions between Aβ and αS, leading to tau aggregates.
Footnotes
ACKNOWLEDGMENTS
The authors wish to thank Tatsunori Oguchi, Pha-rmacological Research Center, Showa University, Tokyo, Japan, for his assistance in toxic Aβ42 con-former assay. This work was supported by Grants-in-Aid for Young Scientists (Kakenhi) from the Japan Society for the Promotion of Science (JSPS) under Grants JP20K19111 (A.F.), Grants-in-Aid for Scientific Research (Kakenhi) from JSPS under Grants JP19H00921 (K.I.), JP26461266 and JP19K07965 (K.O.), Research and Development Grants from the Japan Agency for Medical Research and Development (16dk0207021h0001) (K.O.), and AMED under Grant Number JP20dm0207073 (T.I.).