
Abstract
As an established treatment for movement disorders, deep brain stimulation (DBS) has been adapted for the treatment of Alzheimer’s disease (AD) by modulating fornix activity. Although it is generally regarded as a safe intervention in patients over 65 years of age, the complex neurophysiology and interconnection within circuits connected to the fornix warrants a careful ongoing evaluation of the true benefit and risk potential of DBS on slowing cognitive decline in AD patients. Here we report on a patient who died long after being implanted with a DBS device who donated her brain for neuropathologic study. The autopsy confirmed multiple proteinopathies including AD-related change, diffuse neocortical Lewy body disease, TDP-43 proteinopathy, and a nonspecific tauopathy. We discuss the possible mechanisms of these overlapping neurodegenerative disorders and caution that future studies of DBS for AD will need to take these findings into consideration.
INTRODUCTION
Deep brain stimulation targeting the fornix (DBS-f) as a treatment of Alzheimer’s disease (AD) is emerging as a safe intervention with the potential to slow the rate of decline of patients over 65 years of age. In a Phase II study, a lower average score on the Clinical Dementia Rating Sum of Boxes (CDR-sb) was observed in the subgroup of patients > 65 years after 12 months of stimulation (n = 15) compared to sham stimulation (n = 15). After DBS-f was activated in both groups followed for up to two years, there was evidence that DBS-f may have slowed the rate of decline for all patients≥65 [1–3].
METHODS
A 72-year-old woman initially presented with mild cognitive impairment in 2006 with subtle memory loss and disorganization. In 2008, she became noticeably less verbal and began to experience progressive memory impairment, difficulty making decisions, and mood instability. In 2012, she entered into the DBS-f trial for AD [1]. Shortly following entrance into the trial, she moved to assisted living and withdrew from the trial in 2015. However, her family elected to continue DBS-f stimulation due to the pro-mising initial trial results. In 2018, she became septic due to a urinary tract infection and died.
Her son consented to an autopsy limited to a neuro-pathological evaluation; this was performed at Johns Hopkins Hospital. Microscopic examination was co-nducted using hematoxylin and eosin (H&E), Hirano silver staining, and immunohistochemistry for alpha-synuclein, beta-amyloid 4E10, nonphospho-specific TDP-43, ubiquitin, and phospho-tau as previously described [4].
RESULTS
The patient was cachectic at the time of autopsy, a common finding in patients with neurocognitive decline. The brain was extracted and weighed 1,150 g (with a reference range of 1050–1550 g). The external surface of the brain had bilateral deep brain sti-mulator leads, mild symmetric frontal and parietal atrophy, and severe temporal atrophy. There was no atherosclerotic disease. The brain was coronally sectioned to reveal sulcal widening and loss of white matter in conjunction with moderate symmetric hydrocephalus. The deep brain stimulator electrode tracks were mostly evident in the distal part, with the leads appropriately located adjacent to the bilateral columns of the fornix, just proximal to the mamm-illary bodies. There was no grossly apparent hemorrhage or softening along the two electrode tracks. There was severe atrophy and softening of the amyg-dalae, entorhinal cortices, and hippocampal formati-ons. The brainstem and cerebellum were transversely sectioned to reveal pallor of the substantia nigra and locus coeruleus.
Microscopic examination demonstrated pseudol-aminar necrosis in the temporal lobe, hippocampal sclerosis, and severe neuronal dropout in the amygdala. The substantia nigra and locus coeruleus dem-onstrated severe neuronal dropout with pigment incontinence and a reactive gliosis.
Amyloid-β immunohistochemistry and silver sta-ining (Fig. 1) demonstrated findings consistent with intermediate level Alzheimer’s disease related neuropathologic change (ADRNC) (A3, B2, C3) acc-ording to the 2012 NIA-AA criteria [5]. In addition, tau immunohistochemistry demonstrated high concentrations of neurofibrillary tangles and neurites in the striatum, subthalamic nucleus, substantia nigra, locus coeruleus, and medulla, and low concentrations of neurofibrillary tangles and neurites in the pallidum and basis pontis.
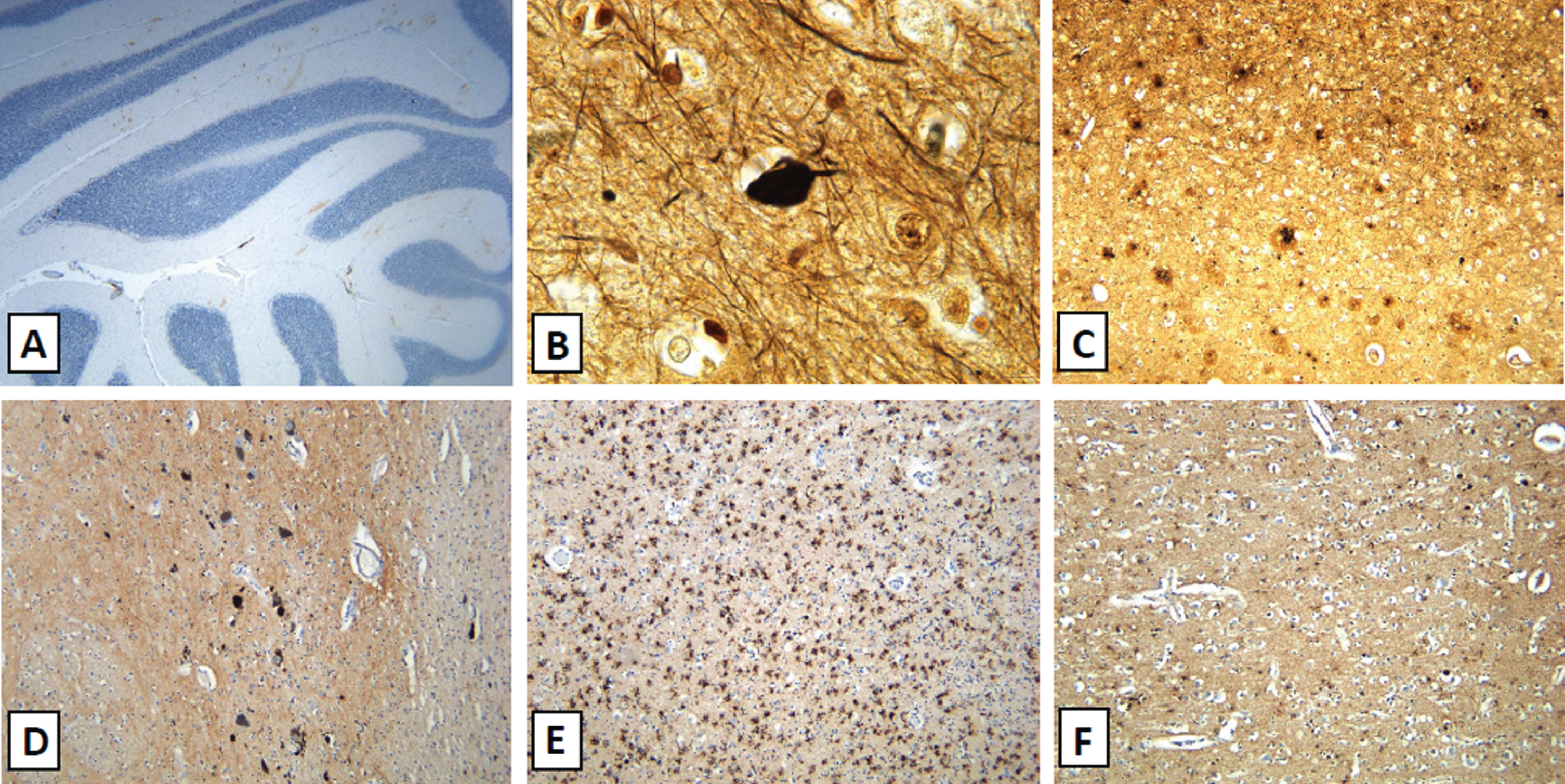
ADRNC and diffuse Lewy body disease neuropathological findings. The brain demonstrated extensive positive beta-amyloid staining in the molecular layer of the cerebellum (A), neurofibrillary tangles extending into the temporal lobe (B), and frequent neuritic plaques in the inferior parietal lobule (C). These findings are associated with intermediate level Alzheimer’s disease related neuropathologic change. There was extensive involvement of the substantia nigra (D) by Lewy bodies shown on alpha synuclein staining. There was also extensive involvement of both neurons and glial by alpha synuclein in the amygdala (E) and the middle frontal gyrus (F) shown by alpha synuclein immunohistochemistry. These findings were compatible with diffuse (neocortical) Lewy body disease).
Immunohistochemistry for alpha-synuclein dem-onstrated a neocortical distribution of Lewy body disease according to consensus criteria [6]. TDP-43 immunohistochemistry (Fig. 2) demonstrated marked neuronal cytoplasmic inclusions and nuclear clearance in the amygdala, entorhinal cortex, granule cell layer of the dentate gyrus, transentorhinal cortex, occ-ipitotemporal gyrus, and middle frontal gyrus. There were sparse interspersed short neuritic processes. The pattern of TDP-43 staining in the dentate gyrus was consistent with ubiquitin staining. This pattern of sta-ining can be seen in frontotemporal lobar deg-eneration-TDP-43 (FTLD-TDP) Type A [7]. This staining in the presence of hippocampal sclerosis and advanced age fulfils diagnostic criteria for limbic-predominant age-related TDP-43 encephalopathy neuropathologic change (LATE-NC) [8].
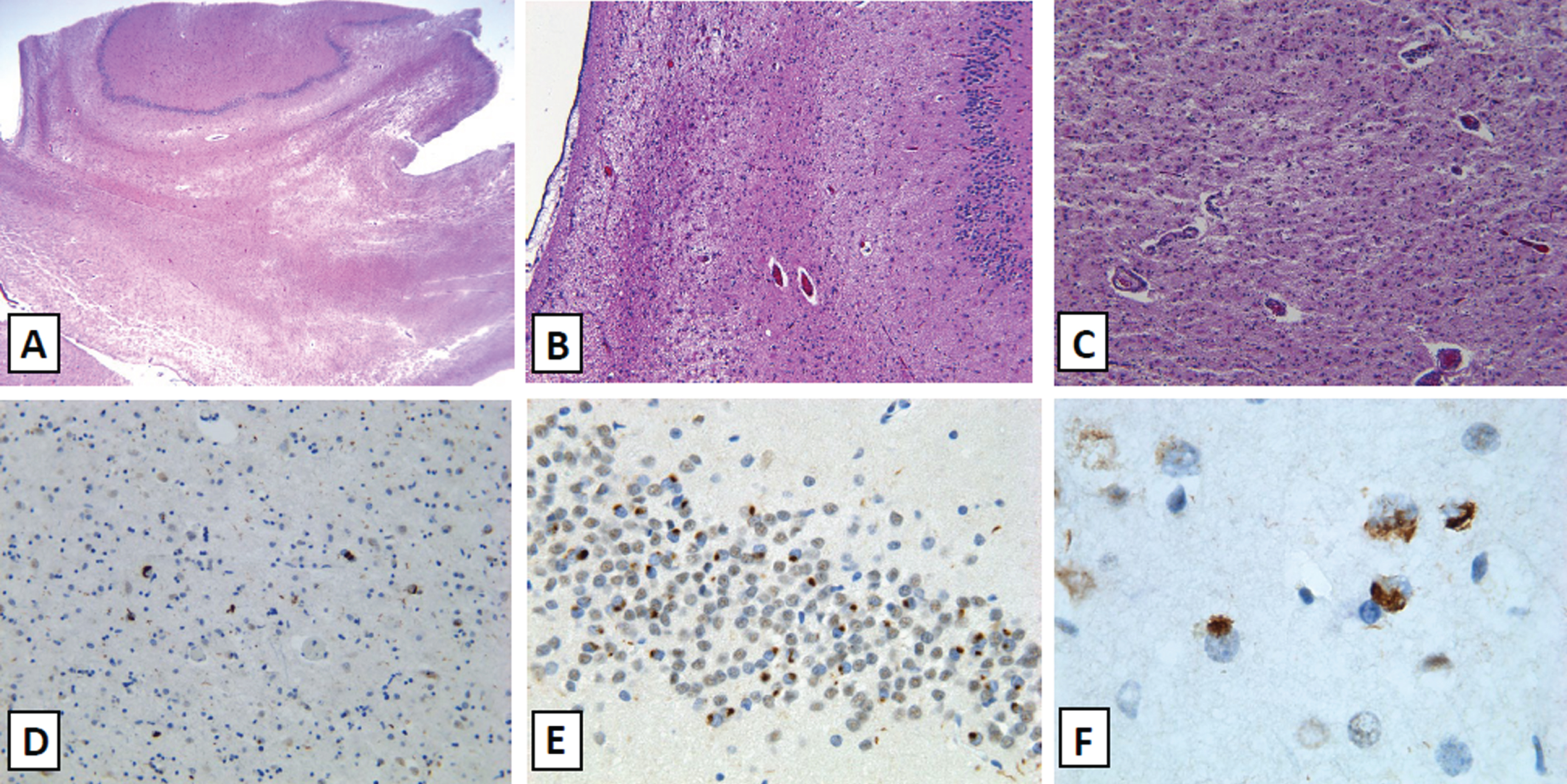
TDP-43 associated neuropathological findings. The patient demonstrated extensive neuronal loss in the mesial temporal lobe (A) with severe hippocampal sclerosis and near complete neuronal loss in the cornu ammonis (B). There is also extensive neuronal dropout in the amygdala with an associated gliosis and neuropil rarefaction (C). TDP-43 immunohistochemistry demonstrates extensive neuronal involvement by cytoplasmic inclusions and negative nuclear expression in neurons with sparse neuritic processes in the amygdala (D), granule cell layer of the dentate gyrus (E), and the middle frontal gyrus (F).
DISCUSSION
DBS-f was trialed to modulate neuronal activity within memory circuits with the goal of stabilizing or restoring cognitive function [1]. Although underlying mechanisms are still being elucidated, DBS-f is considered to be safe and well tolerated in AD patients≥65 years old. When used in clinically diagnosed patients with AD aged < 65 years, this treatment had variable effects that may have been complicated by multiple underlying brain pathologies [3]. Despite the use of DBS-f, the patient’s husband/caregiver felt that she declined significantly during and after the trial. Her Mini-Mental State Examination (MMSE) score was 23/30 prior to DBS-f implantation, which declined to 15/30 and becoming dependent on activities of daily living (ADLs) within three years. She died six years after the implantation. Clinically the patient had findings diagnostic of AD with notable progressive nonfluent aphasia early in the disease course. She was diagnosed with multiple proteinopathies postmortem including ADRNC, diffuse neocortical Lewy body disease, FTLD-TDP Type A or LATE-NC, and a nonspecific tauopathy.
The correlation between the clinical presentation and the pathologic findings is complicated by multiple competing disease criteria and nomenclatures. The clinical picture correlates well with the neuropathologic findings of ADRNC and neocortical (diffuse) Lewy body disease. These diseases are often seen together and commonly involve TDP-43 pathology [9]. Recent diagnostic criteria for limbic-predominant age-related TDP-43 encephalopathy also fit the clinical findings (78 years old with severe amnestic dementia) and pathological findings (stage 3 spread of TDP-43 with hippocampal sclerosis) [8]. Despite these associations, the extent and distribution of neuronal TDP-43 involvement was more typical of FTLD-TDP Type A. This is associated with behavioral variant frontotemporal dementia and progressive nonfluent aphasia; only the latter was noted in this patient [7]. The clinical significance of the tauopathy is uncertain.
Although the rate of cognitive/functional decline in this patient appears only slightly faster than what is expected for AD patients, the extent of neuropathological disease is unexpected. As we continue to evaluate the utility of DBS-f, it is important to keep in mind the potential effects of TDP-43 proteinopathies. This case also serves to show the diagnostic difficulty of separating LATE-NC from FTLD-TDP in both clinical and pathological realms. Though clinically this case fits well with Alzheimer’s disease, it also fulfils diagnostic criteria of LATE-NC. Yet, the extent and severity of disease with the history of progressive nonfluent aphasia might make FTLD-TDP43 better diagnostic fit.
It is increasingly clear that patients diagnosed clinically with AD often suffer from several neurodegenerative diseases which often overlap [10]. It is most remarkable that this patient’s neurodegeneration involved at least three: Alzheimer’s disease typified by neurofibrillary tangles and neuritic plaques, dementia with Lewy bodies, and either LATE-NC or FTLD-TDP43 (or both). Notably, the pathological changes associated with these mechanisms were relatively advanced. The diagnostic criteria for LATE-NC have only recently been published [8] and appears to be a particularly prevalent cause of dementia in older persons such as this participant. It is possible that there are other non-AD pathologies waiting to be discovered, and sobering to note how many non-AD pathologies were present in this participant who presented clinically with AD. It is possible that this overlap of mechanisms is coincidental. However, a question also arises that whether DBS could contribute in an unknown way to one or more of these mechanisms of neurodegeneration. Such an effect of DBS has not been reported in autopsy studies of DBS for Parkinson’s disease [11], and has not been observed in animal models treated with DBS [12, 13]. Future studies of DBS for AD will need to take these possible safety concerns into consideration.