
Abstract
Background:
Previous studies suggested a link between various infectious pathogens and the development of Alzheimer’s disease (AD), posing the question whether infectious disease could present a novel modifiable risk factor.
Objective:
To assess whether infectious disease burden due to clinically apparent infections is associated with an increased risk of AD.
Methods:
We conducted a population-based nested case-control study using the United Kingdom Clinical Practice Research Datalink. We included all dementia-free subjects ≥50 years of age enrolling in the database between January 1988 and December 2017. Each case of AD identified during follow-up was matched with up to 40 controls. Conditional logistic regression estimated adjusted odds ratios (ORs) with 95% confidence intervals (CIs) of AD associated with ≥1 infection diagnosed > 2 years before the index date compared with no infection during the study period. We further stratified by time since first infection and cumulative number of infections.
Results:
The cohort included overall 4,262,092 individuals (mean age at cohort entry 60.4 years; 52% female). During a median follow-up of 10.5 years, 40,455 cases of AD were matched to 1,610,502 controls. Compared with having no burden of infectious disease, having a burden of infectious disease was associated with an increase in the risk of AD (OR, 1.05; 95% CI, 1.02 to 1.08). The risk increased with longer time since first infection, peaking after 12–30 years (OR, 1.11; 95% CI, 1.05–1.17). The risk did not increase with cumulative number of infections.
Conclusion:
The overall risk of AD associated with infectious disease burden was small but increased gradually with longer time since first infection.
INTRODUCTION
Dementia currently affects around 50 million people globally with nearly 10 million cases being newly diagnosed every year [1]. Alzheimer’s disease is the most common form of dementia contributing up to 70% of cases [1]. Given the ageing population and increasing life expectancies, the burden of Alzheimer’s disease is projected to dramatically increase in the following decades. Thus, ongoing research has been dedicated toward understanding the pathology of this disease in order to develop effective treatment and prevention strategies.
To date, several modifiable risk factors for Alz-heimer’s disease have been identified including smo-king, obesity, or arterial hypertension [2]. However, randomized controlled trials studying the effects of multimodal interventions targeting several of these risk factors showed little [3] or no efficacy [4, 5]. Interestingly, evidence from many pre-clinical, serological, and postmortem studies has suggested a link between various infectious pathogens and the development of Alzheimer’s disease [6–13], posing the question whether infectious disease could present a novel modifiable risk factor. Moreover, studies assessing the association between clinically apparent infections and the risk of Alzheimer’s disease or overall dementia reported increased risks of up to 260% [14–18]. However, these studies had methodological limitations including reverse causality, selection bias, and important residual confounding, which render the interpretation of their findings difficult [14–18]. In addition, the role of cumulative infectious disease burden and timing of infections with respect to Alzheimer’s disease remains poorly understood.
Taken together, current literature lacks robust epidemiological evidence on the potential association between infectious disease burden and the risk of Alzheimer’s disease. Thus, our population-based nested case-control study assessed whether infectious disease burden, defined by clinically apparent infections easily detectable in routine clinical practice and related to pathogens previously linked to dementia, is associated with an increased risk of Alzheimer’s disease.
METHODS
Data source
We conducted a population-based nested case-control study using the United Kingdom (UK) Clinical Practice Research Datalink (CPRD) Gold. The CPRD contains the medical records of over 11 million patients enrolled across 700 UK general practices and is one of the largest databases of longitudinal medical records from the primary care setting in the world [19]. Age, sex, and ethnicity distributions of patients in the CPRD are broadly representative of the UK population [19]. In addition, because general practitioners in the UK serve as a first point of contact for non-emergency health-related issues, the database contains useful information on routinely recorded symptoms, laboratory tests, diagnoses, therapies, health-related behaviors, and referrals to secondary care [19]. Medical diagnoses and procedures are recorded using the Read code classification, a hierarchical coding system containing over 80,000 terms encompassing the various aspects of a patient’s health status [20]. The CPRD undergoes regular quality controls, and its valid and high-quality health data makes it a favorable data source for epidemiological research covering a vast range of health outcomes [19].
Study population
We included all subjects at least 50 years of age enrolled in the CPRD between January 1, 1988 and December 31, 2017. Cohort entry date was defined as the date of the 50th birthday of the subject or one year after their date of enrolment in the CPRD, whichever occurred later. We then excluded subjects with a prior diagnosis of any dementia, including mild cognitive impairment, and those with early symptoms suggestive of dementia (e.g., memory impairment, aphasia, apraxia, or agnosia) at any time before cohort entry. We also excluded subjects treated with medications indicated for dementia including acetylcholinesterase inhibitors (i.e., donepezil, rivastigmine, or galantamine) and N-methyl-D-aspar-tate receptor antagonists (i.e., memantine) at any time before cohort entry. Cohort members were followed from the date of cohort entry until the date of the first outcome event (defined below), end of registration with the general practice, death from any cause, or the end of the study period (i.e., 31 December 2019), whichever occurred first.
Case definition
Within the study cohort, we identified all subjects with a first-ever diagnosis of Alzheimer’s disease at any time after cohort entry. We defined Alzheimer’s disease based on a modified algorithm initially developed and validated by Imfeld and colleagues [21], which has previously been used by our group [22]. Using this algorithm, Alzheimer’s disease was defined by meeting at least one of the following criteria: 1) a diagnosis of Alzheimer’s disease with at least one prescription of a medication for dementia, 2) a diagnosis of unspecified dementia with at least two prescriptions of a medication for dementia, 3) at least two diagnoses of Alzheimer’s disease, 4) a diagnosis of Alzheimer’s disease after a dementia test (e.g., Mini-Mental State Examination, abbreviated mental test) or a referral to a specialist (e.g., neurologist, psychiatrist, geriatrician, psychogeriatrician) or a neuroimaging assessment (e.g., magnetic resonance imaging, computed tomography, single-photon emission computed tomography), or 5) a diagnosis of Alzheimer’s disease with any dementia symptoms (e.g., memory impairment, aphasia, apraxia, agnosia) in any sequence. The index date (i.e., date of Alzheimer’s disease diagnosis) was defined as the date of the last event contributing to the definition. The quality of the recording of Alzheimer’s disease in the CPRD has been shown to be high, with a positive predictive value of 83% [23].
Control selection
Each case of Alzheimer’s disease was matched with up to 40 controls who belonged to the risk set defined by the case (i.e., those subjects still at risk of the event at the index date) on age (±1 year), sex, cohort entry date (±1 year), and duration of follow-up. The high number of controls was chosen to minimize feasibility issues in secondary analyses related to the potential scarcity of matched controls. The date resulting in the same duration of follow-up for the case and controls was set as the index date for the controls. Controls could contribute to different risk sets and could subsequently become a case. For our analyses, we only used cases and controls with at least two years follow-up given the use of a two-year lag period in the assessment of exposure (see below).
Exposure definition
For cases and controls with at least two years of follow-up, we identified all diagnoses of clinically apparent infections potentially involving pathogens which have previously been linked to the pathophysiology of Alzheimer’s disease regardless of the proposed mechanism. These infections included herpes labialis or genitalis (Herpes simplex virus) [9], cytomegalovirus related hepatitis, retinitis, colitis, mononucleosis, or other infections [10], Lyme disease (Borrelia burgdorferi) [11, 24], gingivitis (Porphyromonas gingivalis) [12], urinary tract infections (Escherichia coli) [13], gastritis (Helicobacter pylori) [8], pneumonia (Chlamydophila pneumonia) [7, 25], and candidiasis (Candida albicans) [6]. Infections due to pathogens with no potential link to the pathophysiology of Alzheimer’s disease (e.g., influenza, common cold) were not considered in the analyses. Subjects with a clinical diagnosis of any of these infections two years or more before the index date were considered as having a burden of infectious disease, while those without a diagnosis of any of these infections during that time period were considered as having no burden of infectious disease. Subjects with a diagnosis of any of these infections only within the 2-year period before the index date were also considered as having no burden of infectious disease. This 2-year ‘lag period’ was introduced given the insidious (i.e., non-acute) nature of the study outcome, and also to account for the delays associated with the diagnosis of Alzheimer’s disease [2, 26].
Statistical analysis
Conditional logistic regression was used to compute odds ratios of Alzheimer’s disease associated with infectious disease burden, compared with no infectious disease burden. Odds ratios are unbiased estimators of hazard ratios, with little or no loss in precision [27, 28]. In addition to the matching factors, estimates were further adjusted in the regression model for the following potential confounders associated with Alzheimer’s disease, measured at any time before cohort entry: body mass index category (< 25 kg/m2, 25–29 kg/m2, ≥30 kg/m2, unknown; last measurement before cohort entry), smoking sta-tus (ever, never, unknown), alcohol-related disorders (including alcoholism, alcoholic cirrhosis, or alcoholic hepatitis), arterial hypertension, atrial fibrillation, congestive heart failure, coronary artery disease, stroke or transient ischemic attack, peripheral vascular disease, dyslipidemia, diabetes mellitus, chronic kidney disease, liver disease, depression, epi-lepsy, Parkinson’s disease, traumatic brain injury, osteoporosis, hypothyroidism, and cancer. We also included the use of the following drugs in the two years prior to the index date: oral anticoagulants, antiplatelet agents, opioids, lipid-lowering drugs, beta-blockers, thiazides, angiotensin-converting en-zyme inhibitors, angiotensin II receptor blockers, calcium channel blockers, antipsychotics, non-steroidal anti-inflammatory drugs, and antidepressants. In the case of missing data (expected for the covariates body mass index and smoking), a separate category (‘unknown’) was created to classify this missing information.
Secondary analyses
We conducted five exploratory secondary analyses. First, to examine a potential ‘dose-response’ relation between infectious disease burden and the risk of Alzheimer’s disease, we estimated odds ratios for each of the following categories: 1, 2-3, and > 3 infections. Second, to examine a potential ‘time-response’ relation between infectious disease burden and the risk of Alzheimer’s disease, we estimated odds ratios for each of the following categories: 0–4.9, 5–7.9, 8–11.9, and 12–30 years since the time of the first infection (first infection after the 50th birthday; cut-offs for the different categories were based on the distribution of durations of follow-up among the controls). To account for the scenario of a non-linear association, we also modeled time since first infection as a continuous variable using restricted cubic splines with five interior knots [29]. Third, we examined the association by specific type of infection (i.e., herpes, cytomegalovirus related infection, Lyme disease, gingivitis, urinary tract infection, gastritis, pneumonia, candidiasis). Finally, we stratified by age (< 65 years versus≥65 years) and sex to assess a potential effect modification, since advanced age and female sex are established risk factors of Alzheimer’s disease [30, 31].
Sensitivity analyses
We also performed several sensitivity analyses to assess the robustness of our findings. First, given the uncertainty regarding the latency of a potential association between infectious disease burden and the development of Alzheimer’s disease, we repeated the primary analysis after increasing the lag period to 3, 5, and 10 years. Second, we censored follow-up at dementia diagnoses of non-Alzheimer’s disease etiology (e.g., vascular dementia, alcoholic dementia). Third, we restricted the medical codes for pneumonia to those with a clear link to Chlamydophila pneumonia to reduce exposure misclassification due to pneumonia caused by other infectious pathogens (e.g., pneumococci, viruses; medical codes for other infections remained unchanged). Finally, given that some of the previous studies assessed the association between infectious disease burden and the risk of overall dementia (instead of Alzheimer’s disease specifically) [16, 17], we repeated the analyses after expanding our outcome definition to include any diagnosis of dementia (see Supplementary Table 1 for Read codes). All analyses were conducted with SAS version 9.4 (SAS institute, Cary, NC).
Standard protocol approvals, registrations, and patient consents
The study protocol was approved by the Independent Scientific Advisory Committee of the CPRD (protocol 19_236R) and by the Research Ethics Board of the Jewish General Hospital, Montreal, Canada. Written consent from participants was not required due to use of anonymized data and no direct patient involvement.
RESULTS
The study cohort included a total of 4,262,092 dementia free individuals who were at least 50 years of age and enrolled in the CPRD between January 1, 1988 and December 31, 2017 and followed until December 31, 2019 (Fig. 1). Mean (standard deviation) age at cohort entry was 60.4 (11.5) years, and 52.1% were female. During a median (interquartile range) follow-up of 10.5 (6.2 to 14.6) years, 42,912 individuals in the study cohort were diagnosed with Alzheimer’s disease (crude incidence rate, 2.3 per 1000 person-years). Most diagnoses were based on the combination of a diagnostic code of Alzheimer’s disease accompanied either by respective symptoms, or tests for dementia, referrals to specialists, and neuroimaging assessments (Supplementary Table 2).

Flowchart showing the construction of the study cohort. CPRD, Clinical Practice Research Datalink.
We matched 40,455 cases of Alzheimer’s disease with at least two years of follow-up to 1,610,502 controls from the study cohort. Characteristics of cases and their matched controls are presented in Table 1. Cases were similar to controls except that they were more likely to have previously used antipsychotics or antidepressants. Compared with having no burden of infectious disease, having a burden of infectious disease was associated with a small increase in the risk of Alzheimer’s disease (odds ratio, 1.05; 95% confidence interval, 1.02 to 1.08) (Table 2). There was no evidence of a dose-response relation, with the risk of Alzheimer’s disease not significantly changing with cumulative number of infections (Table 2). However, there was a suggestion of a time-response relation, with the risk of Alzheimer’s disease gradually increasing with longer time intervals since the first infection (peak after 12–30 years: odds ratio, 1.11; 95% confidence interval, 1.05 to 1.17; p for trend = 0.0003) (Table 2, Fig. 2).
Baseline characteristics of cases of Alzheimer’s disease and their matched controls*
S = Cells with less than 5 counts are suppressed as per the confidentiality policies of the Clinical Practice Research Datalink. *Numbers are presented as n (%) unless otherise specified. aFor controls, means and percentages were weighted by the inverse number of controls matched to each case. bMeasured in the two years prior to index date as a surrogate measure of overall health. cIncludes beta-blockers, thiazide diuretics, angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, and calcium channel blockers.
Crude and adjusted odds ratios for the association between infectious disease burden and the risk of Alzheimer’s disease (overall and stratified by cumulative number of infections and time since first infection)
OR, odds ratio; CI, confidence interval. *Given the use of a 2-year lag period in the definition of exposure, the minimum time since first infection was 2 years. **Matched on age, sex, date of cohort entry, and duration of follow-up. ***Adjusted for body mass index, smoking, alcohol-related disorders, arterial hypertension, atrial fibrillation, congestive heart failure, coronary artery disease, stroke or transient ischemic attack, peripheral vascular disease, dyslipidemia, diabetes mellitus, chronic kidney disease, liver disease, depression, epilepsy, Parkinson’s disease, traumatic brain injury, osteoporosis, hypothyroidism, cancer, oral anticoagulants, antiplatelet agents, opioids, lipid-lowering drugs, beta-blockers, thiazides, angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, calcium channel blockers, antipsychotics, non-steroidal anti-inflammatory drugs, and antidepressants. ±p value for trend was 0.13 in the dose-response analysis. ±±p value for trend was 0.0003 in the time-response analysis.
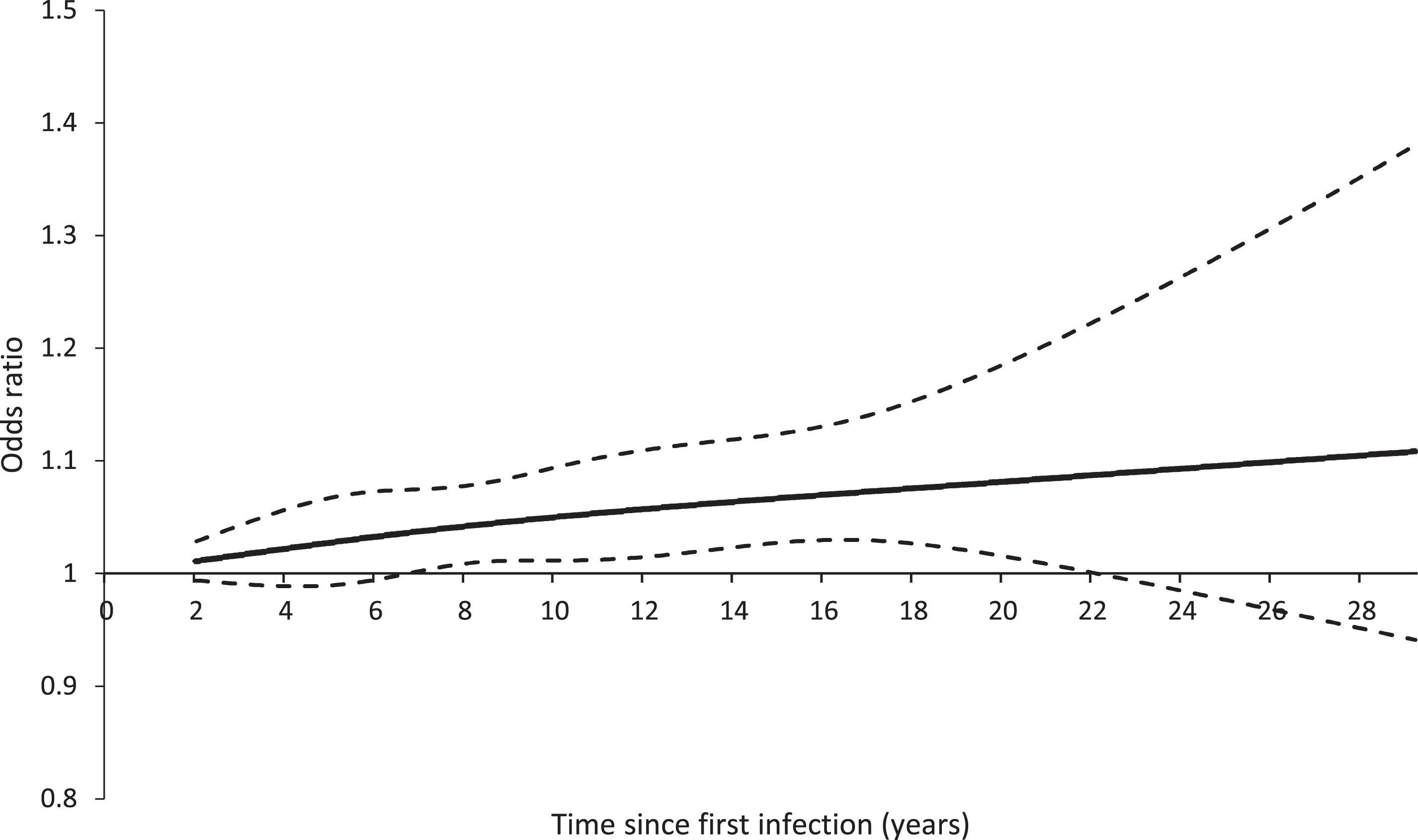
Restricted cubic spline of time since first infection on the risk of Alzheimer’s disease. The solid line shows the odds ratio and the dashed lines show the lower and upper bound of the 95% confidence interval. The curve begins at 2 years given the use of a 2-year lag period in the definition of exposure.
Stratifying by specific type showed an increased risk for gastritis (odds ratio, 1.08; 95% confidence interval, 1.03 to 1.13) but not for other infections (Table 3). Age did not seem to modify the association; however, the risk of Alzheimer’s disease was only increased among female patients (odds ratio, 1.08; 95% confidence interval, 1.04 to 1.11) and not among male patients (odds ratio, 0.99; 95% confidence interval, 0.94 to 1.04) (Supplementary Table 3).
Crude and adjusted odds ratios for the association between infectious disease burden and the risk of Alzheimer’s disease (stratified by specific type of infection)
OR, odds ratio; CI, confidence interval. S = Cells with less than 5 counts are suppressed as per the confidentiality policies of the Clinical Practice Research Datalink. ¥Cytomegalovirus related infections are not included in the analysis due to a very low number of exposed events. *Non-mutually exclusive categories. **Matched on age, sex, date of cohort entry and duration of follow-up. ***Adjusted for body mass index, smoking, alcohol-related disorders, arterial hypertension, atrial fibrillation, congestive heart failure, coronary artery disease, stroke or transient ischemic attack, peripheral vascular disease, dyslipidemia, diabetes mellitus, chronic kidney disease, liver disease, depression, epilepsy, Parkinson’s disease, traumatic brain injury, osteoporosis, hypothyroidism, cancer, oral anticoagulants, antiplatelet agents, opioids, lipid-lowering drugs, beta-blockers, thiazides, angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, calcium channel blockers, antipsychotics, non-steroidal anti-inflammatory drugs, and antidepressants.
Finally, the sensitivity analyses using extended lag periods, censoring follow-up at non-Alzheimer’s disease dementia diagnoses, and restricting pneumonia diagnoses to those with a clear link to Chlamydophila yielded results that were highly consistent with those of the primary analysis (Supplementary Table 4). The results also did not change substantially after expanding our outcome definition to include any dementia (characteristics of cases of dementia and their matched controls are presented in Supplementary Table 5; the results of the primary, secondary, and sensitivity analyses are presented in Supplementary Tables 6–9 and Supplementary Figure 1). For example, similar to the analyses on the risk of Alzheimer’s disease, the increased risk of any dementia associated with infectious disease burden was not accompanied by a dose-response relation but a possible time-response relation. However, there was an increased risk associated with pneumonia, which was not observed in the Alzheimer’s disease specific analyses.
DISCUSSION
Our large population-based nested case-control study showed a small increase in the risk of Alz-heimer’s disease associated with infectious disease burden. This effect was not augmented with cumulative number of infections, but there was a suggestion of a gradual increase in the risk with longer time since the first infection. Focusing on specific types of infections, we identified a small increase in the risk associated with gastritis. Moreover, sex seemed to modify the association, with the risk of Alzheimer’s disease being increased only among female patients. The results remained consistent in sensitivity analyses addressing different sources of bias.
Despite the rapidly increasing numbers of individuals diagnosed with Alzheimer’s disease and the devastating course of the disease, the efficacy of available pharmacologic treatments is modest at best [2]. Moreover, multimodal interventions targeting several modifiable risk factors of Alzheimer’s disease and dementia have yielded sobering findings [3–5]. As a result, there is an ongoing search for novel angles in the area of Alzheimer’s disease prevention, with one of the most promising approaches in the past years being the ‘infectious hypothesis’ [32]. According to this hypothesis, hallmarks of Alzheimer’s disease such as the deposition of amyloid-β peptide or abnormal forms of tau protein in the brain are indicators of an infectious etiology [32]. Of note, these pathological changes may occur up to 20 years prior to the onset of symptoms [33]. The obvious and extremely intriguing consequence, should the infectious hypothesis be proven, would be that by reducing the burden of infectious diseases (e.g., via preventive treatments or vaccination programs) we could also potentially reduce the burden of Alzheimer’s disease.
Several pre-clinical, serological, and postmortem studies have supported this hypothesis linking various infectious pathogens to Alzheimer’s disease [6–13]. Moreover, epidemiological studies have uniformly shown an increased risk of Alzheimer’s disease or overall dementia associated with clinically apparent infections (e.g., pneumonia, septicemia, gingivitis, or overall infections), which ranged from 20% up to 260% [14–18]. However, the quality of these studies could be affected by reverse causality [15, 18], selection bias [14, 17], and important residual confounding [15, 18]. Reverse causality in particular can lead to spuriously increased effect estimates in this setting, since patients with early symptoms of Alzheimer’s disease could be at a higher risk of infections, or they could be followed-up more closely by the treating physician increasing the probability of infectious disease reporting [2]. Of note, the study with the highest quality included almost exclusively male individuals, which could compromise external validity [17].
Our study also showed a statistically significant increase in the risk of Alzheimer’s disease associated with infectious disease burden as defined by clinically apparent infections. Of note, the increase (5%) was much smaller than what has previously been reported, which potentially limits the clinical significance of the association. Moreover, there was no further increase in the risk of Alzheimer’s disease with cumulative number of infections. However, there was a gradual increase in the risk with longer time since the first infection, with a peak (11%) after 12–30 years. The potential time-response relation is intriguing, suggesting that infections occurring many years before the diagnosis of Alzheimer’s disease may contribute to its etiology. This hypothesis is in accordance with the early, pre-symptomatic onset of pathological changes linked to infections such as amyloid-β peptide deposition or tau protein abnormalities discussed earlier. That being said, additional studies are needed in the area to better understand this potential association.
After stratifying by sex, we observed an increased risk of Alzheimer’s disease associated with infectious disease burden only among female patients, a finding that supports previously reported data on the effect modifying properties of female sex [34]. When focusing on specific types of infection, we observed a potential signal for gastritis but not for other entities. Of note, while these analyses were pre-specified and based on a sufficient number of exposed cases, their findings should be considered hypothesis generating given the number of assessed associations. Thus, they require further investigation. Finally, another finding warranting additional research is the increased risk of any dementia associated with pneumonia, which was not observed in the Alzheimer’s disease specific analysis.
Our study has several strengths. First, the popula-tion-based design and the application of few exclusion criteria during the construction of the study cohort likely maximized the generalizability of our findings. Second, the large sample size allowed the calculation of precise effect estimates in the primary analysis and the secondary analyses. Indeed, the secondary analyses assessing potential dose-response and time-response relations between infectious disease burden and the risk of Alzheimer’s disease yielded useful insight regarding aspects of the association that were poorly characterized so far. Finally, the use of a 2-year lag period (and even longer lag periods in sensitivity analyses) minimized the possibility of reverse causality, a well-established challenge when assessing insidious, non-acute outcomes such as Alzheimer’s disease [2].
This study has some limitations. First, given its observational nature, residual confounding is possible. To mitigate this potential limitation, we matched on age and sex and further adjusted for numerous important confounders. Second, misclassification of exposure is possible, since we did not have access to microbiology data to confirm the infection. For example, not every gastritis case is a result of infection, with medications such as non-steroidal anti-inflammatory drugs or stress being possible alternative causes. Moreover, the link between the infectious pathogen with the putative role in the pathophysiology of Alzheimer’s disease and the clinically apparent infection may be weak. For example, pneumococci and viruses are far more common causes of pneumonia than Chlamydophila pneumonia. However, a sensitivity analysis restricting to pneumonias related to Chlamydophila pneumonia yielded highly consistent results. Moreover, we would like to emphasize that the goal of our study was to specifically focus on infections that are symptomatic and thus easily detectable in the natural setting of routine clinical practice. Third, we assessed the infectious disease burden only in patients at least 50 years of age. Thus, infections occurring earlier in life could not be considered in our analyses. Given the observed time-response relation between time since first infection and the risk of Alzheimer’s disease, future studies should assess the potential impact of infectious disease burden in the first decades of adulthood. Fourth, misclassification of the outcome is possible. However, the recording of Alzheimer’s disease and dementia in general in the CPRD has been shown to be good [35]. Moreover, we defined Alzheimer’s disease using a previously validated algorithm which incorporates not only diagnostic codes but also symptoms, diagnostic procedures, and medications, which possibly further improved the accuracy of our outcome definition [21]. In addition, the incidence rate in our study (2.3 per 1000 person-years) was consistent with the incidence rates reported in other population based studies with similar age distributions (from 1.7 per 1000 person-years to 7.1 per 1000 person-years for individuals aged between 65 and 75 years) [36, 37]. Finally, since we did not have access to patients’ vitamin D levels, analyses considering the potential role of vitamin D as a risk factor of infection-associated Alzheimer’s disease were not possible.
Overall, our large population-based nested case-control study identified a statistically significant but small and probably not clinically significant increase in the risk of Alzheimer’s disease associated with infectious disease burden. Given that the risk seemed to gradually increase with longer time since the first infection, peaking after 12 years, the role of infections occurring several years prior to the diagnosis of Alzheimer’s disease warrants further investigation.
Footnotes
ACKNOWLEDGMENTS
This study was funded by a Discovery Proof of Concept Grant of the Alzheimer Society of Canada Research Program (Grant number: 21-13) to P. Brassard.
A. Douros is the recipient of Chercheur-Boursier Junior 1 Award from the Fonds de recherche du Québec –santé (FRQS). C. Renoux is the recipient of Chercheur-Boursier Junior 2 Award from the FRQS. L. Azoulay is the recipient of Chercheur-Boursier Senior Award from the FRQS and a William Dawson Scholar award from McGill University.