
Abstract
Background:
Mitochondrial dysfunction, bioenergetic deficit, and extensive oxidative stress underlie neuronal perturbation during the early stage of Alzheimer’s disease (AD). Previously, we demonstrated that decreased PTEN-induced putative kinase 1 (PINK1) expression is associated with AD pathology in AD-affected human brains and AD mice.
Objective:
In the present study, we highlight the essential role of PINK1 in AD-relevant mitochondrial perturbation and neuronal malfunction.
Methods:
Using trans-mitochondrial “cybrid” (cytoplasmic hybrid) neuronal cells, whose mitochondria are transferred from platelets of patients with sporadic AD, we observed the effect of PINK1 in neuronal-like differentiation and synaptogenesis and mitochondrial functions.
Results:
In AD cybrid cells, the downregulation of PINK1 is correlated to the alterations in mitochondrial morphology and function and deficit in neuronal-like differentiation. Restoring/increasing PINK1 by lentivirus transduction of PINK1 robustly attenuates mitochondrial defects and rescues neurite-like outgrowth. Importantly, defective PINK1 kinase activity fails to reverse these detrimental effects. Mechanistically, AD cybrid cells reveal a significant decrease in PINK1-dependent phosphorylated mitofusin (Mfn) 2, a key mitochondrial membrane protein that participates in mitochondrial fusion, and an insufficient autophagic activity for the clearance of dysfunctional mitochondria. Overexpression of PINK1, but not mutant PINK1 elevates phosphorylation of Mfn2 and autophagy signaling LC3-II. Accordingly, PINK1-overexpressed AD cybrids exhibit increases in mitochondrial length and density and suppressed reactive oxygen species. These results imply that activation of PINK1 protects against AD-affected mitochondrial dysfunction and impairment in neuronal maturation and differentiation.
Conclusion:
PINK1-mediated mitophagy is important for maintaining mitochondrial health by clearance of dysfunctional mitochondria and therefore, improves energy homeostasis in AD.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease and one of the most common forms of dementia. It begins with mild memory loss and predominantly affects the elderly populations. Senile plaques formed by extracellular deposits of amyloid-β (Aβ) and intracellular neurofibrillary tangles (NFTs) composed of aggregations of hyperphosphorylated tau protein are the principal hallmark pathologies of AD [1, 2]. Recent evidence suggests that Aβ and tau pathologies have synergistic effects in the progression of AD [3, 4]. However, there is currently no effective treatment for AD, which may be attributed in part to the lack of a clear underlying mechanism.
Mitochondria constitute a major source of reactive oxygen species (ROS) and have been proposed to integrate the cellular responses to stress. Oxidative stress has been implicated in the pathogenesis of AD [5]. AD has a long latent period before symptoms appear, known as mild cognitive impairment (MCI), an interim phase without significant formation of senile plaques and NFTs [6]. Escalating signs of the antioxidant and oxidant imbalance were detected in subjects with MCI, indicating oxidative stress-related damage is one of the earliest events in the onset and progression of AD [7].
Accumulating studies, including ours, have shown that a reduction in PTEN-induced putative kinase 1 (PINK1) expression is associated with Aβ accumulation and mitochondrial abnormalities in AD-affected human brains and AD mouse model [8–15]. Restoring neuronal PINK1 resume mitochondrial quality control ability through enhanced autophagy signaling in Aβ-rich brains [8]. However, the direct impact of PINK1 on human AD-induced mitochondrial defect remains to be investigated. Using the AD transmitochondrial cytoplasmic hybrid (cybrid) neuronal cell lines incorporated AD platelet mitochondria and cognitively normal aged-matched subjects to model human AD mitochondrial dysfunction, we address an essential question of whether enhancing PINK1 function protects against AD-mediated mitochondrial perturbation and neuronal malfunction in comparison with the non-AD cybrid cells whose mitochondria were transferred from platelets of age-matched subjects with relatively normal cognition. We first determined the expression level of PINK1 and then evaluated the effect of restoration of PINK1 kinase activity on the ability of neuronal differentiation, enzyme associated with mitochondrial respiratory chain, energy metabolism, mitochondrial morphology, and ROS levels in neuronal-like differentiated AD cybrid cells. To explore the potential mechanisms underlying the effect of PINK1, we investigated the modulations of key mitochondrial fission and fusion regulatory proteins and autophagy signaling. The present study provides the direct evidence of the link between PINK1 reduction and mitochondrial dysfunction in AD-affected mitochondria. The specific effect of PINK1 on phosphorylation of mitochondrial fusion protein-mitofusin (Mfn) 2 and activation of autophagy for clearance of defective mitochondria indicates that PINK1 might form the basis of a therapeutic treatment for mitochondrial defects in AD.
MATERIALS AND METHODS
Human subjects and creation of cybrid cell lines
Both AD and non-AD cybrid cells were kindly provided by Dr. Swerdlow from the University of Kansas Alzheimer’s Disease Center (KUADC). AD patients and non-AD controls were recruited from the University of Kansas Alzheimer’s Disease Center (KUADC). Subjects with AD met the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association criteria [16]. This study was approved by the University of Kansas Medical Center (KUMC) Institutional Review Board. Non-AD subjects were cognitively normal and age-matched to AD subjects. All subjects provided written informed consent to participate in the study. We used 5 cell lines per group in this study and the ages of AD and non-AD platelet donors were 73.6±2.96 and 75.8±5.04 years, respectively. AD and non-AD platelet donors were both obtained from two females and three males, respectively. The detailed information about gender, age, and disease status of donors was described in our previous studies [17, 18]. We do not have an interest in making cybrids from mutation subjects because presumably the mitochondrial defects would be corrected in cell culture, since the mitochondria are separated from the presence of the nuclear mutation when we do the transfection into the Rho0 cells.
Cybrid cell lines were created on the human neuroblastoma cell (SH-SY5Y) nuclear background (by the KUADC Mitochondrial Genomics and Metabolism Core) [19]. There are several reasons to select SH-SY5Y cells to create AD cybrid cells: Firstly, they are a commonly used human neuronal line and available in the laboratory when we decided to generate human neuronal cybrid cell line and secondly, they can be differentiated into neuronal-like cells. Importantly, SH-SY5Y cells have been very successfully transmitted by mitochondria derived from human AD and non-AD subject as a human AD cybrid cell lines that recapitulate specific AD mitochondriopathies [18, 20].
To create the cybrid cell lines used for this study, SH-SY5Y cells that were previously depleted of endogenous mtDNA (Rho0 cells), which were fused with platelet cytoplasm from human subjects, and repopulated with mitochondria containing mtDNA from patients or controls as previously described [21]. The quantitative real-time PCR showed that the intact mtDNA copies were present in all cybrids without detectable large scale deletion after many passages of cell proliferation as previously described [18].
Cybrid growth and differentiation
AD and non-AD cybrid cells were grown in T75 tissue culture flasks in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10%characterized fetal bovine serum (FBS; Gibco BRL, Logan, Utah), 100μg/ml pyruvate, 50μg/ml uridine, antibiotic-antimycotic, 100 Units/ml penicillin G, and 100μg/ml streptomycin as previously described [18]. Culture medium was replaced to the differentiation media [neurobasal media supplemented with 1×B27 (Invitrogen, Carlsbad, CA) and 0.5 mM glutamine, and antibiotic-antimycotic] with 10 nM staurosporine (SAT, Sigma-Aldrich Corp, St. Louis, MO, USA). Half of the differentiation media were made fresh with 10 nM SAT and replaced every day as previously described [17, 18]. The differentiated neuronal AD and non-AD cybrid cells were used in this study.
Lentivirus construction of Lenti-PINK1, Lenti-mPINK1, and Lenti-GFP
PINK1 constructs and viral packaging–p FUGW (Addgene, # 14883) were used to generate lentiviral plasmids expressing wild type PINK1 (PINK1) and triple kinase dead mutant of PINK1 (K219A, D362A and D384A, triple-mPINK1) with EGFP at their C-terminus as described previously [8].
Immunocytochemistry
Differentiated cybrid cells cultured on coverslips were fixed in 4%paraformaldehyde and then permeability with 0.1%Triton X-100 in PBS. Cells were incubated in blocking solution (5%goat serum in PBS) for 20 min and then incubated with different primary antibodies: mouse anti-MAP2 (1:2000, #1284959, Chemicon) and rabbit anti-Pink1 (1:1000, sc-33796, Santa Cruz Biotechnology) overnight at 4°C. After three times washed with PBS, cells were incubated with Alexa Fluor® 488 conjugated goat anti-rabbit IgG or 633 goat anti-mouse IgG secondary antibodies at 1:1000 dilutions for 2 h at room temperature. Fluorescence images were acquired and analyzed using Leica LAS AF software (Leica Wetzlar). Fluorescence density was analyzed with NIH Image J software.
Immunoblotting analysis
Samples were lysed in extraction buffer [10 mM Tris-HCl (pH 7.4), 100 mM sodium chloride, 1 mM EDTA, 1 mM EGTA, 1 mM sodium fluoride, 20 mM sodium pyrophosphate, 2 mM sodium orthovanadate, 1%Triton X-100, 10%glycerol, 0.1%SDS, 0.5%deoxycholate, 1 mM PMSF] containing protease inhibitor mixture (set V, EDTA-free; Calbiochem, San Diego, CA), separated by SDS/PAGE (12%Bis-Tris gel; Invitrogen), and then transferred to a nitrocellulose membrane (Amersham, Pittsburgh, PA). After blocking in TBST buffer (20 mM Tris-HCl, 150 mM sodium chloride, 0.1%Tween-20) containing 5%(wt/vol) nonfat dry milk (Santa Cruz) for 1 h at room temperature, the membrane was then incubated and gently shaken overnight (at 4°) with a primary antibody. The primary antibody was rabbit anti-Pink1 (1:1000, sc-33796, Santa Cruz Biotechnology), rabbit anti-synaptophysin (syn, 1:5000, #AB9272, Millipore), mouse anti-MAP2 (1:2000, #1284959, Chemicon), rabbit anti-phospho-Drp1 (Ser616, 1:2000, #3455, Cell Signaling), rabbit anti-phospho-Mfn2 (Ser442, 1:1000, #ABC963, Millipore) and rabbit anti-LC3A/B (1:1000, #12741S, Cell Signaling Technology). The membranes were incubated for 1 h with horseradish (HRP)-conjugated secondary antibodies (Sigma, USA) after washed with TBST 3 times. Final detection of immunoreactive bands was performed by enhanced chemiluminescence (Pierce™ ECL Western Blotting Substrate, Thermo Fisher, USA). Anti-mouse β-actin monoclonal antibody (1:10,000, #A5441, Sigma-Aldrich) was used to ensure equal protein loading of the samples.
Measurement of respiratory chain complex enzyme activity and ATP levels
Activity of Cytochrome c oxidase, a key enzyme of respiratory chain complex IV, and ATP levels were determined as described previously [17]. Briefly, cybrid cells were washed with ice-cold PBS, and then harvested, centrifuged, and suspended in 50μl of isolation buffer containing 250 mM sucrose, 20 mM HEPES, and 1 mM EDTA. Cell suspensions (containing 3-4 mg of protein/ml) were added to a cuvette containing 0.95 ml of 1×assay buffer (10 mM Tris-HCl, pH 7.0 and 120 mM KCl), and the reaction volume was brought to 1.05 ml with the addition of 1×enzyme dilution buffer (10 mM Tris-HCl, pH 7.0 and 250 mM sucrose). The reaction was then initiated by the addition of 50μl of ferrocytochrome substrate solution (0.22 mM). The rate of change in absorbance at 550 nm was recorded immediately using a Shimadzu (Kyoto, Japan) UV1200 spectrophotometer programed for a 5 s delay and 10 s intervals for 6 readings. ATP levels were determined using an ATP Bioluminescence Assay Kit (Roche) following the manufacturer’s instruction as we previously described [8]. Briefly, cells were quickly harvested by ATP lysis buffer, incubated on ice for 30 min, and then centrifuged at 12,000 g for 10 min. ATP levels were measured by Luminescence plate reader (Molecular Devices). A 1.6 s delay time after substrate injection and 10 s integration time were used.
Evaluation of reactive oxygen species
Evaluation of intracellular ROS levels was assessed by EPR (election paramagnetic resonance) spectroscopy as described in our previous study [22, 23]. Cybrid cells cultured on 6-well plates were washed with PBS and then incubated with CMH (Cyclic hydroxylamine 1-hydroxy-3-methoxycarbonyl-2, 2, 5, 5-tetramethyl-pyrrolidine, 100μM) at room temperature for 30 min. After that, cybrid cells were washed with ice-cold PBS, and then harvested with 100μl of PBS for each well. EPR measurements were performed using a Bruker EMX plus X-band EPR spectrometer running Bruker Xenon acquisition/processing software and equipped with a dual mode cavity (ER4116DM) and Oxford ESR900 cryostat. For each sample ∼50μl was loaded into a 1.5 mm O.D. micropipette (Blaubrand intraMARK), sealed with Bruker X-Sealant and placed into a 4 mm O.D. EPR tube (Wilmad Labglass 707-SQ-250M). The EPR spectrometer was operated at 9.63 GHz and 100 kHz field modulation at room temperature. The spectra were recorded with the following parameters: number of scans, 3; magnetic field center, 344 mT; scan range, 10 mT; microwave power, 2 mW; modulation amplitude, and 0.1 mT; time constant, 0.08 s. Signal intensity was determined as the height of the central peak on the up-field side of the midpoint.
Visualization of mitochondria
Cells were stained with Mitotracker Red (MitoRed, 200 nM; Molecular Probes) at 37°C for 30 min prior to fixation. Images were obtained using Leica TCS SPE confocal scanning microscopes with 63×1.4 NA Apochrome objective lens (Carl Zeiss MicroImaging, Inc.) by the mean of excitation wavelength settings at 543 nm for Mitotracker Red. Mitochondrial morphology was visualized by MitoRed staining and post-acquisition processing was performed using NIH Image J software for assessing mitochondrial density and length.
Statistical analysis
Data are presented as mean±SEM. Statistical analysis was performed using Statview software (SAS Institute, Version 5.0.1). Differences between means were assessed by one-way analysis of variance (ANOVA) with Fisher post hoc test. p < 0.05 was considered significant.
RESULTS
Decreased PINK1 level in the differentiated AD cybrid cells
To assess the significance of PINK1 in human neuronal-like AD cybrid cells, we first examined the expression level of PINK1 in differentiated neuronal AD cybrid cells with significant neuronal like morphology. Compared to the differentiated non-AD cells, PINK1 expression in AD cybrids was significantly decreased by 50–60%based on the analysis of immunoblotting (Fig. 1A, B) and immunocytochemistry result (Fig. 1C, D). These results support the association of downregulation of PINK1 with AD-related mitochondrial defect and AD pathology in AD-affected brains and several well-established AD mouse models [8, 24].

PINK1 expression in non-AD and AD cybrid cells during differentiation. A, B) Immunoblotting of protein extracts from non-AD and AD cybrid cells for PINK1 expression. β-actin was used for protein loading control. A) Quantification of immunoreactive bands for PINK1 relative to β-actin. B) Representative immunoblot for PINK1 and β-actin. (n = 5 per group). C) Representative fluorescent images of PINK1 (green), mitochondria (MitoRed, red), merge of PINK1 and mitochondria (yellow), and MAP2 (purple) from non-AD and AD cybrid cells under differentiated conditions induced by SAT (14 days after 10 nM staurosporine [SAT] treatment). Enlarged views of an individual non-AD or AD cybrid cell are shown in the right panels. Scale bars = 50μm. D-F) Quantification of fluorescent intensity of PINK1 (D), MAP2 (E) and mitochondrial density (F, MitoRed staining) of cybrid cells using the image J program. G, H) Analysis of the relationship between PINK1 expression and MAP2 level (G) or mitochondrial content (H). (n = 9–11 cybrid cells of each group).
PINK1 reduction correlates to the defective differentiation and mitochondrial density of AD cybrid cells
We have previously revealed impairment in the capacity of neuronal differentiation with shorter neurites-like processes of AD cybrid cells [17, 18]. Consistent with these results, there was a significant reduced level of microtubule-associated protein 2 (MAP2), a marker of neuronal differentiation [25] in differentiated AD cybrid cells compared to non-AD cybrids (Fig. 1C, E). Accordingly, AD cybrid cells revealed lower mitochondrial density as shown by reduced the intensity of MitoRed staining as compared to non-AD cybrid cells (Fig. 1C, F). The enlarged images of an individual non-AD or AD cybrid cell (right panels, Fig. 1C) clearly show decreased PINK1 expression with lower mitochondrial density in AD cybrid cells.
To determine the relationship of PINK1 expression with differentiation ability and mitochondrial content of AD cybrid cells, we further assessed PINK1 expression with MAP2 level (Fig. 1G) or mitochondrial content (Fig. 1H) by visualizing triple immunocytochemical staining of PINK1 (Green), MAP2 (Purple), and MitoRed (Red) under confocal microscopy (Fig. 1C). Both MAP2 level and mitochondrial density were positively correlated with PINK1 expression (R2 = 0.6528 for PINK1/MAP2 in Fig. 1G, and R2 = 0.6150 for PINK1/mitochondria in Fig. 1H). Therefore, downregulation of PINK1 associates with deficits in neuronal differentiation and AD mitochondrial defects.
PINK1 rescues impairments in the differentiation and synaptogenesis in AD cybrid cells
In view that a reduction in PINK1 expression was associated with mitochondrial defect in AD cybrids, we next determined whether restoring/increasing PINK1 expression could promote neuronal differentiation and synaptogenesis in AD cybrids. AD and non-AD cybrid cells were transduced by lentivirus encoding human PINK1 or lentivirus control vector encoding GFP as a control. Introduction of lentivirus-PINK1 into AD cybrid cells increased the length of neuronal processes (Fig. 2A, B) and the levels of neuronal and synaptic marker proteins, including MAP2 and synaptophysin/syn (Fig. 2C, D) compared to the mock AD cybrid cells transduced by lentivirus-GFP during differentiation. To further validate the role of PINK1 kinase activity on differentiation, we examined the effect of a triple kinase dead mutants of PINK1 (K219A, D362A and D384A, triple-mPINK1) as a classical kinase dead mutant form [8, 26] in AD cybrid cells. Compared to wild-type PINK1 transduced-AD cybrid cells, triple-mPINK1 transduced AD cybrid cells failed to rescue the impairment in the differentiation (Fig. 2A-D) and synaptogenesis (Fig. 2C, D). Therefore, PINK1 kinase activity is required for healthy neuronal differentiation capacity in AD cybrid cells. The efficiency of lentivirus-PINK1 and mPINK1 transduction was confirmed by the immunoblotting showing that PINK1 expression levels were increased by ∼2.5 fold in PINK1-transduced cells as compared to GFP-transduced control cells. Expression level of PINK1 was comparable to mPINK1 in AD and non-AD cybrid cells (Fig. 2E, F).
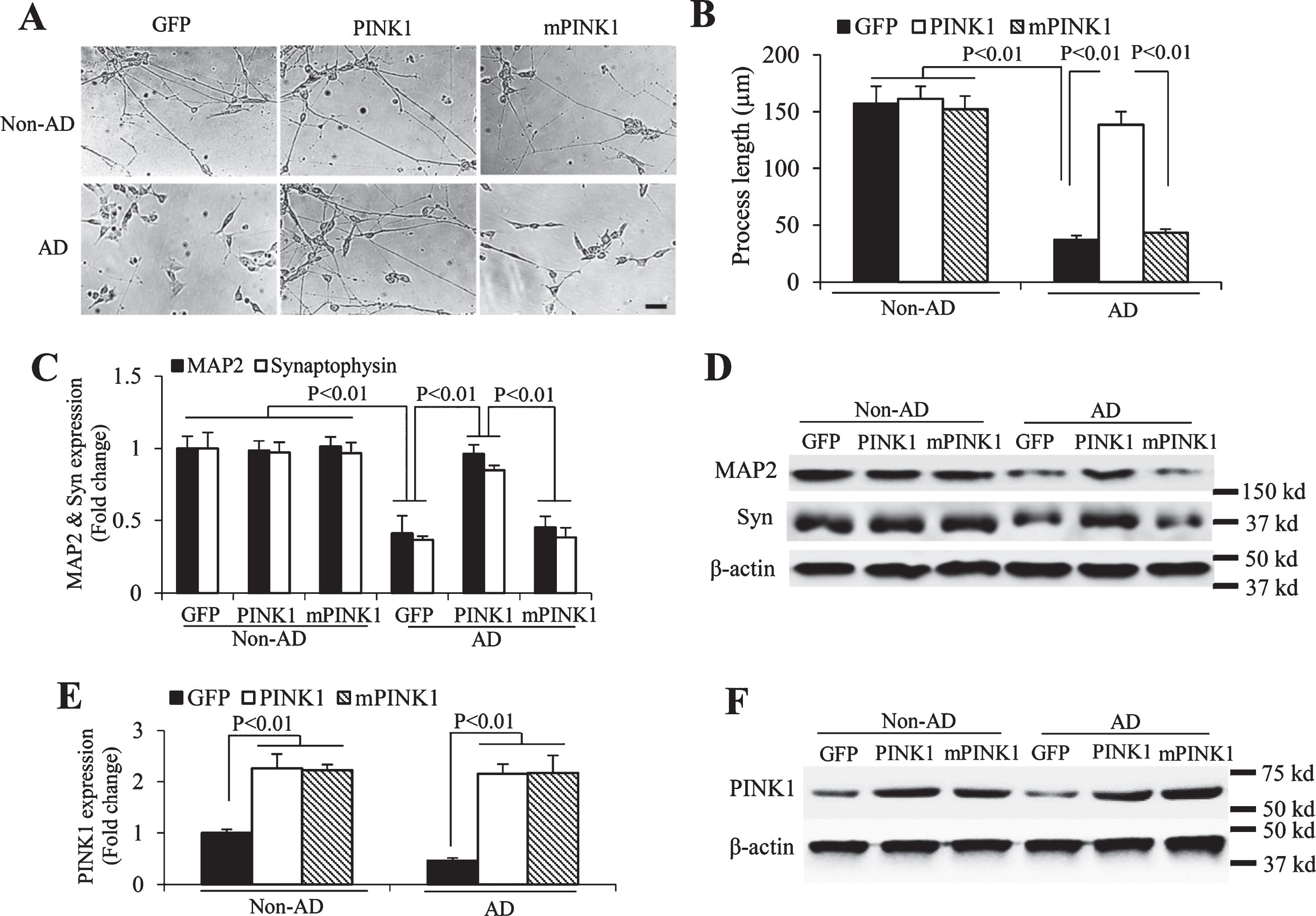
Effect of PINK1 on neuronal differentiation in AD cybrid cells. A) Representative morphological images from differentiated non-AD and AD cybrid cells with or without transducing PINK1 or mPINK1 lentivirus. Scale bar = 50μm. B) Quantification of length of neuronal processes from indicated cybrid cells (12 neuronal processes for each group) using the image J program. C-F) Immunoblotting of protein extracts from non-AD and AD cybrid cells with or without PINK1/mPINK1 lentivirus transduction for MAP2 and synaptophysin (Syn, C and D) and PINK1 (E and F) expressions. β-actin was used for protein loading control. Quantification of immunoreactive bands for the corresponding protein relative to β-actin in C and E. Representative immunoblots for the indicated proteins in D and F. n = 3 per group.
PINK1 attenuates mitochondrial defects and oxidative stress in AD cybrid cells
Next, we determined whether increased PINK1 improves mitochondrial function in AD cybrid cells. We evaluated mitochondrial respiratory function by measuring cytochrome c oxidase (CcO) activity, a key enzyme in mitochondrial respiratory chain complex IV [22, 28]. CcO enzyme activity was significantly decreased in AD cybrid cells compared to non-AD controls, whereas lentivirus-PINK1 transduction restored CcO activity (Fig. 3A). In parallel, the decline in ATP level in AD cybrid cells was reversed by lentivirus-PINK1 transduction (Fig. 3B). Importantly, mPINK1 failed to reverse these detrimental effects in AD cybrid cells (Fig. 3A, B).

Effect of PINK1 on AD-related mitochondrial dysfunction. A, B) Enzymatic activity of cytochrome c oxidase (CcO, A) and cellular ATP levels (B) were determined in cell lysates from non-AD and AD cybrid cells with or without PINK1 or mPINK1 lentivirus transfection. C, D) Generation of ROS in the indicated groups of cells was detected by electron paramagnetic resonance (EPR) spectra. Quantifications of the peak of EPR (C) of the indicated cybrid cells and Representative EPR images (D). n = 5 per group.
Mitochondria are the main source of ROS production. In view of overproduction of ROS in AD cybrid cells, we next evaluated whether increased PINK1 affects ROS levels in AD cybrid cells. Consistent with our previous observation, AD cybrid cells demonstrated a significantly increased ROS levels [18] compared to non-AD cybrid cells (Fig. 3C, D). Introduction of PINK1 but not mPINK1 blunted intracellular ROS levels in the cells detected by highly sensitive and specific EPR spectroscopy (Fig. 3C, D). These results suggest that resuming PINK1 kinase activity alleviates oxidative stress in AD-affected mitochondria.
Given that mitochondrial morphology and distribution are important for maintaining mitochondrial functions, it is logical to examine whether PINK1-mediated mitochondrial dysfunction alters mitochondrial morphology and content. AD cybrid cells displayed shorter mitochondrial length and lower mitochondrial density than non-AD cybrid cells (Fig. 4A-C). Transduction of lentivirus-PINK1, but not mPINK1 restored mitochondrial length and density (Fig. 4A-C), suggesting that resuming PINK1 kinase activity ameliorates alterations in AD-specific mitochondrial morphology and distribution in the differentiated AD cybrid cell model.

Effect of PINK1 on mitochondrial morphology in AD cybrid cells. A, B) Quantifications of mitochondrial length (A) and density (B). Mitochondria are visible by MitoRed staining from differentiated non-AD and AD cybrid cells with or without PINK1 or mPINK1 lentivirus transfection. For the analysis of mitochondrial length, 25 mitochondria in the growth cone of axon-like neurites from 5 cybrids per group were analyzed. 25 cybrid cells of each group were analyzed for mitochondrial density. C) Representative fluoresce images of mitochondria (MitoRed), GFP (green) and nucleus (blue) from non-AD and AD cybrid cells. Scale bars = 10μm.
PINK1 regulates mitochondrial fission/fusion balance through Phosphorylation of Mfn 2 in AD cybrid cells
To explore the potential mechanisms underlying the regulation of PINK1 on mitochondrial morphology in AD cybrid, we investigated the expressions of phosphorylated Mfn2 and Drp1, two major proteins modulating fusion/fission balance. As shown in Fig. 5A and B, AD cybrid cells exhibited a relatively lower level of phosphorylated-Mfn2 (p-Mfn2, Ser442) compared to the non-AD cells. Introduction of PINK1, but not triple kinase dead mutant of PINK1 (mPINK1) completely restored p-Mfn2 (Ser442) levels in AD cybrids relative to GFP-transduced AD and nonAD cybrid cells. Similarly, PINK1 overexpression significantly increased phosphorylated Mfn2 at Ser442 in non-AD cybrids compared to GFP-transduced control non-AD cells (Fig. 5A, B). In contrast, there was no changes in the expression of phosphorylation of Drp1 at Ser616, the phosphorylation site of Drp1 responsible for the fission of mitochondria, in AD cybrids compared to non-AD cybrids with or without transduction of lentivirus-PINK1 (Fig. 5A, C). These results indicate that the reduction in the phosphorylated-Mfn2 level may be responsible for driving mitochondrial fragmentation, which underlies, at least in part, the abnormalities in mitochondrial morphology and distribution observed in AD cybrids. PINK1-dependent phosphorylation of Mfn2 promotes its ubiquitination and restores the fusion/fission balance.

PINK1 phosphorylates Mitofusin 2 (Mfn2) at Ser442 and activates autophagy signaling in AD cybrid cells. A, B) Immunoblotting of protein extracts from non-AD and AD cybrid cells with or without PINK1/mPINK1 lentivirus transfection for detecting phosphorylated-Mfn2 (p-Mfn2, Ser442, A and B), phosphorylated Drp1 (p-Drp1, Ser616, A and C), and LC3II (D and E). β-actin was used for protein loading control. Quantifications of immunoreactive bands for the corresponding protein relative to β-actin in B, C and E. **p < 0.01 versus non-AD transduced by lentivirus-GFP. n = 3 per group.
PINK1 activates autophagy signaling in AD cybrid cells
In view of the significance of autophagy initiated by PINK1 in the clearance of defective mitochondria [8, 29], we finally assessed whether gain of PINK1 function enhances autophagy signaling by assessing the expression level of LC3-II, a marker for activation of autophagy. Levels of LC3-II were significantly increased in both AD and non-AD cybrid cells transduced by lentivirus-PINK1, but not by lentivirus-mPINK1, whereas AD cybrids exhibited considerably higher levels of LC3-II than non-AD cybrids, suggesting the compensatory requirement for autophagy to remove excessive dysfunctional mitochondria in AD cybrids (Fig. 5D, E). These results indicate that PINK1-activated autophagy signaling facilitates disposal of dysfunctional mitochondria and therefore maintains healthy mitochondria and energy homeostasis.
DISCUSSION
Mitochondrial defect is one of the most prominent and early pathological features of AD [30–32]. Cytoplasmic hybrid (“cybrid”) cell models have been used to model human AD mitochondrial defects relevant to the pathogenesis of AD. AD cybrid cells recapitulate AD pathological features, especially mitochondrial dysfunction observed in AD brain, including excessive oxidative stress, cytochrome oxidase defects, decreased membrane potential, impaired energy metabolism, and alterations in mitochondrial dynamics [33–36]. Additionally, compared to non-AD cybrid cells, differentiated AD cybrid cells revealed shorter neurite-like processes and lower synaptic content, indicating the AD-derived mitochondria impair the capacity of neurogenesis and synaptogenesis of cybrid cells.
PINK1 is critical for the maintenance of mitochondrial integrity and function against mitochondrial dysfunction with mitochondrial quality control via mitophagy [8, 37–39]. PINK1 expression increases during early brain and neural stem cells development, while PINK1 deficiency is associated with increased deficits of adult hippocampal neurogenesis [40, 41]. We and other studies have demonstrated downregulation of PINK1 in AD-affected brains and several well-established AD mouse models [8, 12]. In the present study, we first investigated the impact of AD mitochondria on neurogenesis in AD cybrid cells upon neuronal-differentiation induction to gain the maturation of human AD neuronal model. A significant PINK1 reduction was observed in the differentiated AD cybrid cells compared to non-AD cybrids, and importantly, a positive correlation between PINK1 level and MAP2 expression, indicating PINK1 deficiency accounts for the impairment of neurogenesis derived from AD mitochondria induction.
Mitochondria are complex organelles regulating a variety of physiological and pathological processes, from energy production to cell death. Substantial evidence supports the hypothesis that respiratory chain complex dysfunction and impairment of ATP production play a critical role in neurodegeneration relevant to the AD pathogenesis [42, 43]. Oxidative stress contributes importantly to the formation of AD-associated pathology. Defective enzyme activity associated with mitochondrial respiratory chain, impairment in mitochondrial energy production, and excessive ROS accumulation are early functional features of AD. Therefore, mitochondria are considered as potential therapeutic targets in neurodegenerative diseases, including AD. Consistent with our previous studies [17, 18], AD cybrid cells revealed mitochondrial defects as shown by decreased CCO enzyme activity and ATP synthesis, and increased ROS levels. Here, we provide the direct evidence of the effect of PINK1 on AD-related mitochondrial dysfunction on cybrid cell model. Upregulated PINK1 levels rescue mitochondrial respiratory function, increase ATP levels, and attenuate oxidative stress insulted by AD-derived mitochondria.
Mitochondria are constantly undergoing morphological changes driven by fission and fusion, which are necessary for cell survival in response to the altered extracellular conditions [44]. Dysfunctional mitochondria may undergo mitophagy in the brain cells with neurodegenerative diseases [45, 46]. Morphometric studies of the mitochondria in AD exhibit a significant reduction in mitochondrial density with an excessive mitochondrial fission in endothelial cells [47], fibroblasts, and other cells obtained from patients with AD [48]. AD cybrid cells also displayed significant changes in mitochondrial morphology and density [17, 18]. However, the potential mechanism underlying changes in mitochondrial dynamics in AD has not yet been clearly elucidated. PINK1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery leading to neuroprotection, while PINK1 deficiency tips the balance toward fission [49]. We observed a fission-pattern with fragmented and scattered mitochondria in AD cells as opposed to jointed and elongated mitochondria distributed in the differentiated normal non-AD cells. The density and numbers of mitochondria were significantly reduced in the differentiated AD cells, which are correlated with the decreased PINK1 activity. Consistent with our previous results obtained in AD mouse model [8], augmenting PINK1, but not defective PINK1 kinase activity with triple kinase dead mutant of PINK1 (K219A, D362A and D384A, triple-mPINK1), reduced the percentage of fragmented AD-derived mitochondria and restored mitochondrial density in AD cybrids. These results demonstrate that PINK1 attenuates fission-dominated mitochondrial morphological change observed in AD mitochondria.
The balance of mitochondrial fusion and fission is important for maintaining proper mitochondrial structure and function. Mfn2 and Drp1, two key regulatory proteins of mitochondrial fusion and fission, are essential for the maintenance of mitochondrial morphology and the operations of the mitochondrial network and metabolism [50]. The involvement of Mfn2 in the PINK1-dependent ubiquitination of mitochondrial proteins correlates the induction of mitophagy [51, 52], while Drp-1 was not found to be ubiquitinated [51]. Compared to non-AD cybrids, the expression level of phosphorylated Mfn2 at Ser442 was significantly reduced in AD cybrids. Accordingly, AD cybrid cells displayed abnormal mitochondrial morphology as shown by shorter and fragmented mitochondria and reduced mitochondrial density. These observations suggest that a significant reduction in p442-Mfn2 associated with downregulation of PINK1 expression contributes to the abnormal mitochondrial morphology toward fission-dominated morphology observed in AD cybrids. To support this, the restored expression level of phosphorylated-Mfn2 by PINK1 overexpression almost abolished fission-dominated mitochondrial morphology, as shown by increases in mitochondrial length and density and reduction in mitochondrial fragmentation observed in AD cybrids. The inactivated mutant PINK1 lacking kinase activity was not able to rescue mitochondrial morphology and functional change in AD cybrids containing AD-derived mitochondria. It is noted that no changes of the levels of p616-Drp1 were observed in differentiated AD cybrids compared to non-AD cybrids. Thus, PINK1-mediated Mfn2 phosphorylation is responsible for the restoration of mitochondrial fission/fusion balance in AD cybrids. The observation of the elevated level of p442-Mfn2 in the PINK1-overexpressing non-AD cybrids further supports that the effect of PINK1-targeted mitochondrial fusion is through phosphorylation of Mfn2 under pathophysiological conditions, such as mitochondrial stress.
In view of the involvement of autophagy in maintaining mitochondrial integrity and function through clearance of damaged mitochondria and the essential role of PINK1 activity in recruiting autophagy signaling [29], we finally assessed whether gaining PINK1 function would activate autophagy signaling in AD cybrids. A significant up-regulation of autophagosome marker-LC3-II, the active form of LC3, was detected in AD cybrids overexpressing PINK1, suggesting enhancement of autophagy to promote the removal of the defective mitochondria in AD cybrids triggered by PINK1. Consistent with our result obtained in AD mouse brain [8], the elevated level of LC3-II in AD cybrids suggests that autophagy signaling is attempting to, but insufficient for the clearance of the defective mitochondria, specifically engulfed by autophagosomes and targeted for degradation in AD. Thus, efficient and stable PINK1 expression is necessary to sufficiently induce mitophagy to eliminate the excessive dysfunctional mitochondria that occur in AD mitochondria, thereby ameliorating mitochondrial pathology and maintaining mitochondria quality control.
In summary, we provide the first direct evidence that defective PINK1 function contributes to mitochondrial defects and impaired neurogenesis in differentiated human neuronal cybrids carrying AD-derived mitochondria. Restoring PINK1 expression/activity not only restores mitochondrial function and suppresses oxidative stress, but also promotes the neuronal differentiation and synaptogenesis in AD cybrid cells. Thus, enhancing PINK1 function could be a therapeutically intervention by improving mitochondrial function and enhancing neurogenesis, thereby defeating AD at an early stage.
Footnotes
ACKNOWLEDGMENTS
This study was supported by NIH/NIA (R37AG037319, R01AG044793, R01AG053041, and RF1AG054320) and Alzheimer’s Association Research Grant (AARG, 2018-AARG-592230). We thank Dr. Swerdlow for providing cybrid cells for our study and Generation and characterization of cybrid cells were supported by the University of Kansas Alzheimer’s Disease Center (NIA P30AG035982). We thank Justin T. Douglas for assistance in using EPR instrumentation.