
Abstract
Mutations in ITM2B have been found to be associated with familial Danish dementia (FDD) and familial British dementia (FBD). Here, we describe a patient with dementia caused by a novel ITM2B p.*267Leuext*11 mutation. The patient presented with dementia, ataxia, deafness, and paraplegia. Amyloid PET and Tau PET showed abnormal deposition of amyloid and tau protein in brain. Summarized from previous 26 FBD and FDD cases, the clinical phenotype of ITM2B; p.*267Leuext*11 mutation in ITM2B is different from the features of FBD and FDD. Our findings increased genetic knowledge of familial dementia and extend the ethnic distribution of ITM2B mutations.
INTRODUCTION
Familial Danish dementia (FDD) and familial British dementia (FBD) are rare autosomal dominant neurodegenerative disorders that share features of progressive ataxia and dementia associated with ITM2B/BRI2 mutations [1, 2]. FDD is also clinically characterized by cataracts and deafness. FBD also involves spastic paresis.
The ITM2B is located on 13 chromosome and en-codes the BRI2 protein. In normal individuals, BRI2 is synthesized as an immature type-II membrane protein (imBRI2) that is cleaved at the C-terminus by a proprotein convertase to produce mature BRI2 (mBRI2) and a 23 amino acid soluble C-terminal fragment (Bri23) [3]. This cleavage is particularly important because the mutated forms of BRI2 in FDD/FBD are cleaved in the same way to release a longer C-terminal fragment, which has amyloidogenic properties, aggregates in the patient’s brain (ADan and ABri peptide, respectively), leads to corresponding clinical symptoms [4]. Another pathogenic mechanism, proposed for FBD and FDD, is that both pathogenic mutations alter maturation of ITM2B resulting in reduced levels of functional mature BRI2 protein at synapses [5].
To date, several reviews on FBD and FDD cases have been published, but there are no reports of familial dementia caused by ITM2B gene mutations in the Chinese population. Here we describe a familial Chinese dementia caused by a novel mutation of ITM2B c.800G > T; p*267Leuext*11, which manifests as deafness, ataxia, spastic paraplegia, and progressive dementia, and summarized clinical features, pathology, and genetic characteristics of 22 FBD cases and 4 FDD cases.
METHODS
Clinical evaluation
Detailed medical history and pedigree information were collected. Neurologic examination and ancillary tests, such as brain magnetic resonance imaging (MRI), neuropsychological assessment, and [18F] -FDG/amyloid (Florbetapir, [18F] AV45)/Tau ([18F] PM-pyridinyl-butadienyl-benzothiazole 3, [18F] PM-PBB3) PET were performed in the proband. The patient signed an informed consent document.
Genetics analyses
Whole-exome sequencing (WES) was performed on the proband using the Illumina Hiseq sequencing platform. Variant filtering of WES was performed based on the guidelines of the American College of Medical Genetics and Genomics for sequence variant interpretation. Sanger sequencing was performed on the proband (III-8), his father (II-12), and cousin (III-1).
Search strategy and study selection for literature review
We carefully searched articles published in English from the PubMed from January 1933 to December 2019. The following terms were used as keywords: “familial British dementia AND (ITM2B or BRI2)”, “familial Danish dementia AND (ITM2B or BRI2)”. We selected all types of case reports and some studies proven ITM2B gene mutation. By hand screening references of literature reviews on this subject, we also identified additional citations. For repeated cases in different articles, preference was given to the one with more comprehensive clinical and pathological data. Clinical information was extracted from each article as followings: diagnosis, clinical phenotype, ITM2B mutation, neuropsychological assessment, pathology, and MRI. The flow chart of the review is shown in Fig. 1.

Flow chart of the article selection procedure.
RESULTS
Clinical findings
The proband was a 44-year-old male from China, with a high school education. In March 2017, he experienced unsteady walking, lower limbs stiffness, and dizziness when standing. In September 2017, the symptoms of instability and stiffness progressed and the patient experienced hearing loss in both ears. Then his disease progressively worsened, and he could not walk independently beginning in 2018. He also developed slurred speech and memory impairment in November 2018.
Neurological examination revealed obvious dysarthria, deafness, and cognitive impairment. No evident weakness of limbs was observed. Finger-nose and heel-shin ataxia were evident. The deep tendon reflexes were increased in lower limbs and Babinski’s sign was positive bilaterally. No sensory disturbances were observed. He had a broad-based gait and ataxia was obvious.
The proband scored 26 on the Mini-Mental State Examination (MMSE) and 18 on the Montreal Cognitive Assessment (MoCA). Pure tone threshold showed severe neurological deafness in both ears and the electromyogram was normal. Brain MRI showed hyperintensity signals in cortical and subcortical areas on T2-weighted image (Fig. 2A), and no cerebral microbleeds on susceptibility-weighted imaging (Fig. 2B). Cervical MRI was normal. Brain [18F]-FDG PET scan showed bilateral hypometabolism in the cerebellum (Fig. 2C). Amyloid PET showed bilateral cerebellar amyloid deposition, and bilateral frontal, parietal, temporal lobe, and posterior cingulate gyrus slightly amyloid deposition (Fig. 2D). Tau PET showed abnormal deposition of tau protein in the bilateral cerebellar cortex, occipital, and medial temporal lobes (Fig. 2E).
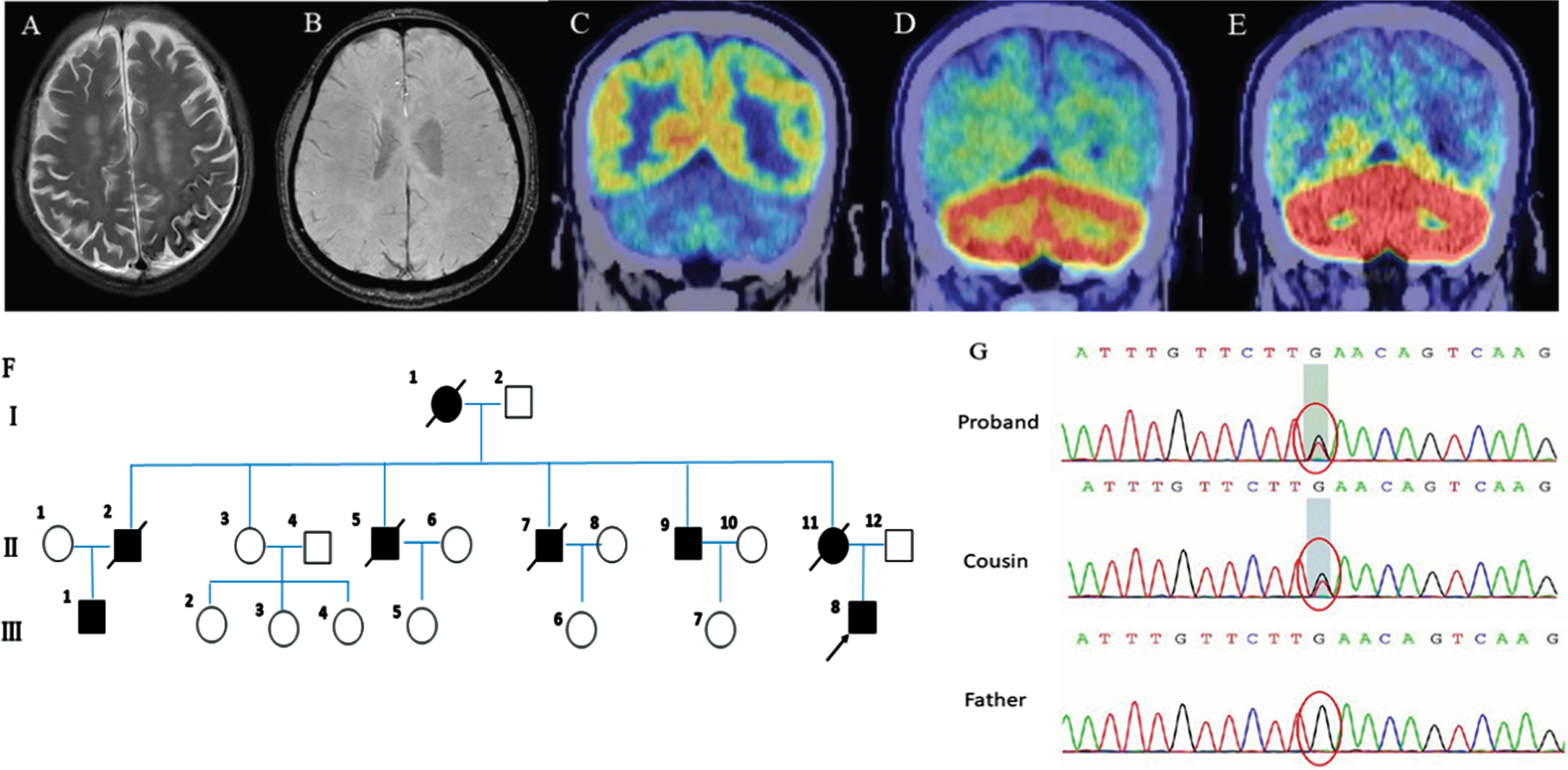
Clinical features of the proband and segregation analysis of the ITM2B p*267Leuext*11mutation. A) MRI scan showing white matter lesions on both frontal and parietal lobes and lateral ventricles on T2-weighted image. B) Susceptibility-weighted imaging showing no cerebral microbleeds. C) 18F-FDG PET scan showing bilateral hypometabolism in the Cerebellum. D) Amyloid PET showing bilateral cerebellar amyloid deposition. E) Tau PET showing abnormal deposition of tau protein in the bilateral cerebellar cortex, occipital and medial temporal. F) The pedigree of the patient’s family. G) Segregation analysis of the ITM2B p*267Leuext*11 mutation.
The proband’s grandmother (I-1), mother (II-11), four uncles (II-2, 5, 7, 9), and cousin (III-1) all had similar medical history (Fig. 2F). The age at onset was between 40s–50s, and the age at death was 60s–70s. They spent their last time in bed, and the cause of their death is unknown. They all manifested as ataxia, deafness, spastic paraplegia, and dementia.
Genetics findings
WES revealed novel heterozygous mutation in ITM2B, c.800G>T, p*267Leuext*11 in the proband (III-8). This mutation was absent from the control databases (gnomAD and ExAC). Subsequently, Sanger sequencing demonstrated that his cousin (III-1) also carried the p*267Leuext*11 mutation while it was absent in his father (Fig. 2G).
Literature review
Here we summarized clinical data of 26 patients with probable or definite FBD or FDD; 11 cases underwent pathological testing (autopsy or tissue biopsy) and 2 cases had genetics studies. We carefully summarized the clinical, pathological, and genetic characteristics of these cases to analyze the similarities and differences between the proband and FBD and FDD.
A total of 4 FDD and 22 FBD patients were in-cluded in this review. The common clinical manifestations were cerebellar ataxia, language disorder in the early stage of the disease, and dementia and mental problems in the later stage. The patients with FDD manifested cataracts and deafness in the early stage of the disease, while the patients with FBD suffered from spastic paraplegia. They all had pathological changes of extensive amyloid angiopathy, neurofibrillary tangles, and amyloid plaque formation in the hippocampus.
However, co-deposition of Aβ in a proportion of blood vessels affected by ADan has been a constant finding in FDD cases whereas Aβ immunostaining was never observed in any of the FBD specimens analyzed so far (Table 1).
Features in FDD and FBD with ITM2B mutation
NO, number; FBD, familial British dementia; FDD, familial Danish dementia; F, female; M, male; NA, not available; RMT, recognition memory test; VIQ, verbal intelligence quotient; PIQ, personal intelligence quotient; MMSE, Mini-Mental State Examination; NFT, neurofibrillary tangles; WMHs, white matter hyperintensity.
DISCUSSION
Regarding the similarities and differences between FBD, FDD, and the proband, they all had cerebellar ataxia and progressive dementia. Both the proband and patients with FBD had spastic paraplegia, language problem, but patients with FDD and the proband had deafness. Compared to FBD, the proband did not have personality problems and also did not have cataracts compared to FDD. The age of onset of FDD is earliest in three of them (Table 2).
Main clinical, pathological and genetic differences between FBD, FDD, and the proband
FBD, familial British dementia; FDD, familial Danish dementia; WMHs, white matter hyperintensity.
Vidal proposed that ITM2B mutations were associated with FBD and FDD in 1999 and 2000 [1, 2]. Patients with FDD have a 10-nt duplication of the DNA sequence encoded between nucleotides 786 to 795 of the wide-type precursor cDNA sequence (795–796ins TTTAATTTGT), which produces a longer C-terminal fragment causing an amyloid precursor protein with 277 amino acids (ADanPP) [2]. In FBD, a point mutation (T for A) at codon 267 in gene ITM2B changes the normal stop codon into an arginine BRI(Stop-2673Arg), and as a result, the precursor protein extends to 277 amino acids (ABriPP) [1]. Both proteins are cleaved by furin, resulting in the release of the secreted peptides ADan and ABri with 34 amino acids, which, in contrast to the normal peptide, are amyloidogenic and form deposits in the brains [6]. Then researchers found that ADan and ABri deposited most obviously in the cerebellar cortex and hippocampus [7]. The main neuropathology was that extensive parenchymal deposition of cerebral amyloid angiopathy (CAA) and ADan of the Tg-FDD rat model, with rats gradually showing abnormal behavior, bending backwards, and wide gait [8].
Comprehensive analysis of the cases in the previous literature reveals that the neuropathological changes also showed extensive CAA, amyloid deposition in the hippocampus and cerebellar cortex, but tau deposition in hippocampus and other parts of limbic system. It is also possible that mutations in ITM2B affect the function of the presynaptic and postsynaptic of hippocampal neurons, which leads to dementia in patients with FBD and FDD [9]. In the pedigree we described, we found that the c.800G>T (guanine > thymine) mutation of ITM2B caused the amino acid change p*267Leuext*11 (the termination mutation became leucine and then extended by 11 amino acids and then terminated), which also caused the BRI2 turned into a 277 amino acids protein. We speculate that a 34-amino acid peptide was released which was cleaved by furin and might cause extensive deposition of amyloid and tau in the brain.
Amyloid PET of the proband indicated bilateral cerebellar amyloid deposition, and bilateral frontal, parietal, temporal lobe, and posterior cingulate gyrus slightly amyloid deposition. Tau PET showed abnormal deposition of tau protein in bilateral cerebellar cortex, bilateral occipital lobe, and medial temporal lobe, which was different from FBD and FDD. We speculated it may be a new subtype of familial dementia caused by mutation of ITM2B, which was characterized as ataxia, and the pathology of the cerebellum was more obvious. Welzel et al. proposed that ADan and ABri were involved in the formation of neurofibrillary tangles in the brain, but all these suggest that the mutation of the ITM2B gene in this family can also cause changes in the function of the BRI2 protein with neuropathological changes similar to FBD and FDD.
We identified a novel mutation in ITM2B the familial dementia related gene in Chinese population. The clinical manifestations of the affected patients described are slightly different from those previously reported in FBD and FDD. Our findings increased genetic knowledge of familial dementia and extended the ethnic distribution of ITM2B mutations.