
Abstract
Recent evidence suggests that about 30%of patients with mild to moderate Alzheimer’s disease (AD) without a known diagnosis of epilepsy may display epileptiform spikes during electroencephalographic (EEG) recordings. These abnormal discharges occur predominantly during sleep and may be associated with accelerated disease progression. Subclinical spikes may represent a relevant target for clinical drug interventions, and there is a clear unmet need for preclinical testing of novel disease modifying agents in suitable animal models. Transgenic rodent models of AD pathology exhibit various forms of epileptiform EEG activity related to the abnormal levels of amyloid species in the brain. Among them, large-amplitude cortical and hippocampal EEG spikes in mouse and rat AD models may be reminiscent of the subclinical epileptiform EEG spikes recorded in some AD patients. This article reports the recommendations of a multidisciplinary panel of experts on optimal EEG markers and experimental designs to measure and report epileptiform activities and their response to symptomatic and disease-modifying drugs in transgenic AD model rodents. These recommendations may harmonize future preclinical EEG studies in the drug discovery research and may increase the comparability of experimental outcomes and their translational clinical value.
INTRODUCTION
Background
Alzheimer’s disease (AD) has long been associated with an increased risk of epileptic seizures. Epidemiological studies have shown that unprovoked epileptic seizures may occur in 0.5–64 %of patients with dementia due to AD [1–3]. Furthermore, these seizures are more frequent in patients with early-onset familial AD than in the common late-onset sporadic form [2].
Although epileptic seizures in AD patients may be sporadic without dramatic clinical manifestations, recent studies suggest that they may represent only the tip of the iceberg of larger pathophysiological phenomena. These patients may suffer from a background central nervous system hyperexcitability associated with electroencephalographic (EEG) epileptiform discharges, possibly having subtle but significant effects on AD patients’ cognitive functions [2, 4]. Two prospective video-EEG studies on patients with early stages of dementia due to AD found epileptiform spiking without motor manifestations (also known as “subclinical epileptiform activity”) in 22%[5] –42%[6] of patients. This percentage may still be an underestimate since local temporal electrophysiological activity does not always show up in routine scalp-recorded EEG, appearing only in intracranial EEG recordings [7]. Furthermore, this epileptiform spiking occurs almost exclusively during sleep [6, 8] and can be entirely missed during routine, daytime EEG recordings. These features of epileptiform activity may explain its low incidence in earlier reports [9]. On the other hand, a recent video-EEG study did not find a significant overexpression of epileptiform discharges in more elderly AD patients during sleep [10]. Nevertheless, subclinical epileptiform activity in AD patients does not appear to be a benign condition, as over a 5-year period, AD patients with dementia and subclinical epileptiform activity declined more than twice as fast on cognitive testing compared to AD patients with no such detected activity [6]. At baseline, the two groups of AD patients did not differ clinically or in terms of their APOE ɛ4 carrier frequency, medication, disease duration or brain atrophy (measured in 81%of the subjects) by structural magnetic resonance imaging. Taken together, previous field studies suggest that subclinical epileptiform activity may be clinically relevant in AD patients with both mild cognitive impairment (MCI) and dementia [2, 4]. However, more research is needed about the disease onset and progression to ascertain whether the subclinical epileptiform activity is always associated with a more aggressive AD form.
As mentioned above, the frequent occurrence of epileptiform activity in AD patients has been recognized for a long time and it is considered as a risk factor for accelerated disease progression [4, 6]. However, the impact of current medications widely used for the treatment of AD (e.g., cholinesterase inhibitors and the NMDA receptor antagonist memantine) on AD-related epileptiform activity has not been yet investigated to our knowledge. Furthermore, only a few prospective clinical trials have tested the effects of antiepileptic symptomatic drugs (AEDs) in AD patients with epileptiform activity [11]. The review of these clinical trials is beyond the scope of this article, so we will just summarize their main findings in the following. Previous studies tested AEDs of both first (e.g., carbamazepine, valproic acid, phenobarbital, and phenytoin) and second (e.g., levetiracetam, lamotrigine, and gabapentin) generations [2, 4]. Among them, lamotrigine, levetiracetam and phenobarbital AEDs were able to reduce seizures by 60–70%during the observation period of 12 months with relatively favorable tolerability and clinical effects [12–14].
Several studies have also tested the cognitive effects of levetiracetam based on the idea of treating network hyperexcitability in AD patients with dementia or MCI. Those clinical trials have showed promising results in terms of improved attention and verbal fluency [13] as well as improved recognition memory and normalization of the task-related functional MRI activation in the dentate gyrus/CA3 and entorhinal cortex [15, 16]. Notably, those patients received just 250 mg per day of levetiracetam well below 1000–3000 mg per day typically used for the treatment of epilepsy [17], to explore the mere properties as cognitive enhancer of that drug. Finally, the administration of a single dose of levetiracetam in AD patients with dementia and epileptic seizures was associated with normalizing resting state EEG rhythms by decreasing the spectral coherence at the delta band (1–4 Hz) and an increasing that at the beta band (13–30 Hz) [18]. However, despite increasing interest in levetiracetam and its atypical synaptic vesicle glycoprotein (SV2A) modulating antiepileptic mechanism, only the last study included EEG recordings, but even that one did not specifically look at silent epileptiform activity. This important issue will be clarified by several ongoing clinical trials on cognitive effects of levetiracetam in AD, which include EEG as an outcome measure in some of them, such as “Levetiracetam for Alzheimer’s Disease-Associated Network Hyperexcitability (LEV-AD; https://clinicaltrials.gov/ct2/show/NCT02002819)”, and “Levetiracetam for Alzheimer’s Disease Neuropsychiatric Symptoms Related to Epilepsy Trial (LAPSE, https://clinicaltrials.gov/ct2/show/NCT04004702”.
Validity of rodent models to assess drug treatments against AD-related epileptiform activity
Validity of an animal disease model has been traditionally evaluated in terms of construct, face, and translational predictive validity. In this line, transgenic AD rodent models have
Aim of the article
Since two seminal papers described a high prevalence of spontaneous seizures in common APP [19] and APP/PS1 [20] transgenic AD mouse models about 15 years ago, the effect of the most common anticonvulsive drugs has been tested on these models [25, 26]. Albeit seizures (whether convulsive or nonconvulsive) are easy to detect on a routine chronic epidural EEG, their occurrence in amyloid plaque forming transgenic mice is so rare (one seizure per three weeks of 24/7 on average in a continuous video-EEG, [20]) that it is very difficult to obtain sufficient statistical power in those studies without really large study groups. Therefore, when considering spontaneous epileptic events, the focus of the preclinical studies on AD related epilepsy for years has been on nonconvulsive epileptiforms events, spike-wave discharges and single spikes, which occur at the rate of tens to hundreds of events per hour. Keeping in mind the recent findings of high incidence of subclinical epileptiform spiking in AD patients and above-mentioned clinical trials, preclinical studies on AD rodent models investigating nonconvulsive epileptiform activity are needed more and more. They may provide new insights into the early evaluation and selection of the most promising new drugs targeting subclinical epileptiform activity in AD patients with MCI and dementia.
At present, there are no generally accepted standards on how spontaneous epileptiform activity in rodent AD models should be recorded, analyzed and reported. This complicates the comparison across studies and thereby may seriously slow down ongoing drug development for subclinical epileptiform activity in AD. This article aims at providing recommendations to harmonize future preclinical EEG studies in the drug discovery research and to increase the comparability of experimental outcomes and their translational clinical value.
METHODOLOGY
The present multidisciplinary Working group of experts surveyed and discussed recent literature on EEG findings on nonconvulsive epileptiform activity in AD rodent models in detail. This resulted in recommendations about optimal EEG markers and experimental designs to measure and report this kind of epileptiform activity and its response to symptomatic and disease-modifying drugs in AD rodent models. Expertise in the Working group covers several relevant disciplines such as Pharmacology, Pharmaco-EEG, Preclinical and Translational Neurosciences, Clinical Neurophysiology, and Quantitative Analysis of EEG activity. The recommendations by the Workgroup may be helpful to harmonize future preclinical EEG studies in transgenic AD rodent models showing epileptiform activity in drug discovery research. They also aim to foster the comparability of novel findings and their translational value to clinical research. Of course, the present recommendations will have to be revised based on the results produced by future studies.
The present Workgroup formulated the recommendations based on a comprehensive review of the field literature performed through Web of Science, Scopus, and MEDLINE. This review was performed using several appropriate combinations of the following key words.
For the “Transgenic AD rodent models” the following key words were used: “Alzheimer” AND ”Mouse OR Mice” OR “Rat OR Rats”. This search term was combined with either epilepsy related or drug of interest related keywords listed below. The numbers in parentheses indicated the number of hits found.
For the “Epileptiform activity,” the following key words were used: “Epilepsy”(489), “Epileptic” (535), “Epileptiform” (64), “Seizure (354)”, “Spike (189)”, “Hyperexcitability” (143), “EEG OR Electroencephalography (278)”.
For the “Drugs of interest,” the following key words were used: “Antiepileptic (242)”, “Anticonvulsive (237)”, “Levetiracetam (24)”, “Brivaracetam (1)”, Lamotrigine (9)“, “Phenytoin (10)”, Carbamazepine (10)”, “Valproic acid OR Valproate (54)”.
Only papers published in international journals with peer-review and an impact factor were considered (–2020). We first collected all hits with the search terms above. Second, we removed all duplicate or multiple hits and review articles, which yielded in total 1,139 articles. Third, we excluded all studies where no EEG recording was made resulting in 247 articles left. Finally, we excluded studies where reported epileptiform activity was not spontaneous and not recorded in freely moving animals. Some full papers could not be downloaded from available Internet resources or obtained by the authors and were consequently not considered. This procedure left us with 37 original publications.
Concerning the general procedure at the basis of this article, all selected abstracts/papers were critically reviewed by two members of the Expert Panel (N.J. and H.T.). Afterward, three members of the Expert Panel (i.e., N.J., C.B., and H.T.) produced a first draft of the article. This draft was sent to the other co-Authors for further discussions, amendments, and serial revisions. As a result, the Workgroup reached a consensus about all contents of this article, including all recommendations provided. The last version of the article was finalized in January 2021.
The results of some relevant papers reviewed are shortly reported and discussed in the following sections with the aim to derive some general caveats and recommendations for future preclinical research in transgenic AD rodent models showing epileptiform activity.
NOTIONS AND RECOMMENDATIONS ON TRANSLATABLE FEATURES OF EPILEPTIFORM ACTIVITY WITHOUT MOTOR MANIFESTATIONS IN TRANSGENIC AD RODENT MODELS
To date, only few long-term EEG studies have been conducted in AD patients with MCI or dementia based on a primary intention to investigate features of the epileptiform activity. Nevertheless, some consistent features can be found in those reports. They are summarized in the following as a basis for understanding the most translatable results of the preclinical EEG studies in transgenic AD rodent models showing epileptiform activity. In AD patients, 1) subclinical epileptiform activity often manifests as single high-amplitude spikes or sharp waves [6–8, 24], or as brief (3–12 s) paroxysmal epochs of delta-theta (2–8 Hz) activity [8, 27]; 2) the most common locations for the epileptiform spiky activity are frontal and temporal cortices in EEG recordings from the scalp [6, 24], while invasive intracranial recordings via the foramen ovale reveal medial temporal epileptiform spike waveforms not visible in the routine scalp EEG activity [7]; and 3) epileptiform spikes and sharp waveforms occur predominantly (80–90%) during sleep, mainly in the NREM sleep [5, 8].
Although some standard seizures lasting > 5 s were reported to be revealed by epileptiform activity with no motor manifestations in preclinical EEG studies in transgenic AD mouse models [19, 20], we did not consider them here when the authors did not make a distinction whether these seizures were linked to convulsions or not.
In the reviewed preclinical EEG studies, the seizures without motor manifestations were associated with epileptiform activity classified into two main classes named “spike-wave discharges, SWDs” (Table 1) and “Single spikes” (Table 2). Features and recommendations for the evaluation of that epileptiform activity are reported in the following.
Definition of spike-wave discharges (SWDs) in EEG recording in AD rodent models
Definitions of single epileptiform spikes in AD model mice
Spike-wave discharges (SWDs)
In epidural preclinical EEG recordings, SWDs are characterized by surface-negative spikes alternating with surface-positive waves at 7–10 Hz [28] (Fig. 1A). Their typical duration in mice is 0.5–2 s, in rare cases up to 5 s (Table 1). Notably, SWDs have been also presented in the literature under names such as “Nonconvulsive spontaneous spindle-shaped discharge” and “Epileptiform discharge” (Table 1). In older EEG studies in rats, SWDs were also reported as “High-voltage spindles” [29].
Examples of (A) a spike-wave discharge and (B) a sleep spindle with spiking recorded from a 5-month-old APPswe/PS1dE9 mouse. The spike-wave discharge is characterized by a lower oscillation frequency (∼10 Hz), regular spiking, which gives an asymmetric impression across the 0-line. The sleep spindle is a faster oscillation (∼12 Hz) and symmetric across the 0-line. In this case, four single surface-negative spikes coincide with the troughs of the sleep spindle. Recordings with skull screw electrodes on the frontal bone with a reference over the cerebellum.
The SWDs in transgenic AD mouse models closely resemble those in genetic rat models of absence seizure, such as the Wistar-derived albino inbred lines WAG/Rij [28] and GAERS [30], although SWDs are much shorter in duration in transgenic AD mouse models than those found in rat models. Recently, profound SWD (referred to as high-voltage spindles) activity has been reported in an AD-related transgenic rat, TgF344-AD [31, 32]. The underlying circuitries and neurotransmission of SWDs have been intensively studied over the past three decades [28, 33]. The current dominant view is they are predominantly generated within the motor and somatosensory cortices but modulated by inputs from ventral posterior (VP), posterior (Po), and reticular (RT) thalamic nuclei [33].
While in absence epilepsy rat models the SWDs predominantly occur during transitions from wake to non-REM sleep with a clear circadian pattern [34, 35], it has been consistently reported that in transgenic AD mouse models, the SWDs occur mostly during drowsy wakefulness and light sleep. Importantly, previous preclinical studies [21, 36] showed that SWDs occur > 10 times more often in APP(Swe)/PS1dE9 transgenic mice producing abundant amyloid accumulation in the brain than their wild-type littermates. The frequency of this abnormality in EEG activity is so high that it may be a convenient biomarker in studies testing new pharmacological interventions to mitigate epileptiform activity in those rodent models characterized by AD-related amyloid pathology. However, the relationship between the generation of SWDs and AD-related soluble or plaque deposited amyloid species is still an open issue in preclinical studies and requires more research.
In rats, SWDs have been considered as genetic models of human absence epilepsy, but they differ from the human condition in two notable aspects. First, absence epilepsy is typically a clinical manifestation observed in human childhood [37]. In contrast, SWDs reach their peak occurrence only in the adulthood in the rat absence models [28]. Second, the dominant frequency of SWDs is around 3 Hz in humans, but 7–10 Hz in the rat absence models. Notably, the EEG pattern of rodent SWDs resembles more the human EEG μ-rhythms recorded over Rolandic somatomotor cortical areas during limb muscle relaxation [38] and in certain types of epilepsies (e.g., progressive myoclonus epilepsy) [39] than human SWDs. To our knowledge, no EEG study in AD patients without a diagnosis of epilepsy has discovered SWDs like those recorded in transgenic AD mouse models and rat absence models.
In transgenic AD mouse models, SWDs are interesting but challenging as a readout (i.e., endpoint marker) in testing drug effects on epileptiform activity. Indeed, SWDs typically occur in the light sleep and may to the non-trained eye be confused with sleep spindles typically observed in a bit deeper sleep stage [40]. Notably, SWDs and sleep spindles are generated by oscillatory activity of cortical pyramidal neurons modulated by very similar thalamocortical/corticothalamic circuits and only slightly differ in their oscillatory frequency [40]. However, epileptic SWDs display less symmetry around the 0-line, no typical spindle-morphology related amplitude waxing-and-waning, and -depending on electrode montage- an overall lower amplitude [41]. Further, epileptic SWD and sleep spindles have been reported to be differentially sensitive to pharmacological manipulation. Nevertheless, when physiological sleep spindles are accompanied by spiking, their appearance can be difficult to distinguish from SWDs, especially in EEG activity recorded from skull electrodes (Fig. 1). This makes it extremely difficult to find quantitative parameters of EEG activity that allow a valid software-based automated detection of SWDs. An algorithm for automatic detection of SWD (high-voltage spindles) has been applied previously in rat EEG [32], and it is expected that validated, new mathematical detectors of SWDs will be developed based on advanced deep learning algorithms. Until such developments become further available and more reliable,
Single spikes
The majority of preclinical intervention studies in transgenic AD mouse models investigating epileptiform activity have used single spikes as a readout (Table 2). In this literature, epileptiform single spikes are generally defined as distinct sharp deflections of ongoing EEG activity that stand out from the baseline voltage (Table 2, Fig. 2). However, there is little consensus on the exact definition of this EEG waveform in terms of spike amplitude and duration.

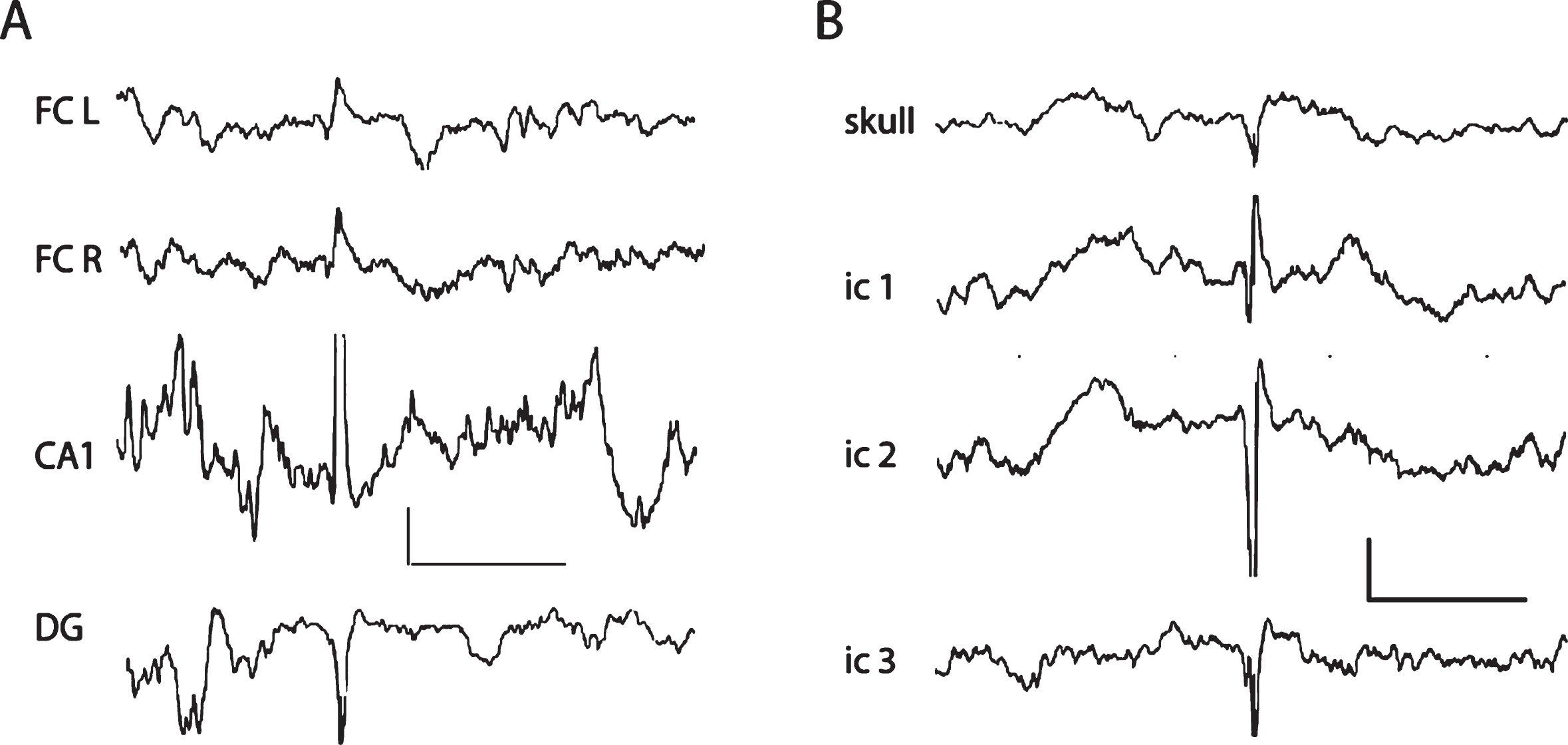
A) Reflections of a large hippocampal spike on the cortical screw channels in an APPswe/PS1dE9 mouse. Note the polarity reversal between hippocampal CA1 and DG recorded with a triple staggered wire electrode. The hippocampal spike is seen as a surface-positive reflection on the cortical screw channels bilaterally on the frontal cortex. Scale 0.5 mV/ 500 ms. FC L and FC R, left and right frontal screw electrode channels; DG, dentate gyrus. B) A cortically generated large epileptic spike in a 5xFAD mouse recorded with screw electrodes and a triple staggered wire electrode spanning the cortical thickness. Note the polarity reversal between intracortical (ic) electrodes 1 and 2. The epidural recording reflects the largest extracellular negative population spike on ic2 channel. All electrodes are referred to a common reference above the cerebellum. Scale 0.5 mV/500 ms.
Voltage threshold of the EEG single spikes
In the preclinical EEG studies performed in transgenic AD mouse models, the overwhelming majority employed skull screw electrodes with very low impedance for the recording and analysis of cortical EEG waveforms. Among them, some EEG studies used
The majority of mouse EEG studies employed a
Two reviewed EEG studies in transgenic AD mouse models simultaneously detected high-voltage epileptiform spikes in cortical and hippocampal recording channels [21, 23]. Specifically, these very large hippocampal spikes (> 7 SD or > 10 SD) were observed in transgenic AD mouse models with APP or APP/PS1 mutations, whereas those spikes were practically absent in wild-type littermates [21, 23]. These EEG waveforms were called “inter-ictal spikes” [23] or “giant spikes” [21]. The amplitude of these spikes were similar in the two brain locations in APP(Swe) Tg2576 mice [23], whereas the hippocampal “giant spikes” were 2–5 times larger in amplitude than the cortical spikes in APP(Swe)/PS1dE9 mice [21]. Furthermore, the hippocampal “giant spikes” were characterized by an afterhyperpolarization and an attenuation of background field potentials that could extend up to 1-2 s [21] (Fig. 3).
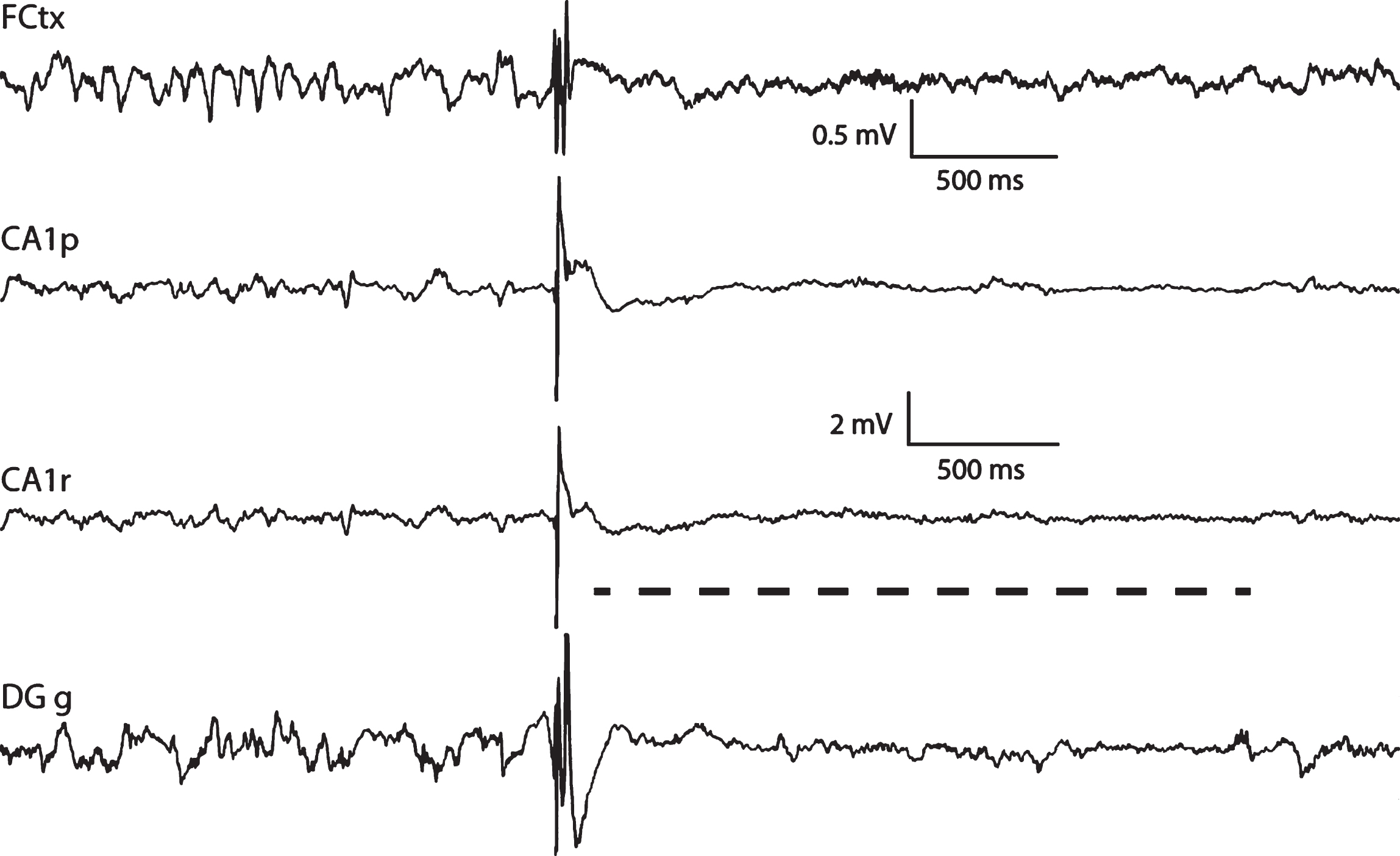
A giant spike recorded from an APP/PS1dE9 mouse. It is characterized by a complex large-amplitude sharp waveform that occurs simultaneously in hippocampal and cortical channels and is followed by suppression of neural activity up to 2 s after the spike (gray dashed line). A sleep spindle in the beginning of the cortical trace reminds that this giant spike occurred during NREM sleep. CA1p. CA1, pyramidal layer; CA1r, stratum radiatum; DGg, granule cell layer on the upper blade of the dentate gyrus. The upper scale bars refer to frontal cortical channels and the lower scale bars to the three hippocampal channels.
As a source of variability, the referred studies did not report whether the baseline mean voltage was computed in a short EEG epoch preceding the epileptiform EEG spike or by averaging the voltage during the whole recorded EEG activity. Furthermore, none of the referred studies revealed the baseline mean voltage and its SD. Let us assume that a mean negative baseline voltage of ∼50μV is measured in cortical screw channels and ∼150μV in hippocampal wire electrode channels (Ø 50μm stainless steel), and that the SD is ∼10%smaller than the mean. With these values, an amplitude of 200μV on the cortical screw channels would correspond to 4 x baseline or 4.5 SD, while an amplitude of ∼250μV would correspond to 5 SD. Based on this computation, the absolute and the relative voltage thresholds of cortical and hippocampal epileptiform spikes in several studies fell into the same relatively narrow range of voltages. However, the studies using a voltage threshold of 2 x baseline mean voltage (∼100μV) in cortical screw channels defined epileptiform spikes in a significantly different way.
Another source of variability may be the polarity of the epileptiform spikes. Most of the reviewed studies considered both positive and negative polarity of the cortical epileptiform spikes in the analysis. This choice may be problematic with EEG recordings using epidural exploring electrodes referenced to a distant reference electrode placed in the cerebellum. In case of tangentially oriented bipolar neural sources, the positive and negative components of the epileptiform spikes may be recorded from far epidural exploring electrodes (Fig. 2).
Keeping in mind the field literature data and the above considerations,
Duration threshold of the EEG single spikes
In the preclinical EEG studies performed in transgenic AD mouse models, the epileptiform spike width (duration) can vary a lot, with the maximum period ranging from 15–150 ms with a median value of 70 ms (Table 2). This width is one important measure to disentangle between cortical epileptiform spikes and sleep spindles. In mice, cortical sleep spindles have a dominant frequency in the EEG power spectrum between 10–15 Hz [40], which correspond to cycles lasting ∼80 ms and half-cycles of ∼40 ms in the range of epileptiform single spikes. To reduce the chance that a prominent wave of a sleep spindle may be classified as a ‘single spike’,
Density of the EEG single spikes
In the preclinical EEG studies performed in transgenic AD mouse models, a relevant question is whether epileptiform single spikes may be observed more frequently (i.e., higher spike density) in transgenic AD mouse models than littermate controls. In the papers reviewed, only six preclinical EEG studies addressed such a question with converging evidence that cortical epileptiform spikes are at least 10-fold more frequent in transgenic AD mouse models than in littermate controls. Notably, this evidence was obtained in studies using both a relatively low (2–2.5 x baseline) relative voltage threshold to define an epileptiform single spike [43, 44] and those with higher voltage thresholds (4–8 x baseline or 7 SD or ∼240μV) [22, 46].
All the above studies were performed in transgenic AD mouse models carrying only human APP mutations which typically require substantial hAPP overexpression to induce the production of amyloid plaques in the brain. In contrast, a study on aged APP(Swe)/PS1dE9 mice found no difference in single spikes compared to wild-type control mice [47]. A recent EEG study investigated different types of cortical epileptiform spikes using a high relative voltage threshold (> 6 SD) in young adult mice of the same APP(Swe)/PS1dE9 line [21]. In that study, epileptiform single spikes in cortical EEG recordings were 3 times more common in APP(Swe)/PS1dE9 mice than wild-type littermates. Notably, the occurrence of large cortical spikes riding on sleep spindles did not differ between the genotypes, whereas the number of spikes within SWDs was 10-times higher in the former than the latter [21, 36]. That study highlights the translational importance of making a distinction between single high-voltage EEG spikes of probable epileptic nature vs. spikes detected within a larger EEG oscillation pattern (e.g., sleep spindles).
As mentioned above, over 80%of subclinical epileptiform EEG activity (including large EEG single spikes) in AD patients may occur during sleep [6, 8]. Indeed, over 80–90%of all high-voltage cortical and hippocampal EEG spikes in transgenic AD mouse models may occur during sleep [21, 23]. Furthermore, EEG recordings during sleep typically produce a large amount of EEG activity without irremediable instrumental or biological artefacts related to animal exploratory behavior. In a back-translational line,
The fact that high-amplitude epileptiform spikes in transgenic AD mouse models mostly occur during sleep should be taken into account in reporting experimental results. Reporting the epileptiform spike density over the sleep time can avoid the following two biases. The first bias may occur if a given drug has both antiepileptic and sedative effects. In this case, counting the EEG spike density over total recording time may mask the beneficial reduction in EEG spike rate during the sleep paradoxically due to the prolonged sleep time. Indeed, even with that reduction, the prolonged sleep time may show a significant number of high-amplitude epileptiform spikes. Special attention is to be paid when studying effects of for example benzodiazepines, as these agents may induce sleep with physiologically aberrant EEG oscillatory correlates (e.g., concomitant beta activity rather than physiological correlate of non-REM sleep delta activity). The second bias may occur if a given drug prolongs the wakefulness time without a significant antiepileptic effect. In that case, one can get a false impression of an effective antiepileptic action merely due to a shorter sleep period associated with less sleep-related high-amplitude epileptiform spikes (but unaffected density of those spikes over the sleep period).
Translational significance of epileptiform activity without motor manifestations
As described above, SWDs are frequent in transgenic AD mouse and rat models but have not been detected in EEG recordings performed in patients with MCI-AD and AD dementia. Nevertheless, SWD activity in rats is enhanced by scopolamine [29] and significantly diminished by cholinesterase inhibitors in aged rats [48] and TgF344-AD rats [31] showing some predictive validity for the beneficial cognitive effects by cholinesterase inhibitors in AD patients (without necessarily being the mediating mechanism itself). In contrast, high-voltage single spikes in ongoing EEG recordings may represent a suitable translational candidate. As mentioned above, those high-voltage spikes may be overrepresented in transgenic AD mouse models over controls and may match subclinical epileptiform activity recorded in AD patients. In both transgenic AD mouse models and AD patients, epileptiform spikes were recorded from frontoparietal cortical areas or medial temporal lobe as a possible reflection of hippocampal EEG spike [7, 24]. Furthermore, they were more frequent and of highest amplitude during sleep [21, 23].
Single brain spikes (particularly giant hippocampal spikes) may have good translatability because may be specific to AD transgenic mouse models as compared wild-type littermates and they resemble epileptiform spikes described in AD patients and occur largely during sleep. However, despite the striking similarities between sleep-related high-voltage spikes in AD mouse models and subclinical epileptiform activity recorded in AD patients, certain limitations should be kept in mind when translating the rodent results to human patients. So far, single high-voltage spikes during sleep have only been reported in transgenic AD mouse models with amyloid pathology; we do not know the contribution of tau pathology in these models. Second, sleep-related epileptiform spiking in mice appears to be largely limited to REM sleep [23, 49], while in AD patients, spikes are predominantly found during NREM sleep [8]. However, this seems to depend on the specific mouse lines, since high-voltage spiking during NREM sleep was found in APP/PS1 mice [21]. Third, the reported spike/sharp-wave frequency in human long-term video-EEG recordings is on average an order of magnitude lower, around 1/h [8, 24], than the reported spike frequency in mouse recordings with a mean around 10/h [21, 46]. This difference may speak for a different underlying mechanism. On the other hand, the individual variability among human AD patients and transgenic AD model mice is substantial and the distribution of spike frequency partially overlapping between humans and mice.
For the purposes of clinical translation ultimately the most important aspect of translatability of epileptiform spikes in AD rodent models is how they impact cognition or disease progression. For instance, does selective treatment of a particular spike type in AD rodent models correlate better with improvement in cognitive performance or reduction in pathology? So far, only two preclinical studies have administered AEDs to AD model mice and assessed responses both in terms of epileptic spiking in EEG and memory. Unfortunately, these papers focused on different types epileptiform spiking. One study compared several AEDs in their efficacy to suppress SWDs and to improve spatial memory in the Morris swim task, but in separate experiments. Whereas both brivaracetam and ethosuximide suppressed SWDs, only brivaracetam improved spatial memory in APPswe/PS1dE9 mice [47]. The other study first compared several AEDs in their ability to suppress single cortical spikes and then assessed behavioral responses to levetiracetam that proved most effective [46]. In this study, chronic low-dose levetiracetam proved effective in suppressing spiking and alleviating spatial memory impairment of APP(Swe,Ind) mice in Morris swim task as well as hyperactivity in the open field and elevated plusmaze. However, a single injection of levetiracetam did not reduce hyperactivity despite effective spike suppression. Neither study reported reduction in the brain amyloid load after a chronic AED treatment [46, 47] suggesting that the observed behavioral effects of AEDs in AD model mice are independent of brain amyloid-β levels. These studies also point to different immediate and long-term functional consequences of epileptiform spiking in AD mouse models, which may be also relevant to AD patients with subclinical epileptiform activity. These are interesting aspects to be addressed in future studies.
Keeping in mind these converging data and considerations,
RECOMMENDATIONS ON TECHNICAL CONSIDERATIONS IN ENSURING TRANSLATIONAL VALIDITY OF SINGLE EPILEPTIC SPIKES
Behavioral state: assessment and reporting of wake-sleep periods
As mentioned above, over 80%of subclinical epileptiform EEG activity (including large EEG single spikes) in AD patients and single high-voltage cortical or hippocampal spikes in AD mouse models may occur during sleep [6, 23]. Given its importance,
Use of EEG biomarker specific for transgenic AD mouse models
EEG data allow for the derivation of multiple readouts based on visual or spectral (e.g. computation of EEG power density, coherence, entropy, etc.) analyses of those data collected in transgenic AD mouse models [50, 51]. These readouts may be sensitive to different neurophysiological mechanisms of brain neural synchronization and functional connectivity underpinning the modulation of neural excitability and inhibition in relation to the progression of cerebral disorders and therapy responses. However, different transgenic AD mouse models may be associated with different pathophysiological manifestations of brain neural hyperexcitability or dysconnectivity. As a consequence, epileptiform activities may be generated in different regions and phases of the sleep-wake cycle. So far, the accumulated data in nonconvulsive epileptiform activity in the literature are limited to amyloid plaque forming genetically modified mice. All these mice show most severe brain amyloidosis in the neocortex and subiculum, followed by the hippocampus, amygdala and thalamus [52–56]. A comparative density of epileptiform single spikes [22] or SWDs [22, 36] has been detected in transgenic mice with highly variable brain amyloid load and even in young mice before any amyloid plaque formation [22]. It has been claimed that the epileptiform activity in these mice results from overexpression of human APP rather than the amyloid-β accumulation [44]. However, mice with highly variable extent of APP overexpression, ranging from 6- to 8-fold in APP(Swe) Tg2576 mice [23, 54] to no overexpression in APP(NL-F) KI mice [22] show more high-voltage single cortical spikes than their wild-type littermates. Therefore, comparison of different pathologies in different brain regions between AD pathology rodent lines has so far indicated but not explained the exact etiological relationship between the pathological features and epileptiform activity.
Use of hippocampal electrodes
The overwhelming majority of EEG studies for epilepsy in AD rodent models have employed cortical screw electrodes. These are the gold standard, robust and reliable. However, adding simple hippocampal wire electrodes yields a lot of information about brain neurophysiological oscillatory mechanisms at the cost of 2-3 channels. An electrode placed anywhere in the hippocampus (but in the middle of the pyramidal cell layer) is able to record movement and REM sleep associated theta oscillations far better than epidural cortical electrodes [57]. Furthermore, as discussed above, high-amplitude hippocampal spikes during sleep that are synchronous with cortical spikes are specific to APP or APP/PS1 transgenic mice. They also closely resemble epileptiform spikes reported in invasive foramen ovale recordings performed in AD patients [7] and thereby have potential high translational value. Moreover, having hippocampal electrodes in addition to cortical ones also helps artefact rejection. A waveform that has a similar polarity and waveform in all cortical and hippocampal channels is very likely an artefact. However, it is worth noting the risk that hippocampal electrodes may induce microlesions triggering epileptiform activity in the already fragile hippocampal network of AD rodent models.
Keeping in mind the above data and considerations,
Isolation of oscillatory spiking increases the reliability of spike count
Although the translational importance of SWDs as preclinical EEG biomarkers in AD research may be questionable, these epileptiform activities may constitute a significant confound in the assessment of single spike rate. A few-minute epoch with repeating SWDs can easily contain tens of sharp surface-negative spikes that would be indistinguishable from single spikes if only single spike parameters are considered. Since SWDs may be generated by a thalamo-cortical loop instead of local cortical mechanisms, they can differ radically in responses to anti-epileptic medication from single cortical spikes. Therefore, it will be highly important to screen the isolated single spikes for regularly repeating patterns at the typical 7–10 Hz and exclude them from the total count.
As another potential confound, the sleep-spindle associated spikes (Fig. 1) should be treated separately from the single cortical spikes. As SWDs, spikes occurring during the troughs of the sleep-spindle may be generated by a different mechanism that single spikes outside the sleep-spindle. Indeed, in APP/PS1 mice levetiracetam tends to decrease the rate of occurrence of single cortical spikes and SWDs, but increase that of spindle-associated spikes [21].
Keeping in mind these considerations,
Spectral non-stationary analysis of epileptiform activity
Studies in transgenic AD rodent models and AD patients have reported that AD neuropathology may cause disconnection among neural cells, damage to cortico-cortical and cortico-subcortical pathways, and loss of myelinated axons, possibly associated with cortical neural hyperexcitability and hypersynchronization, as well as reduced neurotransmission, neural signaling, and synaptic activity [58]. These pathophysiological processes may be reflected in epileptiform activity and can be investigated by quantitative procedures of signal analysis. An extensive evaluation of those procedures is beyond the scope of this article. However, they represent a new frontier in the analysis of epileptiform activity in preclinical studies, so we will mention some basic concepts in the following that may help quantitative analysis of epileptiform activity (e.g., SWDs, giant spikes, etc.) in transgenic AD rodent models.
Ongoing EEG activity may derive from an ensemble of linear and nonlinear processes in brain circuits generating that activity in the wake-sleep cycle. Specifically, EEG signals are linear when any linear combination of inputs to neural system generating those signals does produce the same linear combination of them. Ongoing EEG activity was found to fit assumption of linearity in healthy brains, but it may violate that assumption during epileptic processes [59]. There are several nonlinear EEG measures such as global correlation dimension, entropy indexes, mutual information indexes [60]. These measures are typically tested using phase-randomized surrogate rsEEG-like data constructed by the well-known Theiler method and should be used considering their possible sensitivity to measurement noise [61].
EEG signals are stationary when they show constant statistical features (e.g., mean, variance, and autocorrelation function, power spectra) over time [63].
CONCLUSIONS
We strongly recommend the consideration of the following aspects when designing and reporting video-EEG studies to detect spontaneous epileptiform spiking in AD rodent models.
A) Detection and description of epileptiform spiking:
B) Relation between epileptiform spiking and sleep
C) Testing anti-epileptic medications in mouse AD models
Footnotes
ACKNOWLEDGMENTS
The present paper is part of a Mini Forum facilitated by the Alzheimer’s Association International Society to Advance Alzheimer’s Research and Treatment (ISTAART), through the Electrophysiology professional interest area (PIA). EPIA is committed to (1) exploit EEG biomarkers for improving the understanding of neurophysiological mechanisms underlying Alzheimer’s disease and age-related dementing disorders at micro, meso, and macro spatial scale and (2) promoting clinical applications. Of note, the views and opinions expressed by authors in this publication represent those of the authors and do not necessarily reflect those of the PIA membership, ISTAART, or the Alzheimer’s Association.
We thank Dr. Iris Oren for helpful comments to the manuscript.
Dr. Claudio Babiloni is partially supported by European Committee (H2020-EU.1.3.1.H2020-MSCA-ITN-ETN-2016 project with short title “BBDiag”).