
Abstract
Almost 115 years ago, Alois Alzheimer described Alzheimer’s disease (AD) for the first time. Since then, many hypotheses have been proposed. However, AD remains a severe health public problem. The current medical approaches for AD are limited to symptomatic interventions and the complexity of this disease has led to a failure rate of approximately 99.6%in AD clinical trials. In fact, no new drug has been approved for AD treatment since 2003. These failures indicate that we are failing in mimicking this disease in experimental models. Although most studies have focused on the amyloid cascade hypothesis of AD, the literature has made clear that AD is rather a multifactorial disorder. Therefore, the persistence in a single theory has resulted in lost opportunities. In this review, we aim to present the striking points of the long scientific path followed since the description of the first AD case and the main AD hypotheses discussed over the last decades. We also propose insulin resistance as a common link between many other hypotheses.
INTRODUCTION
Since the first description of Alzheimer’s disease (AD) in 1906 [1], researchers have coursed a long scientific path seeking a better understanding of this neurological disorder. Many hypotheses have been proposed over the last decades [2, 3]; however, AD has remained an enigmatic and complex disease with etiopathogenetic mechanisms yet to be elucidated. Currently, besides neurodegeneration, AD is mainly characterized by the accumulation of the amyloid-β peptide (Aβ), which tends to aggregate and form Aβ plaques, and by the presence of tangles, caused by the accumulation of hyperphosphorylated forms of tau protein [4].
Alzheimer had not only reported the first case of AD but had also described one important stage in the physiopathology of this disorder [5]. These findings laid the foundations for the most traditional and accepted theory of AD, the amyloid cascade hypothesis [6]. Nevertheless, the original debate on whether the amyloid was the cause or the consequence of the disease actually dates back to the time of Alois Alzheimer [7].
Worldwide, there are approximately 50 million people living with AD or other dementias [8]. AD is a progressive neurodegenerative condition and the most frequent type of dementia, corresponding to 60–80%of the cases [9]. In the United States, it is estimated that one in 10 people age 65 and older have AD, a total number of 5.8 million Americans [9, 10]. Furthermore, epidemiological data suggest an increasing trend in prevalence with estimations being 40 million patients suffering from AD in 2016 [11] and 131 million in the year 2050 [12]. Due to its complexity, AD is usually divided into hereditary AD (hAD) and sporadic AD (sAD). hAD accounts for approximately 1–5%of all cases, and it is usually caused by autosomal mutations in the amyloid precursor protein (APP), presenilin 1 (PS1), and/or presenilin 2 (PS2). Conversely, sAD, responsible for a majority of the cases (approximately 95–99%), does not present a well-defined etiology. It is believed that an interplay of genetic, environmental, behavioral, and metabolic factors might be responsible for the development of the sporadic form of this disorder [13].
Nowadays, more than one century after the discov-ery of this disorder [14], AD is still a chronic condition with no cure or effective interventions to delay its progression [15]. The current medical approaches for AD are limited to symptomatic interventions and the complexity of the disease has led us to constant failures in clinical trials [16]. The pharmacology of AD is currently limited to cholinesterase inhibitors (rivastigmine, galantamine, and donepezil) and memantine, which is an N-Methyl-D-aspartate (NMDA) receptor antagonist. These treatments are not able to prevent or reverse the progression of AD and are often accompanied by many adverse effects. In fact, no new drug has been approved for AD treatment since 2003 [17].
Some authors believe that AD drugs have failed mainly because of an inadequate target since the majority of studies have focused on the drugs targeting amyloid [18, 19]; however, other factors such as the stage of the disease at the moment of therapy initiation and the heterogeneity of factors implicated in AD pathophysiology should also be considered [20, 21]. Consequently, the multifactorial hypothesis of AD has been proposed by some to ponder divergent thinking and investigate multiple and diverse etiological factors that might be converging in a common brain pathology [22]. In this review, we aim to present the striking of the long scientific path since the first description of an AD case and the main AD hypotheses proposed over the last decades. Additionally, the “type 3 diabetes” hypothesis is discussed as accumulated evidence points toward insulin as an important factor implicated in the etiopathogenesis of AD, and dysfunctional insulin signaling in the brain provides a common link between other proposed hypotheses.
THE AMYLOID CASCADE HYPOTHESIS OF ALZHEIMER’S DISEASE
The Aβ, a peptide derived from a larger protein known as the amyloid-β protein precursor (AβPP), was isolated in 1984, by Glenner and Wong at the University of California, in San Diego [23]. The mech-anism responsible for the cleavage of AβPP and the production of Aβ is now well known [24]. AβPP is first cleaved off by the enzyme β-secretase (BACE 1), giving rise to two fragments: sAPP-β (N-terminal fragment), being released in the extracellular space and C-terminal fragment β (CTFβ, CT99, or CT89), which remain bound to the cell membrane. Then, the γ-secretase complex (Nicastrin, Anterior Pharynx defective 1, Presenilin enhancer 2, Presenilin 1, and or Presenilin 2) cleaves the remaining membrane-bound portion of the protein releasing the extracellular fragment Aβ (Fig. 1). Due to heterogeneous γ-secretase cleavage, γ-secretase can cut AβPP at different sites, producing a 37 to 49 amino acid residue peptide. Therefore, Aβ can vary in length [25]. On the other hand, in the non-amyloidogenic pathway, AβPP is firstly cleaved by α-secretase within Aβ sequence, producing soluble α-APP fragments (sAPPα) and C-terminal fragment α (CTFα, CT83), and, posteriorly, CTFα is cut by γ-secretase, releasing non-toxic fragments [26].
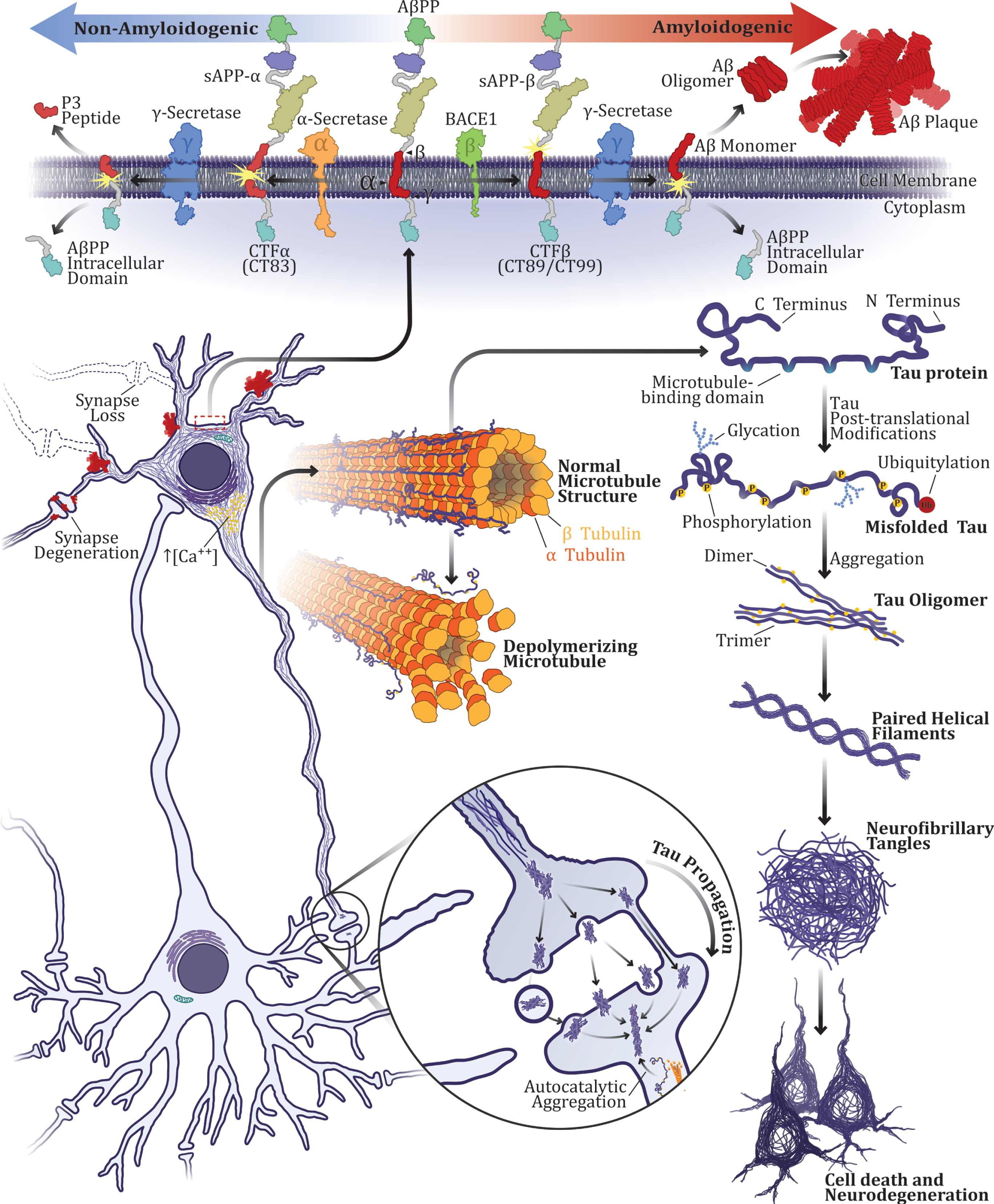
The traditional triad in Alzheimer’s disease pathogenesis: amyloid-β peptide, hyperphosphorylated tau protein, and neurodegeneration. Aβ plaques, neurofibrillary tangles, and cell death have been seen as the main neuropathological hallmarks and major factors in AD pathogenesis for more than a century. According to the amyloid cascade hypothesis, the most traditional hypothesis of AD, disturbances in AβPP metabolism are the triggering event in AD. In the amyloidogenic pathway, AβPP is first cleaved off by the enzyme β-secretase (BACE 1), giving rise to two fragments: sAPP-β (N-terminal fragment) and CT99 or CT89. Then, the γ-secretase complex cleaves the remaining membrane-bound portion of the protein releasing the extracellular fragment Aβ. On the other hand, in the non-amyloidogenic pathway, AβPP is firstly cleaved by α-secretase within Aβ sequence, producing soluble α-APP fragments (sAPPα) and C-terminal fragment α (CTFα, C83), posteriorly, CTFα is cut by γ-secretase, releasing non-toxic fragments (P3 peptide and AβPP intracellular domain). Alterations in AβPP processing usually result in increased Aβ production in the amyloidogenic pathway. Excessive Aβ production, aggregation and deposition into plaques, in turn, lead to intracellular Ca2+ dysregulation and induce tau protein hyperphosphorylation and cell death. Tau is highly abundant in neurons and interacts with tubulin to promote microtubule polymerization and stabilization. However, when hyperphosphorylated, the ability of tau to interact with microtubules is impaired. Hyperphosphorylated tau undergoes conformational changes and self-aggregates into oligomers. Elongated oligomers usually form paired helical filaments, which culminate in neurofibrillary tangles formation, inducing neurodegeneration. Moreover, hyperphosphorylated tau can sequester normal tau into filamentous tau aggregates.
In 1991, a group led by John Hardy demonstrated that mutations in the APP gene could cause the development of AD [27]. Subsequently, in 1996, mutations in PS1 and PS2, two genes that code for proteins from the γ-secretase complex, were found to be implicated in hAD [28]. These autosomal dominant mutations result in increased production and longer variants of Aβ associated with aggregation and formation of oligomers. Further agglomeration promotes the formation of insoluble amyloid fibrils, which tend to deposit in plaques. Although both types of aggregates are involved in the pathogenesis of AD [29], soluble oligomers are considered to be more toxic [30, 31].
The gene encoding AβPP was found to be located on chromosome 21 [32, 33], and individuals with trisomy 21 known as Down’s syndrome, seem to be at an increased risk of developing AD, due to an extra copy of the gene and consequent overexpression of AβPP [34]. These factors laid the groundworks for the amyloid cascade hypothesis, proposed by Hardy and Higgins in 1992 [6]. According to this theory, an acute noxious stimulus, such as head trauma, triggers the pathophysiological cascade that induces disturbances in AβPP metabolism by altering production, clearance, and deposition of Aβ. The Aβ protein, in turn, leads to intracellular calcium (Ca2+) dysregulation, inducing neurofibrillary tangle formation and cell death [35]. Consequently, Aβ has been implied as a triggering factor in both forms of the disease: hAD and sAD.
On the other hand, it has already been demonst-rated that Aβ plaques deposition can be present in elderly individuals without cognitive impairment [36–44], and it is still a matter of debate whether or not this reflects a predisposition or a preclinical state of AD [45]. Additionally, elevated Aβ and the presence of plaques in individuals with Down syndrome do not always lead to the development of dementia [46]. Furthermore, the severity of dementia in humans is not proportional to the quantity of Aβ plaques, but it is in positive correlation with the formation of neurofibrillary tangles in the neocortex, which can occur even when no plaques are present [47, 48]. In fact, it has already been demonstrated that the removal of Aβ plaques from the brain does not prevent AD progression and the propagation of tau pathology [49].
Additional information casting doubt on the amy-loid hypothesis is the fact that, although several pre-clinical studies using transgenic mice overexpressing human mutant APP/Aβ have been successful, the failure rate in AD clinical trials is approximately 99.6%[50, 51]. These failures indicate that, because we still do not fully understand the pathophysiology of AD, we are failing in mimicking this disease in experimental models, or, at least, its sporadic form. Besides the fact that genetic mutations have not been sufficient to mimic sAD [52], the non–deterministic genes related to the development of the sporadic form of AD are related to lipid and glucose metabolism and not to Aβ production [53].
In this context, despite promising results in experimental studies with animal models, many anti-amyloid drugs have failed over the years [15, 54]. In the enzyme inhibitors group, γ-secretase inhibitors, such as Semagacestat from Eli Lilly, failed mainly because of the numerous other cellular substrates of γ-secretase, which ended up worsening the cognitive impairment and increasing skin cancers and infections cases [55, 56]. BACE inhibitors, such as Verubecestat (MK-8931), have also been developed and, although they seemed to be safer than γ-secretase inhibitors, these drugs were not able to promote any improvement in cognitive function [57, 58].
Active and passive immunization against Aβ have also been tested [15, 59]. In humans, active immunization against aggregated human Aβ1–42 (AN1792, Elan Pharmaceuticals) that demonstrated desirable effects on plaque burden and cognitive performance in transgenic AD mice [60], resulted in the removal of amyloid plaques in a few patients but provoked aseptic meningoencephalitis in others [61–63]. Due to this adverse effect, the study was interrupted with drug dosing terminated in January 2002. Nevertheless, thorough clinical follow-up and monitoring of the non-affected patients continued under blinded conditions enabling retrospective analyses [61]. In one such retrospective analysis of the cohort, Nicoll and colleagues (2019) executed a 15-year postmortem neuropathological follow-up of individuals who participated in the first trial of Aβ immunotherapy [49]. The authors concluded that, although clear evidence of plaque removal was observed, most patients progressed to severe cases of dementia, possibly due to propagation of tau as extensive distribution of tangles (Braak V/VI) was found in a substantial number of patients [49].
Other Aβ-targeting antibodies (Solanezumab, Cre-nezumab, Gantenerumab, Bapineuzumab) were also tested, but although some positive effects have been observed, the results were not always replicable [64–68]. Defenders of the amyloid cascade hypothesis believe that the failures in these trials occurred because of difficulties in establishing adequate pro-tocols. Problems of inappropriate dosing and administration of the drug in the late irreversible stages of the disease could explain the failure rate of clinical trials [69]. Indeed, the stage of the disease in which the drugs have been administered may have a huge impact on AD progress, since alterations in Aβ production, clearance, and aggregation might start decades before the appearance of the first cognitive symptoms [70].
On the other hand, critics of this hypothesis argue that, besides the fact that AD is a heterogeneous disorder, the relationship between Aβ and AD is at least indirect. In this sense, Aβ might represent an end-stage of the condition rather than a cause. For them, persisting in this theory may result in a loss of opportunity to consider other options, since numerous alternative hypotheses have been proposed all over the years and did not receive equal attention [18, 21].
Nowadays, the two anti-Aβ antibodies Aducanumab and BAN2401 have shown benefits, but are still on trial [71]. Aducanumab was discontinued after a phase III futility analysis. However, after Biogen’s request, the U.S. Food and Drug Administration (FDA) approved a re-dosing study [72, 73]. Aducanumab has given not just support for the amyloid cascade hypothesis but also hope to society, because, if approved, this drug will be the first medication with the ability both to remove the amyloid and slow down the cognitive decline.
ALZHEIMER’S DISEASE AS A MULTIFACTORIAL DISORDER: PROPOSAL OF OTHER HYPOTHESES
Despite all the attention the amyloid hypothesis received in recent years, other important theories have been proposed (Table 1; Fig. 2). One example is the
Main hypotheses on the pathogenesis of sporadic Alzheimer’s disease and associated therapies
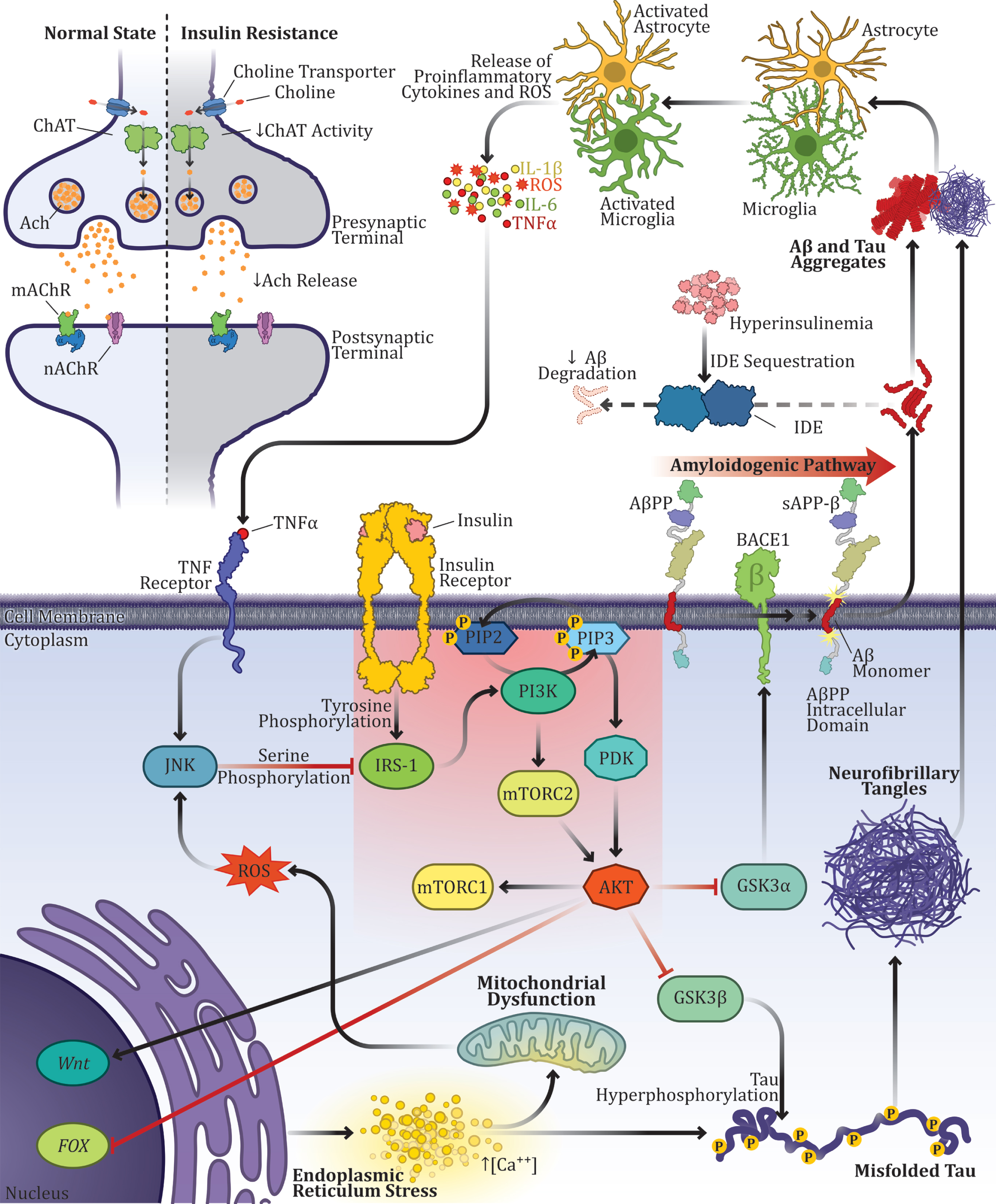
Alzheimer’s disease as a multifactorial disorder: multiple factors converging into a single disease. Interactions between different events proposed in AD pathogenesis over the last decades: Aβ overproduction, tau hyperphosphorylation, neuroinflammation, alterations in the cholinergic system, mitochondrial dysfunction, oxidative stress, calcium imbalance, and insulin resistance as the main link connecting different factors. The PI3K-Akt signaling pathway is particularly important in AD pathogenesis due to its numerous interactions with AD events, but mainly because of its modulation of Aβ production and tau protein hyperphosphorylation through GSK-3 activity. Insulin binds to the alpha subunits of the insulin receptor (IR) by inducing autophosphorylation of its beta-subunit on tyrosine residues. Then, the signal is transduced through the phosphorylation of insulin receptor substrates (IRS), also on tyrosine residues. Phosphorylation of the IRS promotes conformational changes that enable the binding between IRS and another enzyme known as phosphoinositide 3-kinases (PI3K). PI3K activation, in turn, phosphorylates phosphatidylinositol (4,5)-bisphosphate (PIP2) in the cell membrane and results in phosphatidylinositol (3,4,5)-trisphosphate (PIP3) formation. Then, PIP3 enables protein kinase (AKT/PKB) signaling pathway, which regulates the activation of many intracellular proteins in pathways related to cell proliferation and survival, such as the mammalian target of rapamycin (mTOR), forkhead box (FOX) proteins and Glycogen synthase kinase-3 (GSK3). There are two isoforms of GSK-3 in mammals, the isoforms α and β. While GSK-3α regulates Aβ production, GSK-3β modulates tau phosphorylation. Besides GSK-3 activity, alterations of many other proteins in the insulin signaling cascade have also been reported in AD.
Shortly after, Whitehouse and colleagues observed a substantial loss of neurons in the nucleus basalis of Meynert, the source of cortical cholinergic inn-ervation in the brain [83]. Ever since the neurodegeneration of cholinergic projections from the nucleus basalis of Meynert to the neocortex and the hippocampus has been considered as one of the main events in the pathophysiology of AD [84].
The cholinergic hypothesis was a stepping stone in the process of development of most drugs approved to treat AD—the acetylcholinesterase inhibitors (tacrine, rivastigmine, galantamine, donepezil) [85]. These drugs ameliorate cognitive symptoms, but, unfortunately, they are not able to decrease the risk, slow up the onset, or stop the progression of AD. Moreover, individual responses to these drugs may vary. Tacrine, the first drug approved by the FDA and introduced in US marketing in 1993 [86], had quite poor adherence and presented many adverse effects, including hepatotoxicity, which lead to its discontinuation in 2013 [87]. Donepezil, the second drug approved by the FDA, was marketed in 1996 for the treatment of mild and moderate AD. However, in 2010, a higher dose was approved to treat more severe cases. Donepezil is accompanied by side effects, such as nausea, diarrhea, dizziness, and insomnia, and cardiac adverse effects have been reported in some rare cases [88, 89].
Galantamine [90] and rivastigmine [91] were both approved in 2000 for the treatment of AD. However, rivastigmine has also been used to treat Parkinson’s dementia [92]. Recently, Ray and colleagues demonstrated that rivastigmine is able to direct AβPP processing away from the amyloidogenic pathway, by promoting α-secretase activity, and, therefore, it might be explored as a disease-modifying treatment [93].
The
The human tau gene is localized on chromos-ome 17. There are six tau isoforms expressed in the adult brain, as a result of alternative mRNA spli-cing [14]. In AD, all six protein isoforms may be abnormally hyperphosphorylated, resulting in the formation of neurofibrillary tangles and destabilization of the microtubule network [98]. Therefore, in AD-impaired neurons, degenerating neuronal microtubules might be gradually replaced by tangles [99].
Tau is an evolutionarily conserved protein, which can be found in different tissues throughout the body, such as salivary glands, skeletal muscle, and the pancreas [100]. Although tau was first identified as a microtubule-associated protein and most of its known functions are related to the regulation of microtubule dynamic instability, many other roles of tau have been described in different subcellular compartments and beyond the central nervous system (CNS) [101]. It has been demonstrated, for example, that neuronal activity increases tau expression and secretion [102] and that tau also seems to regulate neuronal excitability and might be fundamental for enabling seizure activity [103]. Moreover, DeVos and colleagues (2013) have demonstrated in adult mice that reduction of endogenous tau levels using antisense oligonucleotides is protective against seizures [103].
More recently, Kobayashi and colleagues demon-strated through immunohistochemical and immu-noblotting analyses that glutamatergic stimulation enhances tau protein translation and the accumulation of hyperphosphorylated tau in somatodendrites of mouse hippocampal neurons [104] and that neural excitation may upregulate tau mRNA translation in human neuroblastoma cells (SH-SY5Y), with implications in tau accumulation in the somatodendrite [105].
Other studies have identified important roles of tau in neurogenesis and synaptic integration [106, 107], neuronal maturation [108], neuronal development and synaptogenesis [109–111], normal myelination [112–115], synaptic plasticity [116, 117], and iron export [118, 119]. Tau protein may also influence behavior [101]. Besides functions in learning and memory [120], tau seems to influence locomotor activity [120], anxiety-like behavior [120], the regulation of the circadian rhythm [121, 122], and motor function [123, 124]. More diverse tau physiological influences have also been identified, such as DNA and RNA protection [125], maintenance of chromosomal stability [126], protection against tumorigenesis [127], and modulation of neurovascular coupling [128, 129]. In addition, more recently, studies have demonstrated that tau may regulate the transcription of genes involved in neuronal function [130, 131].
Curiously, studies have demonstrated that both Aβ peptide and tau protein present particular functions after lesions in the nervous system [101]. However, while Aβ increase after an injury seems to be related to improved neurological status [132], tau effects may vary and be dependent on the characteristics of the lesion [112, 133–135]. These facts contribute to the debate of whether increases in pathologic tau and Aβ in AD pathogenesis are a cause or consequence of the disease.
Although recent advances in tau research have ena-bled the identification of numerous tau physiological functions, tau’s roles in pathophysiology are still under debate [118]. Diverse effects, such as morphological changes in neurites [136, 137], synaptic alterations [138], and disruption of cellular trafficking [139] have been reported. Studies have also demonstrated that the soluble form of tau is the most cytotoxic constituent and likely the main cause of synaptic deficits observed in AD, rather than its aggregated form, the neurofibrillary tangles. In fact, Menkes-Caspi and colleagues (2015) analyzed the effects of pathological tau on the intact neoco-rtical network [140]. The authors observed that pathological tau is able to impair the activity of single neocortical pyramidal neurons and the ongoing neocortical network, prior to substantial neurodegeneration, in rTg4510 mice, overexpressing human tau with the dementia-associated P301L mutation [140].
In 2017, Zhou and colleagues demonstrated that pathogenic tau is able to lower neurotransmission by binding to presynaptic vesicles via its N-terminal domain and disrupting synaptic vesicle mobility and release rate [141]. These authors also showed that the inhibition of tau-vesicle binding restores normal presynaptic functions [141]. More recently, Com-mans and colleagues (2021) used combined positron emission tomography (PET) and magnetoencepha-lography in amyloid-positive AD patients to demonstrate that tau pathology is associated with reduced synaptic density and synaptic dysfunction [142].
The fact that the severity of AD cases correlates well with tau pathology in the brain has contribu-ted to the confirmation of the tau hypothesis [143]. Tau pathology is usually classified, according to Braak and Braak [144], affecting primarily the tran-sentorhinal region in stages I and II, the limbic system in stages III and IV, and neocortical fields, mainly temporal and parietal areas, in more advanced stages (stages V and IV) [145]. Based on the network model of neurodegeneration [146], neuroimaging studies have used techniques such as functional magnetic resonance imaging (fMRI) and PET to anticipate the spatial pattern of tau pathology [147–149]. These studies indicate that tau usually accumulates within functionally connected brain networks and that the spreading of tau pathology may be predicted by the extent of baseline tau pathology in connected regions [150, 151]. On the other hand, there is evidence showing that different clinical profiles may result in different tau deposition patterns with deviations from the Braak scheme [152–155]. Additionally, Franzmeier and colleagues recently proposed a connectivity-based prediction model that considers interindividual heterogeneity to predict patient-spe-cific tau deposition and spreading [151].
In this sense, tau has been investigated as a potential target in AD treatment. However, similar to the anti-amyloid therapies, strategies focused on tau have also failed in clinical trials [156, 157]. To better understand Aβ and tau’s functions and their implications in AD therapeutics, check the recent review by Kent and colleagues (2020) [101].
Besides the functions and roles described above, abnormal tau phosphorylation promotes defective axonal transport of mitochondria and other organelles [158]. In fact, mitochondrial dysfunction has been frequently reported in AD [159], and, therefore, it has given support to another theory, the so-called
In 1989, Parker suggested that mitochondrial DNA inheritance could influence AD risk [160, 163]. Other authors claimed that somatic mitochondrial DNA mutations were able to influence the aging process [164–167] and, more specifically, AD development [168–171]. Then, in 2004, Swerdlow and Khan proposed that, since the individual’s baseline mitochondrial function is defined by genetic inheritance, interactions between genetic and environmental factors would define the rhythm at which mitochondrial dysfunction accumulates and, therefore, would determine the AD onset [162]. Subsequently, other studies demonstrated that a maternal family history of AD increases the risk of developing the disease when compared to a paternal family history of this disorder, which indicates that the maternally inherited mitochondria might play an important role in mitochondrial dysfunction and mediate the risk for the development of AD [172, 173].
The mitochondrial cascade hypothesis may be linked to other theories [3] especially the amyloid cascade theory as mitochondrial dysfunction affects both AβPP expression and metabolism [174–177].
Besides Aβ accumulation [178], there is evidence that mitochondrial dysfunction promotes tau hyperphosphorylation [179] inflammation [180], and oxidative stress [181]. Moreover, mitochondrial cha-nges, such as decreased rate of metabolism [180], decreased mitochondrial concentration in the cerebrospinal fluid [181], and mitochondrial morphological alterations [182, 183] have also been described in AD [184]. For this reason, mitochondrial enzymes and energy metabolism have been investigated as potential targets of drugs for the treatment of AD [185].
Mitochondrial dysfunction has also been implicated in the pathogenesis of AD through the gen-eration of reactive oxygen species (ROS) [186]. ROS are oxygen-containing chemicals with reactive properties that play a fundamental role in the maintenance of cellular homeostasis. ROS are con-stantly being produced as by-products of non-enzymatic reactions in the respiratory chains, or enzymatically by macrophages upon recognition of pathogen-associated molecular patterns. Physiologically, enzymes and other compounds usually control and maintain ROS at low levels in a defined homeostatic range, as they cannot be totally eliminated because of their function as specific second messengers in signaling cascades related to cell proliferation and differentiation. However, the accumulation of high levels of ROS, usually due to overproduction or inadequate clearance, disrupts cellular homeostasis by a pathophysiological process known as oxidative stress. Since oxidative stress can damage cells, proteins, lipids, Ca2+ homeostasis, and DNA, it is considered harmful to the human body and a strong contributor to the process of aging [187].
The CNS is particularly susceptible to free radical damage due to its large oxygen demand and high mitochondrial respiration rate. Besides that, the CNS is characterized by a high lipid content and low capacity of enzymatic and non-enzymatic antioxidant systems, which may promote cumulative oxidative damage over time and contribute to AD pathogenesis [188].
Corroborating the
Another metabolic condition that has been imp-licated in AD along with mitochondrial dysfunction and oxidative stress is neuroinflammation [191–193]. This pathophysiological process, characterized mainly by the accumulation of glial cells and upregulation of pro-inflammatory cytokines in the CNS, has been investigated as the crucial event in AD pathogenesis for more than two decades [194–197]. The peripheral immune system is linked to the brain through different mechanisms, including direct passage of cytokines from the blood through leaky regions in the blood-brain barrier (BBB), carrier-mediated transport of cytokines into the brain, and stimulation of cytokine synthesis by microglia activation after detection of a peripheral immune response via vagal afferents [198]. The immune system became particularly relevant to AD research once genome-wide association studies (GWAS) discovered that numerous immune genes are risk factors for sAD [199, 200].
According to the
Microglial activation is usually beneficial, as mic-roglia participates in Aβ clearance and degradation [203], but its persistent activation may result in neurotoxic effects [204]. In fact, there is evidence that hyper-reactive microglia is present even in the early stages of sAD [205]. In this sense, studies have dem-onstrated that constant microglial activation stimula-ted by Aβ, increases Aβ production and diminishes its clearance [206, 207]. However, the inflammatory process induced by other agents is also able to increase Aβ production, via β-secretase cleavage [204]. Furthermore, hyperphosphorylated tau leads to the activation of microglial cells and the synthesis and production of pro-inflammatory cytokines [208]. Pathologically changed astrocytes have also been described in AD [209] and, although astrogliosis has been observed in regions without Aβ pathology, in AD brain tissue, astrogliosis is correlated with the degree of cognitive impairment [210].
In summary, the response to inflammatory stress induces hyperphosphorylation of tau and increases Aβ synthesis. In addition, both Aβ accumulation and tau hyperphosphorylation dysregulate the immune system and activate a constant and persistent inflammatory process, leading to a deleterious micro-glial and astrocytic reactivity, and, consequently, trigger a vicious circle of neurotoxic pro-inflammatory response [211].
Although the effects of exposure to anti-inflammatory drugs in AD, especially nonsteroidal anti-inflammatory drugs (NSAIDs), are still controversial, some studies have observed benefits in the use of this type of medication before the onset of AD. Furthermore, it has been proposed that the activation of the innate immune system might act as a disease-promoting factor in which the senescent microglia is the initial trigger of AD pathogenesis [212]. In this case, AD should be considered an immune senescent disease rather than a neuroinflammatory disorder, as stated by the
According to the innate immunity hypothesis, immunoactivity plays a fundamental role in the pathology of AD and microglia have taken center stage in the investigation of innate immune responses in neurodegenerative diseases [216]. In fact, recent evidence identified particular types of microglia, which have been associated with different features or stages of the pathophysiology of neurodegenerative diseases [217]. Moreover, studies have described diverse microglial activity associated with different variables, such as sex, age, and genetic background [218]. Also, interactions between microglia and the adaptive immune system present important roles in the pathogenesis of AD [219]. Recently, Gate and colleagues (2020) identified adaptive immune responses in the blood and cerebrospinal fluid of patients with AD [220].
Studies also indicate that microglia is a dynamic structure responsible for the homeostatic maintenance of the brain parenchyma, besides its functions related to neuronal connectivity, modulation of BBB permeability, neurogenesis, myelination, vasculogenesis, and synapse pruning [221]. Indeed, the expression of pattern recognition receptors (PRRs) has already been identified on the surface of microglia [222]. These receptors are able to detect and respond to a diverse type of molecular threats, including neurodegeneration [223]. When associated with disease, microglia is in an activated state and upregulate PRR expression [224, 225].
Researchers have worked on the identification of critical genes involved in AD pathology. From this perspective, recently, GWAS have reported more than 30 genetic loci associated with AD [226]. Many variants identified in these studies are related to immune response and link mutations in PRRs with increased risk for developing AD risk [227], including the two microglial phagocytic receptors CD33 and TREM2, which have become important targets in the elaboration of therapeutic strategies for AD treatment [227].
The Aβ itself can be recognized by PRRs in the brain. However, the chronic innate immune activation caused by Aβ recognition leads to chronic inflammation and to further propagation of AD pathology [228]. In addition, hyperphosphorylated tau can promote aberrant immune activation, which results in further production of pathogenic tau and its spreading [224]. Curiously, tau phosphorylation can also be induced by the herpes simplex virus (HSV-1), a very popular virus that infects two-thirds of the global population and seems to be related to AD pathogenesis [224, 229]. Moreover, the overproduction of Aβ peptide may act as a protective agent against latent herpes viruses and other infections, which might explain why brain infections contribute to the progression of AD pathology [230]. In this sense, antiviral drugs have been investigated as potential therapeutic strategies to decrease both hyperphosphorylated tau and Aβ overproduction [231, 232].
Recently, the interferon-induced transmembrane protein 3 (IFITM3), an innate immune protein known to restrict viral infections, was found to modulate γ-secretase activity, contributing to Aβ production [233]. This protein seems to increase with aging and in hAD [233]. Also corroborating the innate immunity hypothesis, there are the facts that Aβ has been hypothesized as an antimicrobial peptide [230], and that partial depletion of microglia ameliorates memory and diminishes neuronal damage [234, 235].
Another pathophysiological event that has been proposed as both the cause and the consequence of metabolic, oxidative, and proteotoxic stress in AD is the dysregulation of Ca2+ homeostasis [236]. In this sense,
The Ca2+ ion works as an intracellular messenger in many signal-transducing pathways and as a regulator in diverse physiological functions. Because of the importance of Ca2+ homeostasis, a number of cellular regulatory mechanisms, such as ion channels, buffers, and ATP-dependent ion pumps, are working to keep the level of Ca2+ at low nanomolar concentrations under resting conditions [241]. Homeostasis is particularly important as action potential-regulated influx and efflux of calcium is indispensable for proper neuronal signaling. Consequently, regulation of a complex network of calcium channels and transporters, as well as conserved activity of endoplasmic reticulum (ER) and mitochondria, two main organelles responsible for intracellular buffering, is a prerequisite for the maintenance of the structure and function of the CNS. Failures of this system result in the inability to maintain calcium homeostasis and lead to neurodegeneration [242]. Although astrocytes, the main homeostatic regulatory cells in the CNS, cannot generate action potentials, they sense fluctuations in intracellular concentration of ions, especially Ca2+, in order to respond to neuronal activity [243].
Corroborating the calcium hypothesis, several studies have shown a bidirectional relationship between Ca2+ and the Aβ peptide in the pathogenesis of AD [244]. Tau pathology might also be linked to disruption in Ca2+ signaling once microtubule dysfunction promoted by hyperphosphorylation of tau impairs dynamics and axonal transport of organelles and vesicles, including mitochondria and ER [158]. When these components are affected, they end up directly influencing the calcium signaling pathway, especially in neurons where the communication networks between ER, mitochondria, and plasma membrane are fundamental for the regulation of temporal and spatial aspects of Ca2+ signaling [245].
Recently, Jadiya and colleagues (2019) demonstrated that impaired mitochondrial calcium efflux stimulates disease progression in AD models, by accelerating memory alterations, Aβ pathology, tau hyperphosphorylation, and development of histopathological changes [246]. In fact, some authors believe that, since mitochondrial Ca2+ overload may appear before the typical pathological features of AD, it should be considered a priority among therapeutic targets for AD [247]. Finally, all proposed hypotheses should not be considered individually, but as pieces of the pathophysiological puzzle contributing to the understanding of the etiopathogenesis of AD. Some hypotheses, such as the mitochondrial hypothesis of the disease, and oxidative stress hypothesis as well as the calcium hypotheses are more closely related as mitochondrial dysfunction is often considered as a key contributor to cellular ROS burden, and bidirectional interaction between the ER calcium and mitochondria make it difficult distinguish their cause-effect relationship. Nevertheless, a number of less obvious interconnections exist between all factors proposed as the main drivers of the disease, and current understanding of molecular mechanisms suggests all have the potential to trigger a pathogenic cascade of AD. Recently, accumulated evidence on the importance of metabolic factors in the context of AD provided additional information that, when considered in the context of other hypotheses, might enable a deeper understanding of the pathogenesis of the disease and reveal some links that might have been overlooked.
Due to numerous metabolic alterations described in AD, it was proposed that this disorder contains a significant metabolic component [248]. One of the main features of AD hypothesized as a metabolic disorder is the consistent findings suggestive of impaired insulin signaling in AD brains. In fact, the term “type 3 diabetes” has been proposed in order to englobe the cellular and molecular mechanisms by which insulin plays an important role in the pathology of AD. Interestingly, alterations in the regulation of the insulin signaling pathway, just like Aβ peptide accumulation, seem to be related to many aspects of AD discussed in this review.
ALZHEIMER’S DISEASE HYPOTHESIS OF A “TYPE 3” DIABETES
Although cognitive dysfunction in diabetes mellitus (DM) has been frequently reported over the last decades [249–256], the first study showing worse performance in attention and memory tests in diabetic patients was made in Boston by Miles and Root, almost a century ago [257]. At that time, these findings were not well understood; however, in 1983, Bucht and colleagues found important results suggestive of decreased insulin sensitivity in AD patients [258]. These data implied for the first time that the hormone insulin could somehow be involved in the etiopathogenesis of AD. In 1998, Frölich and colleagues described alterations in the neuronal insulin signal transduction pathway in AD brains [259], which culminated in the proposal that AD is a brain type of non-insulin-dependent DM, made by Hoyer [260] in the same volume. After extensive work, in 2005, a group led by Suzanne de la Monte at the Brown Medical School proposed the term “type 3 diabetes” to refer to AD as a neuroendocrine disorder, similar, but also distinct, from DM types 1 (T1DM) and 2 (T2DM) [261].
More recently, studies have demonstrated that DM is a risk factor for developing dementia [262–265]. According to Chatterjee and colleagues, this risk is approximately 60%greater for diabetic patients compared with those without diabetes [266]. The most prominent factors that seem to be shared by T2DM and AD as common risks could be found often combined, from aging and age-related alterations like metabolic, hormonal, and vascular pathology to environmental factors. Additionally, although the link between the two diseases is still not fully understood, associations have been reported also at the genetic level [267]. Caberlotto and colleagues have recently analyzed transcriptomic data of post-mortem AD and T2DM human brains and identified a central role for the autophagy pathway in both diseases. In addition, the authors used genetically modified animal AD models to confirm the role of autophagy-related genes in AD pathogenesis. These results suggest that autophagy dysregulation might be a common pathophysiological mechanism underlying AD and T2DM [268].
Considering the metabolic factors, particularly glucose metabolism in the brain decreased glucose utilization and altered energy metabolism have been reported since the early stages of AD [266], especially in regions associated with the process of learning and memory [269, 270].
Evidence has gathered supporting the link between AD and T2DM based on the presence of AD biomarkers in the brain tissue of diabetic patients without clinical signs of dementia [271–274] and alterations in insulin signaling pathways found in the brain of both AD patients postmortem and AD animal models [259, 275–277]. The insulin signal transduction pathway is particularly important in the brain because of its functions related to neuronal survival, central regulation of body metabolism, and modulation of memory and other cognitive and emotional processes [278].
Since the connection between AD and T2DM seems to be bidirectional, some studies have focused on the understanding of this link between central and peripheral insulin functions. One of the main mechanisms that might, at least partially, explain the central-peripheral cross-talk in insulin signaling is the regulation of hepatic glucose production by hypothalamic insulin, emphasizing that central insulin interferes with peripheral organs functions [279]. In this sense, individuals with burn injuries present increased susceptibility to DM [280], which, in these cases, is associated with CNS complications, including brain insulin resistance [281]. In turn, dysfunctional central insulin signaling compromises hepatic glucose metabolism, initiating a pathologic cycle [281, 282]. On the other way around, male ischemic stroke rats present hyperglycemia and impaired hepatic insulin signaling [279].
Central insulin signaling seems to present other important systemic functions, mostly related to food intake and body weight [283, 284]. When insulin is administered to the CNS, it acts as an anorexigenic factor, reducing the ingestion of food [285]. Moreover, there is evidence showing that the dysfunction of hypothalamic insulin receptors promotes hyperphagia, increased body fat, and peripheral insulin resistance due to overfeeding [286]. In addition, normal brain insulin receptor function prevents diabetes, and, therefore, central insulin might work as a negative feedback signal in the modulation of reward-related food intake [284].
More specifically, in AD pathogenesis, Aβ olig-omers are able to compromise the hypothalamus and impair peripheral glucose homeostasis [287]. The hypothalamus is also one of the main brain regions affected by tau pathology [288], and, thus, pathogenic tau also impairs the central control of peripheral glucose metabolism in patients with AD [289]. Moreover, both Aβ peptide and tau protein can be found in systemic organs such as the pancreas, liver, and skeletal muscle and, therefore, might present peripheral functions [290]. In fact, the presence of these two AD hallmarks in pancreatic β-cells has already been reported [291, 292], which corroborates the connection between AD and T2DM [290].
The insulin signal transduction pathway in the AD brain
The presence of insulin in the CNS, as well as its origin and functions, have been widely debated over the decades [284, 293–296] mainly because glucose uptake by neurons is not insulin-dependent. After extensive research in this field, it has become evident that both insulin and insulin receptors (IR) are distributed in a region-specific manner in the brain, with the highest density in the hippocampus, cerebral cortex, olfactory bulb, and hypothalamus [297–301]. Moreover, it is now known that glucose uptake in the brain can be influenced by insulin in conditions of high energy demand induced by increased neuronal activity. Increased glucose uptake upon insulin binding is mediated by the stimulation of the translocation of glucose transporters type 3 (GLUT3) and type 4 (GLUT4) to the plasma membrane in the conditions of increased energy demand, such as hippocampal-dependent tasks [302, 303]. Studies have also demonstrated that systemic insulin may be actively transported through the BBB to the CNS [297, 305], but a small portion of insulin can also be locally produced by neurons [298, 306–309]. There is a higher density of IR in neurons as compared to glia, but astrocytes express both IR isoforms (IR-A and IR-B; IR-A, in contrast to IR-B, shows no negative cooperativity, indicating different functional regulation upon insulin binding), while neurons express exclusively the IR-A isoform [310].
The IRs are composed of dimers of alpha (extracellular) and beta (intracellular) subunits joined by disulfide bonds. Insulin, or insulin-like growth factors (IGF), bind to the alpha subunits of IR inducing autophosphorylation of its beta-subunit on tyrosine residues, thereby promoting the transduction of many signaling pathways [311], especially related to cell proliferation and metabolism. Then, the signal is transduced through the phosphorylation of insulin receptor substrates (IRS), which are usually composed of six members (IRS1-6), also on tyrosine residues [312]. The IRS-1 is one of the most well described in humans and it is involved in the modulation of essential functions in the cerebral cortex [278]. Phosphorylation of the IRS promotes conformational changes that enable the binding between IRS and another enzyme, phosphoinositide 3-kinases (PI3K). PI3K activation, in turn, phosphorylates phosphatidylinositol (4,5)-bisphosphate (PIP2) at the cell membrane and results in the formation of phosphatidylinositol (3,4,5)-trisphosphate (PIP3). Then, PIP3 enables protein kinase (AKT/PKB) signaling pathway [313], which regulates the activation of many intracellular proteins in pathways related to cell proliferation and survival, such as the mammalian target of rapamycin (mTOR), forkhead box (FOX) proteins and glycogen synthase kinase-3 (GSK3), besides the facilitation of the translocation of GLUT4 to the cell membrane and glucose uptake into the cell by the metabolic pathway [314].
GSK-3 activity is extremely relevant to AD pathogenesis and has emerged as an important target for AD drug development [315–317]. There are two isoforms of GSK-3 in mammals, the isoforms α and β, encoded by two different genes [318]. While GSK-3α regulates Aβ production [319], GSK-3β modulates the phosphorylation of tau [320]. In fact, GSK-3β is the main kinase of tau protein, and it is able to phosphorylate at least 12 Ser/Thr of its pro-sites [321–323]. Moreover, GSK3-β activity may also be involved in Aβ production through the modulation of AβPP cleavage, as PS1 has been identified as a GSK-3β substrate and GSK3-β over-activation or overexpression stimulates the cleavage of AβPP by BACE1 [318]. The activities of GSK-3 α/β are inhibited through phosphorylation of GSK-3 by AKT at serines 21 and 9, respectively [324].
GSK-3 is expressed in all tissues, but it is particularly abundant in the brain, especially in the hippocampus [316]. It can inhibit insulin signaling through serine phosphorylation of the IRS 1 and 2 [325–327]. It is also the main suppressor of the Wnt signaling pathway, one of the most important developmental pathways that regulate fundamental aspects of cell fate determination, such as cell migration and neural patterning [328], which means that GSK-3 is able to influence cell differentiation and reproduction [316, 330]. GSK-3 is also involved in the regulation of learning and memory functions, and processes of neurodegeneration, neurogenesis, inflammation, and synaptic plasticity, therefore alterations in GSK-3 activity found in AD could provide a molecular background for some of the neuropathological hallmarks of the disease [316, 331]. Since GSK-3 phosphorylation at serine inhibits its activity, it would be expected to observe decreased phosphorylated GSK-3 α/β levels in AD brains. Curiously, AD studies have been contradictory and, while some authors identified increased levels of GSK-3 α/β in its active form [332], others observed increased GSK-3 phosphorylation [333, 334]. Elevated expression and over-activity of GSK-3 have also been reported in T2DM [335] providing further support to dysfunctional IR signaling cascade as an underlying pathology linking AD and T2DM. Furthermore, increased expression and activation of GSK-3 have also been observed in other diseases, such as bipolar disorder [336–338], Parkinson’s disease [339–341], and Huntington’s disease [342, 343].
Therefore, inhibition of GSK-3 has been investigated as a candidate pharmacological target for the treatment of many diseases, especially AD [317]. One of the most well-studied drugs in this field is lithium, a non-selective GSK-3 inhibitor. However, the obtained results are contradictory and inconclusive [344–346]. Similar results were found with a small molecule non-ATP-competitive and irreversible GSK-3 inhibitor tideglusib (NP12) [347, 348]. More recently, a meta-analysis performed by Matsunagaa, Fujishirob, and Takechia suggested that GSK-3 inhibitors might not be effective in AD treatment. However, the protocol established in the analyzed studies might have not been adequate, and non-selective inhibition of GSK-3 in a number of different cell types with consequent modulation of important signaling pathways might account for both ineffectiveness and side-effects of such a treatment. Hence, further studies are needed to obtain final conclusions about GSK-3 inhibitors [317].
Besides changes in GSK-3 activity, other upstream alterations in the insulin signaling cascade have also been reported in AD pathogenesis. For example, decreased levels of insulin, IGF-1, and their receptors have already been identified in AD brains [259, 349]. Lower levels of IRS-1 [350] and increased IRS-1 serine phosphorylation, which disable normal transmission of the signal through the IR-IRS signaling pathway and may result in insulin resistance [351, 352], have been described in AD [353–355], even a decade before the clinical onset of AD [356]. Serine IRS-1 phosphorylation might be associated with tau dysfunction in AD. In tau knockout mice, serine phosphorylation of IRS-1 is increased, and insulin-induced hippocampal tyrosine phosphorylation of IRS-1 is decreased [357]. Decreased levels of PI3K and reduced phosphorylation of Akt have also been reported in AD [304, 350]. Dysfunctional PI3K/Akt pathway has important downstream signaling consequences in AD, since it has been recognized as a molecular regulator of GSK-3, mTOR, glucose transporter trafficking, and autophagy, all recognized to be altered in the process of neurodegeneration.
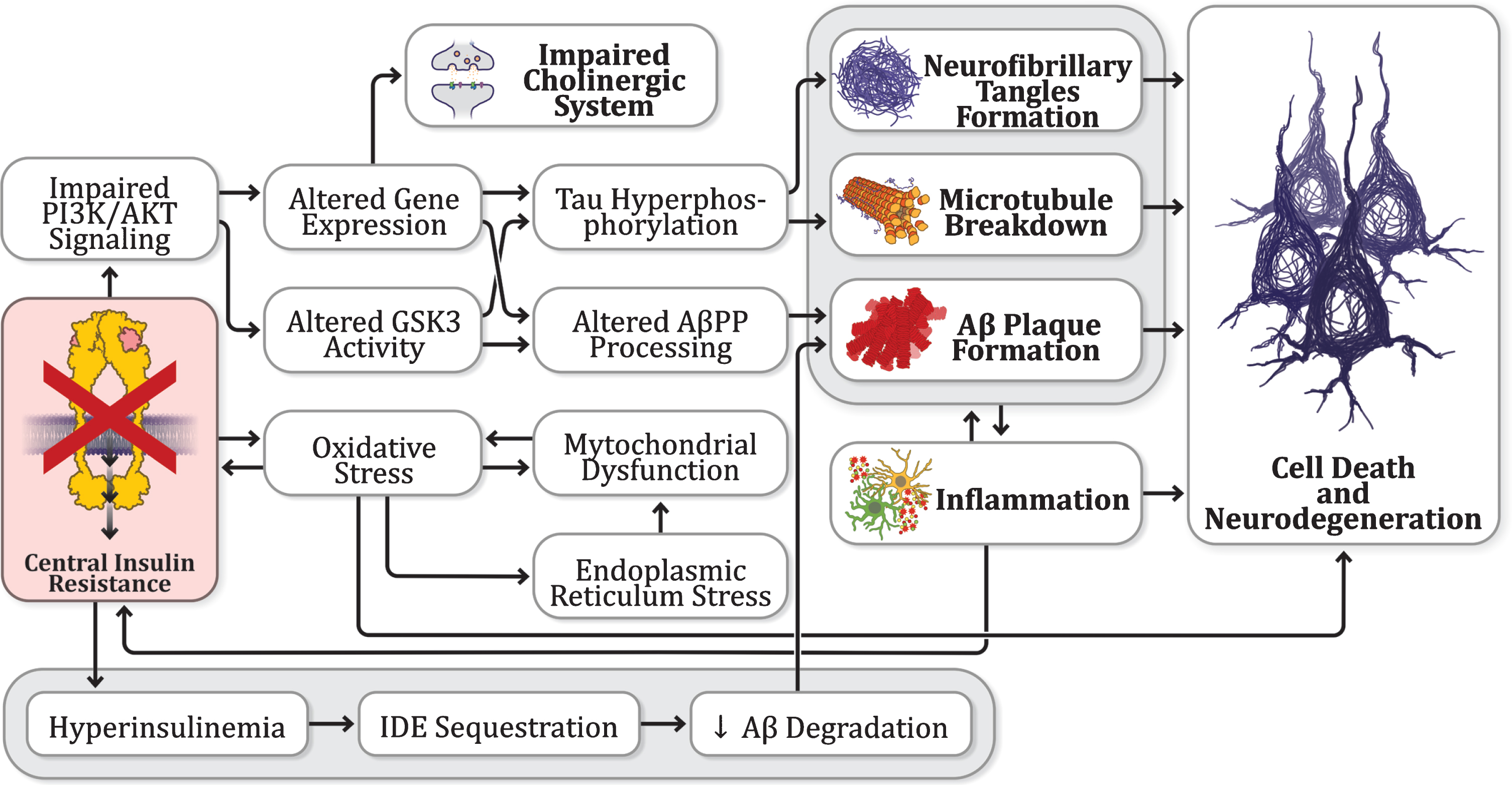
An overview of insulin connections to other AD hypotheses. There are consistent findings pointing toward dysfunctional insulin signaling in the brain as a common link between other proposed hypotheses.
IR signaling pathway also leads to the activation of the mitogen-activated protein kinase (MAPK) pathway, which regulates cell differentiation, proliferation, survival, death, and metabolic activity. The expression of MAPK is increased in AD brain tissue and it is found to also be involved in the process of Aβ plaques formation, tau hyperphosphorylation, neuroinflammation, oxidative stress, and synaptic plasticity. Furthermore, MAPK seems to be involved in the regulation of cognitive function [276, 358]. Consequently, MAPK has also been proposed as a possible therapeutic target in AD [359] and some candidate molecules have been tested in this context. For example, brain-permeable orally bioavailable small molecule isoform-selective inhibitor of p38α MAPK MW181 was reported to improve working memory, reduce tau phosphorylation and inflammation in tau transgenic mouse model of tauopathy [360].
Since many studies have demonstrated that impairment in both peripheral and central metabolism is related to cognitive decline and dementia [361–363] insulin levels and sensitivity became therapeutic targets in AD treatment [364]. In line with that, both insulin secretagogues like glucagon-like peptide 1 (GLP-1) receptor agonists insulin sensitizers, such as thioglitazones and biguanides, have been shown to improve cognitive function in both AD patients and animal models [365–367]. Metformin, a biguanide that decreases gluconeogenesis in the liver and ameliorates insulin resistance, has been associated with a reduced risk of developing AD in older people with DM [368]. Although some results have been contradictory, studies have shown that this drug is able to interfere with the formation of Aβ plaques and neurofibrillary tangles and improve insulin signaling in the brain [276, 369–371].
Many antidiabetic drugs have been investigated in AD treatment. These drugs may present numerous positive effects, such as improvement of insulin res-istance and cell metabolism, which might result in amelioration of cognitive impairment [358]. Rece-ntly, in a literature review, Meng and colleagues summarized the available clinical and experimental studies reporting the effects and the potential mechanism of action for 14 antidiabetic drugs that have been considered for AD treatment [372]. Among them, insulin administration has led to, besides other benefits, significant improvement in cognitive function [373].
The link between diabetes and insulin signaling to other AD hypotheses
Evidence has gathered indicating dysfunctional insulin signaling in the brain of AD patients and animal AD models, which supports the hypothesis of AD as a “type 3 diabetes”; however, alterations in the insulin signaling cascade seem to be shared by other hypotheses of AD as well (Fig. 3). Regarding
Furthermore, the insulin-degrading enzyme (IDE) is one of the main factors responsible for Aβ degradation [382]. However, given its higher affinity, IDE binds preferentially to insulin, when compared to other substrates, including Aβ. Thus, insulin or pathological conditions that affect its levels, such as DM, can indirectly modulate circulating Aβ levels. In this sense, conditions that decrease insulin sensitivity and increase insulin levels may result in greater accumulation of Aβ and, consequently, gradual deposition in senile plaques [383–385]. It has already been demonstrated that IDE’s activity in the brain decreases during the process of aging and is significantly reduced in the early stages of AD [386, 387]. Conversely, insulin can also increase IDE protein levels via the PI3K pathway, and, therefore, deficient insulin signaling is correlated with decreased IDE in AD brains [388].
On the other hand, the Aβ peptide can form oligomers that bind to IRs, acting under certain conditions as insulin antagonists and interfering with the regulation of the insulin signaling cascade through the reduction of Akt activation and the increase of GSK-3 α/β activity [389]. Aβ can also induce serine phosphorylation of IRS-1, which inhibits insulin signaling and initiates a positive feedback loop, leading to an increase in AβPP processing and Aβ production [376]. There is also evidence that Aβ is able to promote the loss and redistribution of neuronal surface IRs, which might be related to the first clinical symptoms of AD [390, 391].
On the other hand, it has already been shown that intranasal insulin administration reduces tau hyp-erphosphorylation in the brain of T2DM rat models induced by a high protein, high glucose, and high-fat diet followed by intraperitoneal injection of streptozotocin (STZ) [395]. STZ is a glucosamine-nitrosourea substance with alkylating properties that destroys pancreatic β-cells and leads to decreased insulin secretion and hyperglycemia, and, consequently, induces diabetes in experimental animals. Curiously, central administration of this com-pound produces memory deficits, impaired insulin signaling, neuroinflammation, neurodegeneration, and other molecular and pathological features that mimic those in patients with sporadic AD, and has, therefore, been considered a model of type 3 diabetes [396–399]. Chronic treatment with intranasal insulin decreases tau hyperphosphorylation, improves cognitive function, ameliorates microglial activation, and increases neurogenesis in this model [334].
Intracerebroventricular injection of STZ also generates oxidative stress and mitochondrial dysfunction, which may contribute to cognitive impairment [400, 401]. Several mechanisms have been proposed to explain STZ-induced oxidative stress, and dysregulation of insulin/IGF signaling, an important regulator of redox homeostasis, provides one possible explanation [402]. Insulin resistance in T2DM is accompanied by hyperglycemia which generates the accumulation of advanced glycation end (AGE) products that promote ROS generation, and increased levels of AGE, as well as overexpression of its receptor (RAGE), have also been observed in AD brains [403]. Besides oxidative stress, an imbalance between pro-oxidants and antioxidants and lipid peroxidation leading to cell damage have been identified in both AD and DM [404]. Moreover, insulin antagonizes the deleterious effects of oxidative stress in the brain. It presents neuroprotective effects against oxidative stress by restoring antioxidants and energy metabolism and modifying anti-apoptotic-associated protein synthesis through the stimulation of the PI3K/Akt pathway and inhibition of GSK-3β [405, 406]. Moreover, it has already been shown that insulin sensitizers are able to protect against mitochondrial dysfunction caused by APOE4, a genetic risk factor for AD [407].
With regards to
Chronic
In addition, it has been shown that TNFα, as well as other inflammatory cytokines and stress-sensitive kinases, can promote insulin resistance [415–417] by stimulating the serine phosphorylation of IRS via the activation of c-Jun N-terminal kinase (JNK) [275, 412] and that the intracerebroventricular administration of an anti-TNF agent is able to ameliorate insulin signaling in rats [418]. On the other hand, the anti-inflammatory cytokine IL-4 increases insulin sensitivity [416, 419]. Najem and colleagues proposed that neuroinflammation, insulin resistance, and Aβ accumulation may act together to drive the pathogenesis of AD [416]. Their proposal was based on findings that insulin signaling modulates Aβ-induced inflammatory response [418] and soluble oligomers of Aβ promote IRS-1 inhibition via TNFα activation [353]. Therefore, they suggested that AD research should focus on understanding the possible link between these three events [416].
Regarding
More recently, it has been shown that acute insulin treatment is able to decrease calcium transients, which may affect intracellular calcium channel functions. These results suggest that insulin-mediated changes in calcium homeostasis may contribute to the positive effects of insulin in the brain [423]. On the other hand, in the CNS, increased levels of intracellular calcium are related to dysfunctional glucose metabolism [424, 425]. Moreover, according to De Felice, aberrant calcium influx may be related to insulin resistance in AD since the neuronal response to insulin can be inhibited by the calcium chelator BAPTA-AM [426].
A possible link between AD and T2DM could also be discussed at the level of cerebrovascular pathology found in diabetic and many AD patients indicating an additive effect on dementia [427, 428]. Bearing in mind the heterogeneous etiopathogenesis of vascular cognitive impairments [429], it could not be excluded that the underlying mechanisms, besides factors like hyperglycemia, maybe also be linked to insulin regulation of vascular function [364]. At normal concentrations, insulin acts as a vasodilator (binding to its receptors on endothelial cells stimulates the release of nitric oxide via the PI3K pathway) while at high concentrations it acts as a vasoconstrictor (stimulation of endothelin-1 production via the MAPK pathway) [430]. In a T2DM condition of chronic hyperinsulinemia due to insulin resistance, the vasoconstrictory role of insulin prevails resulting in reduced cerebral perfusion which may be detected years before the cognitive impairment [364].
Preventive and predisposing factors linked to insulin dysfunction
Mounting evidence suggests that lifestyle choices influence the onset and progression of sAD [431]. Moreover, lifestyle factors are the main contributor to developing peripheral insulin resistance [432, 433]. Although the connection between the style of living and central insulin resistance is not completely enlightened yet, there are some facts that might corroborate this relationship.
In this context, low to moderate consumption of alcohol, a legal drug widely used [434], is related to a lower incidence of cardiovascular diseases [435–437] and T2DM [438–440]. Studies have reported that these protective effects might be due to an increase in systemic insulin sensitivity [441, 442]. Interestingly, low or moderate concentrations of ethanol also seem to be related to a reduction in the prevalence of AD, while higher doses seem to worsen disease prognostic [443–445]. However, this relationship is still under debate [446] and there is no data showing that central insulin signaling may be related to these effects.
Similar to alcohol intake, coffee consumption is associated with a substantially lower risk of both T2DM [447, 448] and AD [449, 450]. Recently, Dong and colleagues (2020) performed a study from the National Health and Nutrition Examination Survey (NHANES), which was conducted by the Centers for Disease Control and Prevention (CDC), and showed that coffee, caffeinated coffee, and caffeine intake from coffee is associated with better cognitive performance in older adults [451]. Studies have also demonstrated that coffee intake is positively related to pancreatic beta-cell function [451] and decreased peripheral insulin resistance [452, 453]. There are recent studies reporting positive effects of green coffee in brain insulin resistance [454, 455].
Physical exercise, one of the most popular potential non-pharmacological therapeutic approaches of AD [456], is also a modulator of peripheral insulin resistance [457], especially in T2DM [458]. Rodent studies have reported improved brain insulin sensitivity after physical exercise [459, 460]; however, there is no clinical data showing the effects of exercise on central insulin sensitivity [364]. On the other hand, sedentary behavior is associated with insulin resistance [432] and a significantly increased risk of dementia [461].
Obesity, one of the major risk factors for T2DM [462], has been shown to also increase the risk for AD [463–465]. Besides peripheral insulin resistance, obesity leads to central insulin dysfunction through several mechanisms, as reviewed by Sripetchwandee and colleagues (2018) [466]. Diets high in simple carbohydrates and saturated fat are also related to increased risk of developing AD [362, 467], and insulin resistance might be one of the main causes of this link [364].
Other factors, such as chronic stress [468] and sleep disturbances [469] are also related to an increased risk for developing AD. Recently, Woo and colleagues (2018) showed that chronic restraint stress promotes hippocampal memory impairment due to dysfunctional insulin signaling in mice [470]. In addition, the authors found that intranasal insulin treatment was able to restore insulin signaling and ameliorate hippocampal deficits after psychological stress [470]. Evidence has also supported a role for sleep disturbances, another kind of psychological stress, as an independent risk factor for the development and exacerbation of insulin resistance [471]. It is already been known that sleep presents modulatory effects on glucose metabolism, insulin sensitivity, and the neuroendocrine regulation of appetite [472]. Moreover, chronic insufficient sleep may disturb the circadian rhythm, impair insulin secretion and increase the susceptibility to T2DM and obesity [473].
CONCLUSIONS
AD is a severe health public problem, with no cure or interventions to delay its progression. Although numerous researchers have focused on the understanding of this disease over the last decades, AD is still a not well-understood disorder, with complex pathogenesis. A lack of perception of AD as a heterogeneous pathological condition with a multifactorial etiology might be contributing to the constant failures in AD clinical trials. After gathering all the main features and hypotheses proposed in the context of the development of AD, we can infer that a single theory that could explain all its enigmas has not yet been proposed. We believe that AD is rather a multifactorial condition that can be influenced by numerous factors and different processes and that an adequate approach to AD should englobe the multiple aspects of this disorder.
Although most studies have focused on the amyloid cascade theory of AD, the metabolic hypothesis of AD, suggesting that AD is a metabolic disorder gained a lot of attention and provided consistent basic and clinical evidence in recent years. Based on this, some authors even proposed that AD should be considered a “type 3 diabetes” to further emphasize the importance of metabolic changes in the context of etiopathogenesis of the disease. The most interesting feature of this hypothesis is the fact that it provides an integrative framework indispensable for understanding individual pathomechanisms proposed by other hypotheses and often considered individually. By adding an additional contextual layer, and providing missing links, this integrative hypothesis of AD taking into account dysfunctional insulin signaling cascade as a missing link between many of the other proposed hypotheses, may help us deepen our understanding of the AD pathophysiology, gain different perspectives, and design better prevention and treatment strategies.
Footnotes
ACKNOWLEDGMENTS
This research was supported by the São Paulo Res-earch Foundation –FAPESP [grant numbers 17/21155-3, 19/02787-4, 19/05957-8 and 19/00849-2], the National Institutes for Science and Technology (INCT) - “Translational Medicine”, FAPESP [grant numbers 14/50891-1] and National Council for Scientific and Technological Development (CNPq) [grant numbers 465458/2014-9], Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001, CNPq [grant numbers 305883/2014-3] (N.G.C.) and Coordenadoria de Aperfeiçoamento de Pessoal de Nível Superior –PROEX-CAPES. NGC holds a CNPq Research Fellowship. This work was also supported by the Croatian Science Foundation [grant numbers IP-2018-01-8938] and the Scientific Center of Excellence for Basic, Clinical and Translati-onal Neuroscience (Project “Experimental and cli-nical research of hypoxic-ischemic damage in perinatal and adult brain”; [grant numbers GA KK01.1.1.01.0007] funded by the European Union through the European Regional Development Fund).