
Abstract
Background:
Dominant missense mutations in the amyloid-β protein precursor (AβPP) cause early-onset familial Alzheimer’s disease (FAD) and are associated with changes in the production or properties of the amyloid-β peptide (Aβ), particularly of the 42-residue variant (Aβ42) that deposits in the Alzheimer’s disease (AD) brain. Recent findings, however, show that FAD mutations in AβPP also lead to increased production of longer Aβ variants of 45–49 residues in length.
Objective:
We aimed to test neurotoxicity of Aβ42 vis-á-vis longer variants, focusing specifically on mitochondrial function, as dysfunctional mitochondria are implicated in the pathogenesis of AD.
Methods:
We generated SH-SY5Y human neuroblastoma cells stably expressing AβPP mutations that lead to increased production of long Aβ peptides with or without Aβ42. These AβPP-expressing cells were tested for oxygen consumption rates (OCR) under different conditions designed to interrogate mitochondrial function. These cell lines were also examined for expression of genes important for mitochondrial or neuronal structure and function.
Results:
The mutant AβPP-expressing cells showed decreased basal OCRs as well as decreased OCRs associated with mitochondrial ATP production, even more so in the absence of Aβ42 production. Moreover, mutant AβPP-expressing cells producing longer forms of Aβ displayed altered expression of certain mitochondrial- and neuronal-associated genes, whether or not Aβ42 was produced.
Conclusion:
These findings suggest that mutant AβPP can cause mitochondrial dysfunction that is associated with long Aβ but not with Aβ42.
INTRODUCTION
Early-onset familial Alzheimer’s disease (FAD) is a rare form of dementia caused by dominant missense mutations in the amyloid-β protein precursor (AβPP) and presenilin-1 and –2 [1]. Other than the completely hereditary nature and mid-life onset, FAD resembles the more common sporadic late-onset Alzheimer’s disease (SAD) in pathology, presentation, and progression. Cerebral plaques composed of the 42-residue amyloid-β peptide (Aβ42) [2] and neurofibrillary tangles composed of the microtubule-associated tau protein [3] are characteristic of both FAD and SAD. The discovery of FAD mutations in AβPP [4] led to formulation of the amyloid hypothesis of AD pathogenesis [5], as these mutations were found in and around the small Aβ region and altered the production or properties of Aβ to render the peptide, particularly the Aβ42 variant, more prone to aggregation [6]. Subsequent discovery of FAD mutations in the presenilins [7, 8] provided further evidence for Aβ42 as the trigger of a cascade of events leading to neurodegeneration. Presenilin is the catalytic component of γ-secretase, a protease complex that produces Aβ peptides of varying lengths [9], and FAD mutations in presenilins increase the critical ratio of Aβ42 to Aβ40 that determines aggregation [10, 11].
Despite the genetic, biochemical, and pathological evidence supporting Aβ42 as critical to AD pathogenesis, the amyloid cascade hypothesis remains controversial [12]. This is due in part to the many clinical failures of drug candidates targeting Aβ, although this may be attributed to testing too late, when the disease process may become Aβ-independent. More concerning is the lack of understanding of how Aβ42 triggers disease at the molecular and cellular level. Critical unknowns about pathogenic Aβ42 include its oligomeric assembly state, its neurotoxic signaling pathways, and its connection to pathogenic tau. Moreover, recent biochemical studies reveal that a number of FAD mutations in presenilin-1 and AβPP do not increase the Aβ42/Aβ40 ratio [13, 14]. γ-Secretase processing to Aβ is complex and involves multiple proteolytic steps, initially forming Aβ48 or Aβ49 and trimming these generally in tripeptide increments [15] (Fig. 1), and deeper analysis has revealed that FAD mutations in presenilin and AβPP reduce trimming and increase production of Aβ peptides of 45 to 49 residues in length [14 , 17]. These findings raise the possibility that longer Aβ peptides—containing most of the transmembrane domain of AβPP and which are membrane-anchored—contribute to pathogenesis [18].

AβPP mutations stably expressed in human SH-SY5Y cells. Non-transfected (NT) control: Expresses wild-type endogenous AβPP. β-secretase cleavage site and Asp1 residue of C99 and Aβ are indicated in purple. γ-Secretase cleavage sites producing Aβ49⟶Aβ46⟶ Aβ43⟶Aβ40 are indicated in blue and Aβ48⟶Aβ45⟶Aβ42⟶Aβ438 are in red. Mutations introduced in and around the Aβ region of AβPP transgenes in stably transfected cells are in green. NL-G: Stably expresses exogenous AβPP containing the FAD KM⟶NL Swedish double mutation (which increases β-secretase processing, C99 levels, and therefore levels of all Aβ) and the FAD E22G mutation (which increases Aβ aggregation). NL-G-F: Addition of the Iberian I45F mutation blocks Aβ46⟶Aβ43 due to phenylalanine mutation placement in the P2’ position of this trimming event, thereby decreasing Aβ40 production. NL-G-FF: Further addition of the V44F mutation blocks Aβ45⟶Aβ42, abrogating Aβ42 production.
In this study, we investigated the neurotoxic potential of longer Aβ peptides produced from AβPP, in the presence or absence of Aβ42 production. To test this, we took advantage of the complete intolerance of phenylalanine in the P2’ position (i.e., second residue C-terminal to the scissile amide bond) with respect to any given cleavage site in the complex processing of the AβPP TMD by γ-secretase [19]. Thus, the I45F FAD mutation (Aβ numbering) dramatically elevates Aβ42/Aβ40 by blocking Aβ46⟶Aβ43 cleavage, increasing Aβ46 and decreasing Aβ40 levels [14, 19]. Further addition of V44F blocks Aβ45⟶Aβ42 cleavage to effectively abrogate Aβ42 production [19]. Human SH-SY5Y neuroblastoma cells lines were generated to stably express AβPP containing I45F and V44F/I45F mutations. As a measure of neurotoxicity, we focused on mitochondrial function, as dysfunctional mitochondria are associated with neurodegenerative diseases in general [20]. In AD in particular, brain imaging reveals hypometabolism [21], and molecular and cellular studies demonstrate increased damage to mitochondrial DNA by reactive oxygen species [22], decreased oxygen utilization [23], and decreased numbers of intact mitochondria [24]. We found that mitochondrial function, as measured by oxygen consumption rates, and expression of certain mitochondrial or neuronal-associated genes are altered by mutant AβPP in an Aβ42-independent manner. These changes were instead associated with AβPP mutations that block γ-secretase processing of Aβ45 and/or Aβ46.
MATERIALS AND METHODS
Cell lines stably transfected with mutant AβPP
SH-SY5Y human neuroblastoma cells were transfected using Lipofectamine 3000 with the cDNA of human AβPP (695-residue isoform) inserted into pCMV vector containing a neomycin resistance gene. Three different mutant forms of AβPP were transfected separately: 1) “NL-G”, containing the KM(-2/-1)NL Swedish FAD double mutation (Aβ numbering) and the E22G Arctic FAD mutation; 2) “NL-G-F”, additionally containing the I45F Iberian FAD mutation; and 3) “NL-G-FF”, further containing an artificial V44F mutation that blocks Aβ42 production. After transfection, cells were passaged multiple times in the presence of geneticin to select for cells stably expressing the exogenous AβPP variants. Mutant AβPP mRNA expression in cultures was measured using SYBR green qPCR with primers directed against the NL and G mutations to confirms stable expression (see Supplementary Figure 1). AβPP expression was determined by western blotting as described below.
Protein expression determination
Cells were grown to confluence in 6-well plates in a 1:1 mixture of DMEM (Sigma) and F12 (Gibco) supplemented with FBS (Gibco) and Pen Strep (Gibco), whereupon the media were replaced with 1 mL per well of fresh media. After 24 h, media were collected, and Aβ40 and Aβ42 were each measured via specific ELISAs (Invitrogen). Cells were lysed in RIPA buffer, protein levels determined via BCA assay (Thermo), and protein-normalized lysates were subjected to SDS-PAGE and transferred to PVDF membranes. Membranes were probed with anti-AβPP antibody C7 (courtesy of D. Selkoe, Harvard) and with anti-GAPDH antibody (Abcam) to verify normalization.
Mitochondrial function assays
Oxygen consumption rates (OCRs) were measured using an Agilent Seahorse XFe96 Analyzer according to the Agilent Mito Stress Kit protocol. Cells were seeded onto 96-well assay plates 24 h prior to assay run. One hour prior to assay, plated cells were incubated in the absence of carbon dioxide. Cells were sequentially treated with the mitochondrial poisons oligomycin (2μM, Sigma-Aldrich), FCCP (Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone; 0.5μM, Sigma-Aldrich), and rotenone/antimycin (0.5μM/0.5μM, Sigma-Aldrich) while measuring oxygen consumption rates (as indicated in Fig. 3A). Basal mitochondrial OCR was determined by subtracting non-mitochondrial OCR (after rotenone/antimycin treatment) from OCR prior to treatment with any mitochondrial poison. Proton leak was calculated by subtracting non-mitochondrial OCR from OCR after ATP synthase inhibition with oligomycin. Hoechst stain (0.1μg/ml, Sigma-Aldrich) was added at the time of the FCCP injection to visualize and count cell nuclei for normalization of OCRs. Profiles of OCRs were determined according to this protocol for each of the three stably-expressing mutant AβPP cell lines as well as the parental SH-SY5Y cell line.
Electron transport chain complex activities were measured using an Agilent Seahorse XFe96 Analyzer according to well-established protocols [25 –27]. Briefly, cells were seeded on 96-well assay plates 24 h prior to assay run. Immediately prior to assay, cells were treated with XF Plasma Membrane Permeabilizer (PMP, 0.5 nM, Agilent) in 1X of nonionic mannitol and sucrose-based MAS buffer (Agilent) containing 10 mM pyruvate/5 mM malate/4 mM ADP (Sigma-Aldrich). Cells were sequentially treated with electron transport chain substrates and inhibitors succinate/rotenone (10 mM/2μM, Sigma-Aldrich), antimycin A (2μM, Sigma-Aldrich), ascorbate/TMPD (N,N,N′,N′-tetramethyl-p-phenylexnediamine, Sigma-Aldrich) (10 mM/100μM), and sodium azide (20 mM, Sigma-Aldrich) while measuring oxygen consumption rates (as indicated in Fig. 4A). OCRs were normalized by protein determination using BCA. Complex I activity was measured by the average OCR prior to complex I inhibition by rotenone (average of time points 1 and 2). Complex II activity was measured by OCR readings after addition of succinate substrate and complex I inhibition by rotenone (average of time points 3 and 4). Complex III activity was measured by the difference in OCR between immediately before and after complex III inhibition by antimycin A (average of time points 3 and 4 minus average of time points 5 and 6). Complex IV activity was measured by the difference in OCR between OCR readings after ascorbate substrate and TMPD addition and after complex IV inhibition by azide (average of time points 7 and 8 minus average of time points 9 and 10).
Cellular ADP and ATP assay
Cells were plated onto a white, opaque-bottom, 96-well plate. After reaching 10%confluence, cells were lysed and ADP and ATP levels were determined via luciferase assays using an ADP/ATP ratio assay kit (Millipore-Sigma) according to the manufacturer’s instructions. Briefly, ATP levels in cell lysates were determined by measuring luminescence (Cytation 5, BioTek) from luciferase turnover of D-luciferin substrate before and after conversion of ADP to ATP. The initial reading establishes the cellular ATP levels, and subtraction of the first reading from the second establishes the cellular ADP levels.
TaqMan arrays gene expression assay
Custom arrays from Applied Biosystems were designed with TaqMan probes for 84 genes important for mitochondria structure and function or associated with mitochondrial-related disorders, and 12 endogenous control genes for normalization. Of the genes selected, 10 are encoded in mitochondrial DNA and 74 are nuclear-encoded. Total RNA was isolated from cells grown from each cell line in a 6-well plate using TRIzol (Invitrogen) and chloroform extraction. RNA was precipitated with isopropanol and washed with 70%ethanol. Total cellular RNA was normalized for concentration and converted to cDNA using reverse transcriptase (Bio-Rad). qPCR was performed using TaqMan reagents (Invitrogen) in custom arrays and a thermocycler equipped with a detector (QuantStudio, Applied Biosystems). Fold change was calculated by the Δ ΔCT method normalizing against 12 different endogenous control genes. The results for the array were visualized using Heatmapper software [28] with downregulation compared to parental untransfected SH-SY5Y cells indicated by red and upregulation indicated by green with varying degrees of intensity based on the fold-change (Fig. 6).
Statistical analysis
The data are shown as the mean±standard deviation. One-way ANOVA was used to analyze differences between more than two groups with a post hoc Tukey test to make comparisons between two means. Differences were considered significant if p < 0.05.
RESULTS
Iberian FAD-mutant I45F AβPP expression generates high levels of Aβ42, while AβPP V44F/I45F double mutation abrogates Aβ42 production
SH-SY5Y human neuroblastoma cells were generated that stably overexpress human AβPP. Three stably expressing lines were produced, all of which contain as background the Swedish and Arctic FAD mutations. The Swedish double mutation (KM⟶NL in positions –2 and –1, Aβ numbering) results in increased cleavage of AβPP by β-secretase [29], which sheds the AβPP ectodomain to leave a remnant 99-residue AβPP C-terminal fragment (C99) in the membrane. As C99 is the substrate for intramembrane cleavage to Aβ by γ-secretase, the Swedish double mutation leads to elevated production of all Aβ peptides. The Arctic mutation (E22G) lies in the middle of the Aβ sequence and increases the aggregation propensity of Aβ peptides [30]. AβPP with both Swedish and Arctic mutations is dubbed here “NL-G”.
To AβPP NL-G was introduced the Iberian mutation (I45F) [31], which resides near γ-secretase cleavage sites in the single transmembrane domain and has the effect of dramatically elevating Aβ42 production [32]. AβPP with Swedish, Arctic, and Iberian mutations is dubbed “NL-G-F”. We have previously shown that phenylalanine mutation two residues C-terminal to any given γ-secretase cleavage site (called the P2’ position in protease terminology) abrogates that cleavage event [19]. Thus, I45F blocks the Aβ46⟶Aβ43 tripeptide trimming step. As a consequence, Aβ46 levels increase, Aβ40 levels decrease, and production is shunted toward the Aβ42 pathway [14]. Given the established effects of phenylalanine mutation, we further introduced a V44F mutation, to also block the Aβ45⟶Aβ42 tripeptide trimming step. This mutation therefore is expected to additionally eliminate Aβ42 production and elevate Aβ45 levels. AβPP with Swedish, Arctic, Iberian, and V44F mutations is dubbed “NL-G-FF”. The stably expressing cell lines with their corresponding mutations and effects on Aβ production by γ-secretase are illustrated in Fig. 1.
Western blotting established that full-length AβPP protein levels were increased in the NL-G, NL-G-F, and NL-G-FF cell lines compared to the non-transfected (NT) parental SH-SY5Y cells and that the three stably transfected cell lines expressed their respective AβPP mutants to roughly equal degrees (Fig. 2A). Levels of Aβ40 and Aβ42 secreted into the media from these cell lines were determined by specific sandwich ELISAs. In the parental SH-SY5H cell line, only low levels of Aβ40 were detectably produced from endogenous wild-type AβPP, with Aβ42 being below the limit of detection. Stable expression of AβPP NL-G led to dramatically elevated (8-fold) Aβ40 and clearly detectable Aβ42 levels (Fig. 2B), with a normal physiological Aβ42/Aβ40 ratio (∼0.05, Fig. 2C). As expected [14, 19], the additional I45F mutation in AβPP NL-G-F resulted in decreased Aβ40 and dramatically increased (∼4.5-fold) Aβ42 levels. Finally, further introduction of V44F in AβPP NL-G-FF led to further reduction of Aβ40 and virtually no detectable Aβ42 levels (Fig. 2B). Introduction of the I45F Iberian mutation in NL-G-F resulted in dramatic elevation (8-fold) of the Aβ42/Aβ40 ratio compared to NL-G, while further introduction of V44F mutation in NL-G-FF led to an Aβ42/Aβ40 ratio that was effectively zero (Fig. 2C). Thus, we had cell lines in which we could compare effects of blocking specific steps in AβPP processing by γ-secretase, with both NL-G-F and NL-G-FF elevating long Aβ peptides Aβ45 and/or Aβ46, while Aβ42 levels were either dramatically increased or eliminated in comparison with the NL-G background mutations.
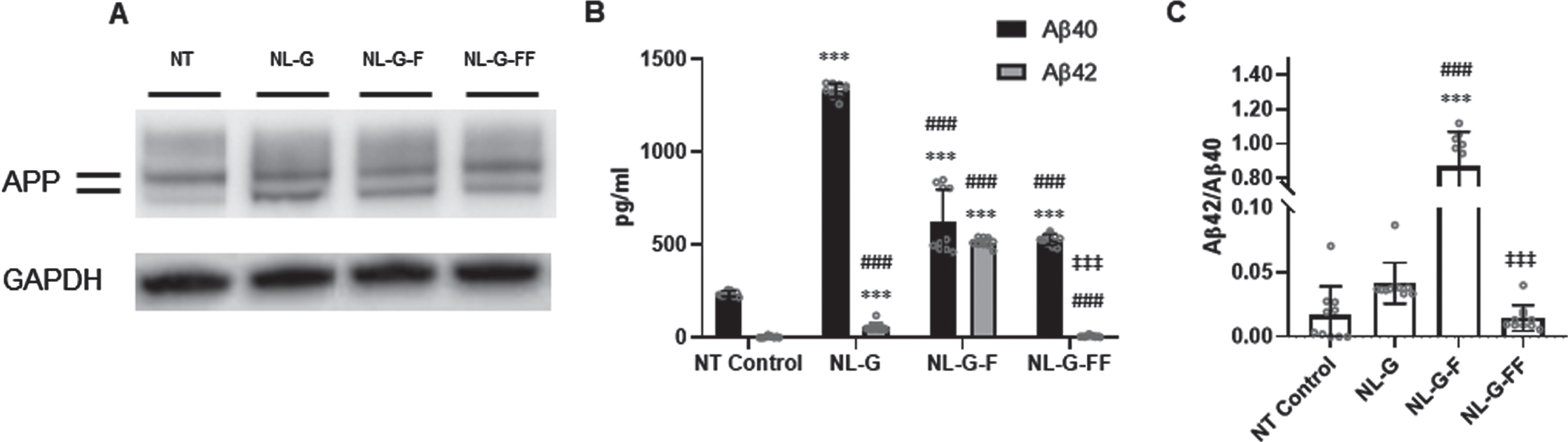
Characterization of SH-SY5Y human neuroblastoma cell lines stably expressing mutant AβPP proteins. A) AβPP and GAPDH protein expression by western blot. Lines near the APP label indicate bands for O- and N,O-linked glycosylated AβPP (lower and upper bands, respectively). B) Secreted Aβ40 and Aβ42 in media determined by ELISA. C) Aβ42/Aβ40 ratio. Values are represented as mean±S.D. (N = 10 for each cell line, Error bars are S.D.). * p < 0.05, ** p < 0.01, *** p < 0.001 between non-transfected (NT) parental SH-SY5H neuroblastoma cells versus the three mutant AbetaPP-expressing cell lines. # p < 0.05, ## p < 0.01, ### p < 0.001 between NL-G and other groups. ‡ p < 0.05, ‡‡ p < 0.01, ‡‡‡ p < 0.001 between NLG-F and other groups.
Mutant AβPP elicits mitochondrial dysfunction independent of Aβ42 production
None of the cell lines expressing AβPP mutants exhibited altered proliferation or morphology in either normal culture conditions or serum starved conditions compared to the non-transfected parental SH-SY5Y cells (see Supplementary Figure 2). To explore effects of these mutant AβPP proteins on mitochondrial function, we measured OCRs using a Seahorse XFe96 analyzer under different mitochondrial stress conditions. OCRs were measured for each cell line at various time points through the course of treatment with mitochondrial poisons (Fig. 3A). In this protocol, plated cells are incubated in CO2-free conditions before a series of measurements to establish the basal OCR. Cells are then treated with the ATP synthase inhibitor oligomycin, and the subsequent reduction in OCR is directly attributed to the inability of mitochondria to consume the oxygen that normally corresponds to ATP production. The uncoupling agent FCCP is then added to allow uninhibited electron flow through the electron transport chain, causing the OCR to increase to its maximal rate and allowing calculation of the spare respiratory capacity, the difference between maximal and basal respiration. Finally, both the complex I inhibitor rotenone and the complex III inhibitor antimycin A are added to fully eliminate electron transport chain oxygen consumption, which leaves only a residual non-mitochondrial OCR. These experiments were performed as three sets of biological replicates (Fig. 3B-D).
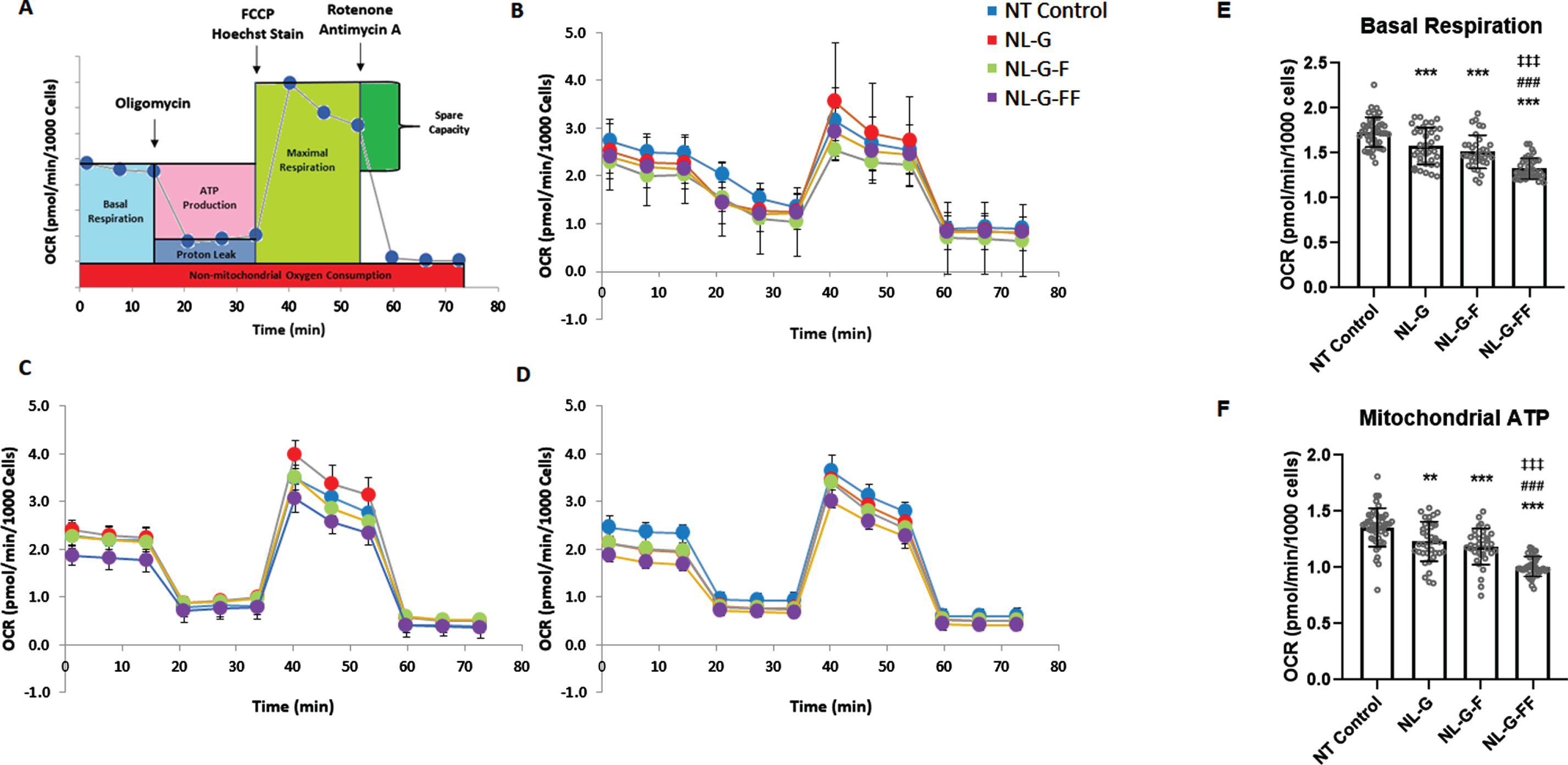
Oxygen consumption rates (OCRs) associated with basal respiration and mitochondrial ATP production in SH-SY5Y neuroblastoma cells stably expressing mutant AβPP. A) General protocol for OCR determination under different mitochondrial stress conditions. After establishing the basal OCR, addition of the ATPase inhibitor oligomycin allows calculation of the OCR associated with mitochondrial ATP production. Subsequent addition of the uncoupling agent FCCP provides the maximal respiration rate. Finally addition of complex I inhibitor rotenone and complex III inhibitor antimycin A shuts down all mitochondrial OCR, providing the non-mitochondrial OCR. B-D) Three biological replicate runs of OCR profile determinations for the three stably expressing mutant AβPP cell lines along with the parental non-transfected SH-SY5Y neuroblastoma cells. E, F) Analyses of the data performed to compare (E) the basal OCR and (F) the OCR associated with mitochondrial ATP production. (N≥35 total combined from the three runs for each cell line. * p < 0.05, ** p < 0.01, *** p < 0.001 between non-transfected (NT) parental SH-SY5H neuroblastoma cells versus the three mutant AβPP-expressing cell lines. # p<0.05, ## p<0.01, ### p<0.001 between NL-G and other groups. ‡ p<0.05, ‡‡ p<0.01, ‡‡‡ p<0.001 between NLG-F and other groups. Error bars are S.D.)
Basal respiration and OCR associated with mitochondrial ATP production are significantly lower in NL-G cells compared to untransfected parental cells after normalization of OCRs to number of cells using Hoechst staining (Fig. 3E, F). This suggests that the presence of the Swedish NL and Arctic G mutations in AβPP may cause mitochondrial dysfunction at these levels. Alternatively, the decreased OCRs may be due to simple over-expression of full-length AβPP and its proteolytic products. Whichever is the case, addition of I45F in NL-G-F cells did not affect basal or mitochondrial ATP-associated OCR compared to NL-G cells, even though the NL-G-F cells produce much higher levels of Aβ42, with a substantially increased Aβ42/Aβ40 ratio. In contrast, despite the absence of detectable Aβ42 production, NL-G-FF cells displayed further decreases in basal respiration and mitochondrial ATP-associated OCR compared to NL-G and NL-G-F cells. Similar analyses of differences in proton leakage and spare capacity between the four cell lines were more variable and not significant (see Supplementary Figure 3). Overall, these results suggest that mutation of AβPP can lead to mitochondrial dysfunction in an Aβ42-independent manner.
To assess differences in the contributions of individual electron transport chain (ETC) complex activities to OCRs, these measurements were determined in the presence of specific ETC complex substrates and/or inhibitors as depicted in Fig. 4A. Normalization was performed using total protein determined by BCA due to difficulties in Hoechst staining in permeabilized cells. Significant decreases in OCRs associated with complexes I, II, and IV were observed for NL-G-FF compared with NL-G-F cells (Fig. 4B). Interestingly, individual ETC complex activities were significantly increased in NL-G cells compared to untransfected parental cells despite overall basal respiration and OCR associated with mitochondrial ATP production being lower compared to control in this cell line.
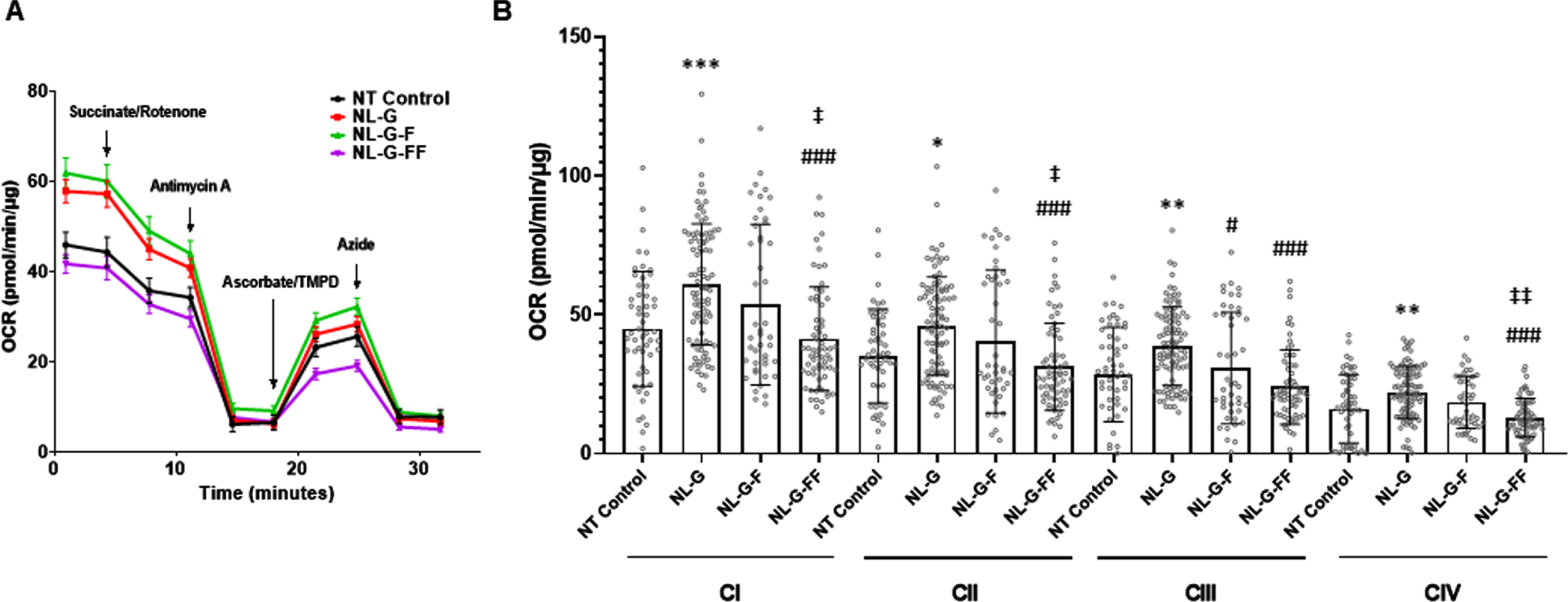
Oxygen consumption rates associated with individual electron transport chain complex activities in mutant AβPP expressing cell lines. A) General protocol for OCR determination under different mitochondrial stress conditions. Plasma membrane permeabilized cells were given complex II substrate, succinate, with complex I inhibitor rotenone to measure complex I and II-linked respiration. Next, complex III inhibitor, antimycin A was added to determine complex III activity. Complex IV activity was driven by oxidation of TMPD by ascorbate, followed by complex IV inhibition by azide. (N≥46 total combined from three runs for each cell line). B) Analyses of the data to compare electron transport chain activities. Values are represented as mean±S.D. * p < 0.05, ** p < 0.01, *** p < 0.001 between non-transfected (NT) parental SH-SY5H neuroblastoma cells versus the three mutant AβPP-expressing cell lines. # p<0.05, ## p<0.01, ### p<0.001 between NL-G and other groups. ‡ p<0.05, ‡‡ p<0.01, ‡‡‡ p<0.001 between NLGF and other groups. Error bars are S.D.)
Total cellular ADP/ATP levels are not changed between AβPP mutants producing long Aβ
Given the effects on OCR associated with mitochondrial ATP production through oxidative phosphorylation, we measured the total cellular levels of ATP and ADP to determine the ADP/ATP ratio in the various cell lines. This ratio is an indication of the balance within cells between production and utilization of the energy currency ATP and of overall cellular health. The ratio also provides an indication of whether cells are undergoing apoptosis (decreased ATP and increased ADP) or necrosis (greatly decreased ATP and greatly increased ADP). Total cellular ATP and ADP were measured via luciferase assay (see experimental methods). While ATP and ADP levels varied between the cell lines, the ADP/ATP ratios were remarkably similar (Fig. 5), which may indicate a compensatory mechanism these mutant AβPP-expressing cells undergo to maintain energy demands despite deficient production of ATP in the mitochondria. The similar ratios provide further validation of overall cellular health.
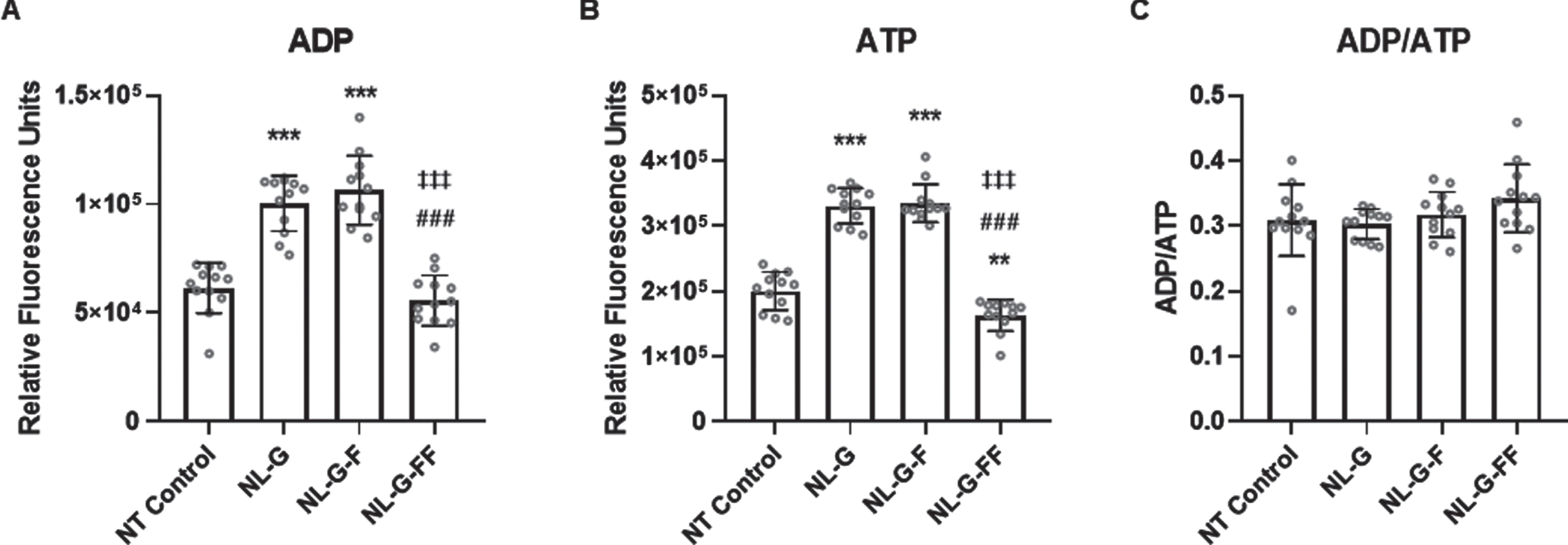
Total cellular ATP and ADP levels of cells expressing mutant AβPP. (A) Relative total cellular ADP levels as measured by luminescence assay, (B) relative total cellular ATP levels, and (C) ADP/ATP ratio. (N = 12 for each cell line, Error bars are S.D.). * p < 0.05, ** p < 0.01, *** p < 0.001 between non-transfected (NT) parental SH-SY5H neuroblastoma cells versus the three mutant AbetaPP-expressing cell lines. # p < 0.05, ## p < 0.01, ### p < 0.001 between NL-G and other groups. ‡ p < 0.05, ‡‡ p < 0.01, ‡‡‡ p < 0.001 between NLG-F and other groups.
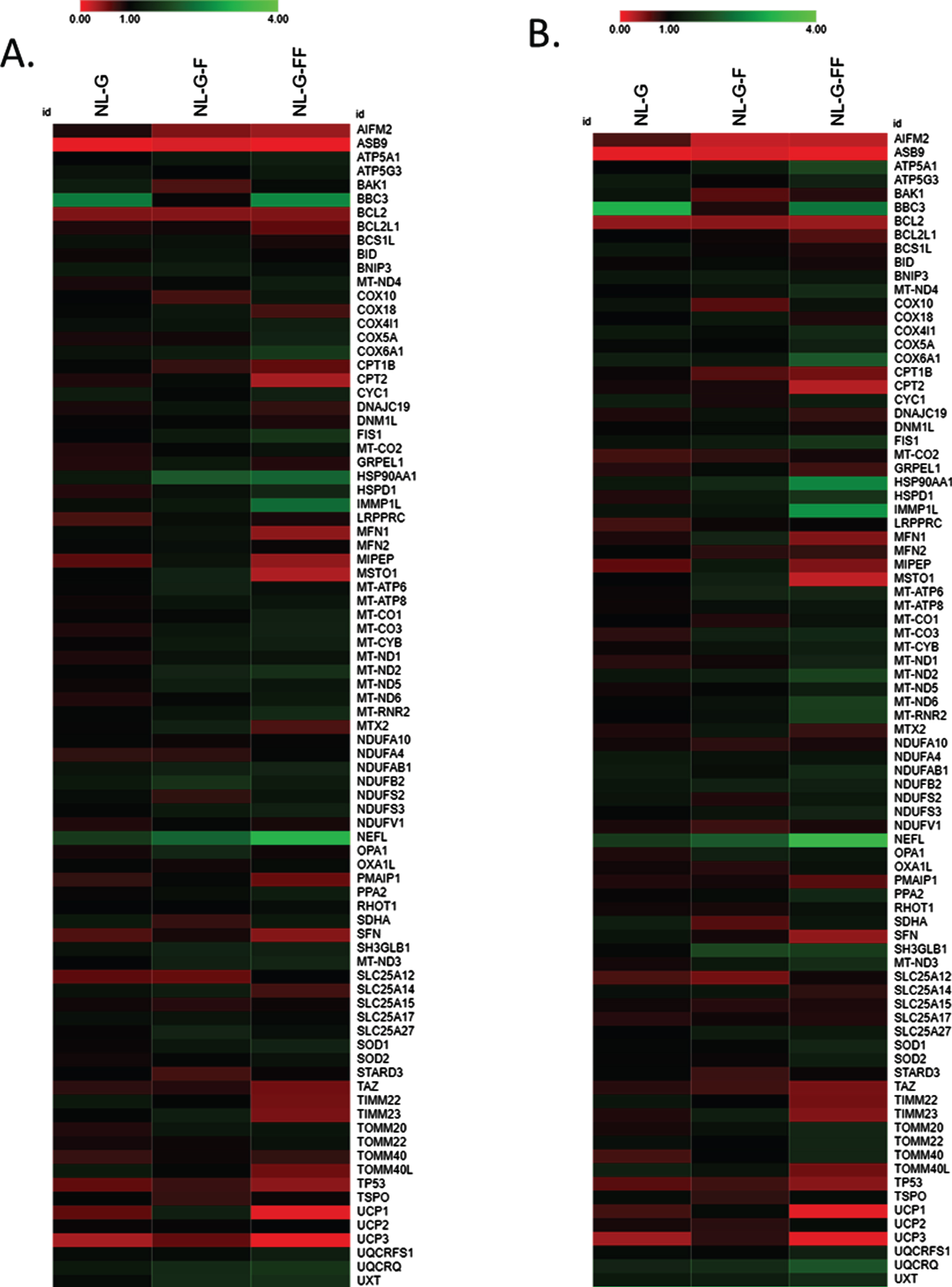
Expression of genes important for mitochondrial and neuronal structure and function are altered by I45F and V44F/I45F AβPP mutations. mRNA levels for a panel of genes were determined by qPCR array for SH-SY5Y cell lines stably expressing mutant AβPP. Expression level fold-change is indicated by heat map, with upregulation compared to control in green, no change in black, and downregulation in red. (A) and (B) are biological replicates, demonstrating reproducibility.
FAD mutant I45F AβPP alters certain mitochondrial and neuronal gene expression patterns independently of Aβ42 production
Custom array plates containing TaqMan fluorescent DNA oligonucleotide probes for expression of genes important for mitochondrial and neuronal structure and function were used to look for changes attributable to the AβPP I45F FAD mutation that are independent of Aβ42 production. In two separate experiments, changes in gene expression patterns were closely consistent (Fig. 6). Notably, AIFM2, an apoptosis-inducing mitochondrial gene [33], is decreased in both NL-G-F and NL-G-FF compared to untransfected cells and NL-G background. NEFL, encoding a structural protein component of the axoskeleton and associated with other neuronal disorders, such as Charcot-Marie-Tooth disease [34], is upregulated in NL-G-F and more intensely upregulated in NL-G-FF compared to untransfected and NL-G background. Also upregulated in this same pattern is HSP90AA1, which encodes a heat-shock protein typically induced in response to elevated levels of misfolded proteins [35]. These changes in AIFM2, NEFL, and HSP90AA1 between the cell lines were each validated in individual tests of gene expression by qRT-PCR.
More directly relevant to our observation of reduced OCR associated with mitochondrial ATP production, expression of a number of genes encoding components of the electron transport chain were altered with a similar pattern (i.e., changes associated with the I45F FAD mutation were not rescued by the Aβ42-blocking V44F mutation). These genes include cytochrome oxidase complex component MT-CO3, a respiratory complex IV subunit, and NADH dehydrogenase component MT-ND2, a respiratory complex I subunit, expression of which were upregulated. Despite AβPP NLG-FF cells not producing Aβ42, expression pattern changes are observed in multiple genes important for mitochondrial and neuronal structure and function just as much or more than with AβPP NL-G-F cells that generate elevated Aβ42 levels.
DISCUSSION
In this study, we generated three SH-SY5Y human neuroblastoma cell lines stably over-expressing AβPP variants carrying FAD mutations to test if an FAD mutation can alter mitochondrial function even in the absence of detectable Aβ42 production. Each of the three cell lines expressed AβPP containing the Swedish KM(-2/-1)NL double mutation (Aβ numbering), which boosts production of all Aβ peptides, and the Arctic E22G mutation, which increases the Aβ aggregation propensity. To this “NL-G” mutant was added the Iberian I45F mutation to generate the “NL-G-F” cell line. The I45F mutation substantially increases Aβ42 production while decreasing Aβ40, resulting in a dramatic increase in the Aβ42/Aβ40 ratio, considered a key indicator of AD pathogenicity [36]. The V44F mutation was further added to the NL-G-F mutant, creating the “NL-G-FF” cell line in which Aβ42 production is abrogated.
The phenylalanine mutations have these effects on Aβ40 and Aβ42 production due to the known intolerance of bulky aromatic residues in the P2’ position of any γ-secretase cleavage event [14, 19]. Thus, I45F blocks the Aβ46⟶Aβ43 carboxypeptidase trimming step, while addition of V44F further blocks the Aβ45⟶Aβ42 trimming step. Along with these effects on secreted Aβ40 and Aβ42, the I45F mutation elevates Aβ46 [14], while adding the V44F mutation should have the further effect of elevating Aβ45 (although note that this has not been directly confirmed in this study). Little is known about the biological or pathological effects of Aβ peptides of 45 residues and longer, which contain most of the transmembrane domain of AβPP and are membrane-anchored [37]. The generation of the NL-G, NL-G-F, and NL-G-FF cell lines thus allows exploring effects of the long Aβ peptides in the presence and absence of Aβ42 production.
We found that NL-G cells have a lower basal OCR and a lower OCR associated with mitochondrial ATP generation in comparison with the parental non-transfected SH-SY5Y cells. While these effects may be due to over-expression of AβPP, the NL-G cell line nevertheless provided a suitable control for testing the effects of adding the I45F and V44/I45F mutations. The NL-G-F cell line did not display lower basal OCR or OCR associated with mitochondrial ATP production in comparison with the NL-G cell line, in spite of the dramatic increase in Aβ42/Aβ40. In contrast, further addition of the V44F mutation in the NL-G-FF cell line, which produces no detected Aβ42, led to lower basal and ATP-associated OCRs as well as decreased electron transport chain complex activity for complexes I, II, and IV as compared to the NL-G-F cell line. Thus, these effects are Aβ42-independent. Instead, the reduced mitochondrial function appears to be associated with increases in Aβ46 and/or Aβ45. Surprisingly, the ADP/ATP ratios were closely similar between all four cell lines, including the non-transfected parental line, suggesting a compensatory mechanism for elevating non-mitochondrial ATP production or decreasing ATP utilization in the lines over-expressing FAD-mutant AβPP.
Gene array analysis of genes important for mitochondrial or neuronal structure and function also revealed several changes in gene expression associated with the AβPP I45F mutation that appear to be independent of Aβ42, as these changes were the same or larger upon further addition of the V44F mutation. Expression of certain genes associated with the electron transport chain were altered, including cytochrome oxidase complex component MT-CO3 and NADH dehydrogenase component MT-ND2. Expression of these genes was upregulated and may be a compensatory response to decreased OCR associated with basal respiration, mitochondrial ATP production, and electron transport chain complex activities. Altered expression of other genes included AIFM2, HSP90A, and NEFL and may also be compensatory in the face of decreased OCR. AIFM2 is an apoptosis-inducing factor released from the mitochondria [33], and downregulation should increase cell survival. The increased expression of an isoform of HSP90, a molecular chaperone [35], may prevent aberrant protein misfolding and aggregation due to long Aβ peptide elevation and/or oxidative damage to other cellular proteins. The increased expression of NEFL, an important structural protein of neurons, may strengthen the cytoskeleton. Intriguingly, NEFL is emerging as an important serum biomarker for FAD [38] and SAD [39].
Further experiments and models are needed to address the limitations of this study. For instance, the SH-SY5Y neuroblastoma cell lines generated and analyzed were undifferentiated and expressed background endogenous wild-type AβPP. Cell lines stably expressing these mutations are currently being generated in a CRISPR/Cas9 AβPP knockout background, and experiments in fully differentiated SH-SY5Y cell lines stably expressing mutant AβPP would provide a more neuronal model better suited to study these observations.
Taken together, these findings suggest that elevated long Aβ peptides such as Aβ45 and Aβ46 may be neurotoxic, resulting in decreased mitochondrial function and leading to changes in gene expression as compensatory responses. In any event, these effects are independent of Aβ42 and suggest that other forms of Aβ or AβPP metabolites are responsible. Further investigation along these lines would seem worthwhile, especially in the wake of recent reports that many FAD mutations do not increase Aβ42/Aβ40 [13, 14] but instead increase long Aβ peptides [14, 16].