
Abstract
Background:
Variants in Niemann-Pick Type C genes (NPC1 and NPC2) have been suggested to play a role as risk or disease modifying factors for Alzheimer’s disease (AD).
Objective:
The aim of this study was to analyze NPC1 and NPC2 variability in demented patients with evidence of brain amyloid-β 1–42 (Aβ) deposition and to correlate genetic data with clinical phenotypes.
Methods:
A targeted Next Generation Sequencing panel was customized to screen NPC1, NPC2, and main genes related to neurodegenerative dementias in a cohort of 136 demented patients with cerebrospinal fluid (CSF) low Aβ levels or positive PET with Aβ tracer and 200 non-demented geriatric subjects.
Results:
Seven patients were carriers of NPC variants in heterozygosis. Four of them displayed pathogenic variants previously found in NPC patients and one AD patient had a novel variant. The latter was absent in 200 non-demented elderly subjects. Five of seven patients (70%) exhibited psychiatric symptoms at onset or later as compared with 43%in non-carriers (p > 0.05).
Conclusion:
The frequency of NPC1 and NPC2 heterozygous variants in patients with CSF evidence of Aβ deposition is higher than in the general population.
INTRODUCTION
Niemann-Pick Type C (NPC) is a rare neurovisceral disease characterized by abnormal lysosomal storage of lipids. It is an autosomal recessive disorder caused by homozygous or compound heterozygous mutations in two genes involved in the cholesterol trafficking: NPC1 (95%of cases) and NPC2, encoding for late-endosomal and lysosomal protein, respectively [1]. The disruption in the lipidic metabolism leads to intracellular accumulation of unesterified cholesterol mainly in spleen, liver, and brain, leading to visceral and neurological symptoms [2]. NPC presents with a highly heterogeneous phenotype for both age of onset and clinical features [3]. Some conditions are indeed considered “clinical niches” for NPC, where patients with symptoms related to the pathology go overlooked for the presence of more relevant or more recognizable features, i.e., movement disorders and early-onset cognitive decline [4]. For this reason, it is thought that the disease could be more frequent than currently estimated (1:120000 live births) [1], since some cases could be misdiagnosed or diagnosed with delay.
As highlighted in a survey by the National NPC Foundation Inc., 50%of NPC cases has a positive family history of neurodegenerative diseases, in particular Alzheimer’s disease (AD) [5]. Besides clinical similarities, NPC shares also neuropathological features with AD, such as the presence of neurofibrillary tangles and increased amyloidogenic processing [6, 7]. A further intriguing link between these two diseases is the cholesterol metabolism, as Apolipoprotein E (ApoE), the main cholesterol transporter in the brain, is the most important genetic risk factor for sporadic AD. Also, endosomes abnormalities and altered endocytic amyloid precursor protein (APP) trafficking are reported in early phases of AD [8, 9].
Recently, a few genetic studies have been carried out to investigate whether genetic variability of NPC1 and NPC2 may influence the risk to develop neurodegenerative disorders. In this regard, a higher frequency of heterozygous carriers was found in cohorts of patients affected by dementia and psychosis [10 –12] as compared with that reported in the Exome Variants Project (EVP, 2%) (https://evs.gs.washington.edu/EVS/). This body of evidence leads to the hypothesis that NPC genes variants are causal of NPC in homozygosis, whereas act as risk factors for dementia and/or psychosis, similarly to glucocerebrosidase gene (GBA) mutations, which are causal for Gaucher Disease in homozygosis, but act as risk factors for Parkinson’s disease (PD) in heterozygosis [5, 13].
In this scenario, the aim of the present study was to evaluate the presence of variants in NPC1 and NPC2 with a targeted Next Generation Sequencing (NGS) panel in a cohort of patients with neurodegenerative diseases with evidence of amyloid deposition in the brain demonstrated by low cerebrospinal fluid (CSF) Amyloid-β 1–42 (Aβ) levels and/or PET with Aβ tracer, and positive family history for dementia and/or psychiatric disorders.
METHODS
Population
Basing on assumption that there is a link between Aβ deposition and NPC, and the evidence that variants in NPC genes may be associated with early onset dementia, we screened for the presence on NPC1 and NPC2 variants a cohort of 136 demented patients with evidence of Aβ deposition in the brain, recruited at the Alzheimer Unit of the Fondazione Ca’ Granda, IRCCS Ospedale Maggiore Policlinico, University of Milan (Milan, Italy). Two inclusion criteria were considered: low CSF levels of Aβ or positivity to PET with Aβ tracer, and a clear anamnestic familiar history for cognitive neurodegenerative diseases (at least one relative, range 1–8 individuals). Eleven patients had also a positive family history for psychiatric diseases.
All patients were referred to our Center in suspicion of dementia. The standard clinical workup comprised detailed medical history, physical, and neurological examination, screening laboratory tests, Mini-Mental State Examination (MMSE); qualitative brain magnetic resonance imaging (MRI) or computed tomography (CT). For a better characterization, 53 patients underwent also fluorodeoxiglucose positron emission computed tomography (FDG-PET). The presence of significant vascular brain damage was excluded (Hachinski Ischemic Score < 4). All patients underwent lumbar puncture for the analysis of CSF biomarkers Aβ, total tau (tau), and tau phosphorylated at position 181 (Ptau). In case of borderline Aβ CSF levels (about±10%of reference value), patients underwent Amyloid-PET (n = 19). The diagnosis of AD was made in accordance with current research criteria [14] (n = 129; of which 106 with typical amnestic presentations and 23 with atypical presentation, i.e., posterior cortical atrophy, frontal variant AD, and logopenic aphasia [14]. Seven patients were diagnosed with different neurodegenerative disorders: Lewy body dementia (n = 4) [15], corticobasal syndrome (CBS) [16] (n = 2), and progressive supranuclear palsy (PSP) syndrome [17] (n = 1). Demographic and clinical data are reported in Table 1 and details of patient selection in Fig. 1.
Demographics and clinical information of the cohort
SD, standard deviation; y, years; AD, Alzheimer’s disease; NA, not available; NP, not performed. *All patients had positive family history for dementia. **Lewy Body Dementia (n = 4), Corticobasal Syndrome (n = 2), Progressive Supranuclear Palsy (n = 1).
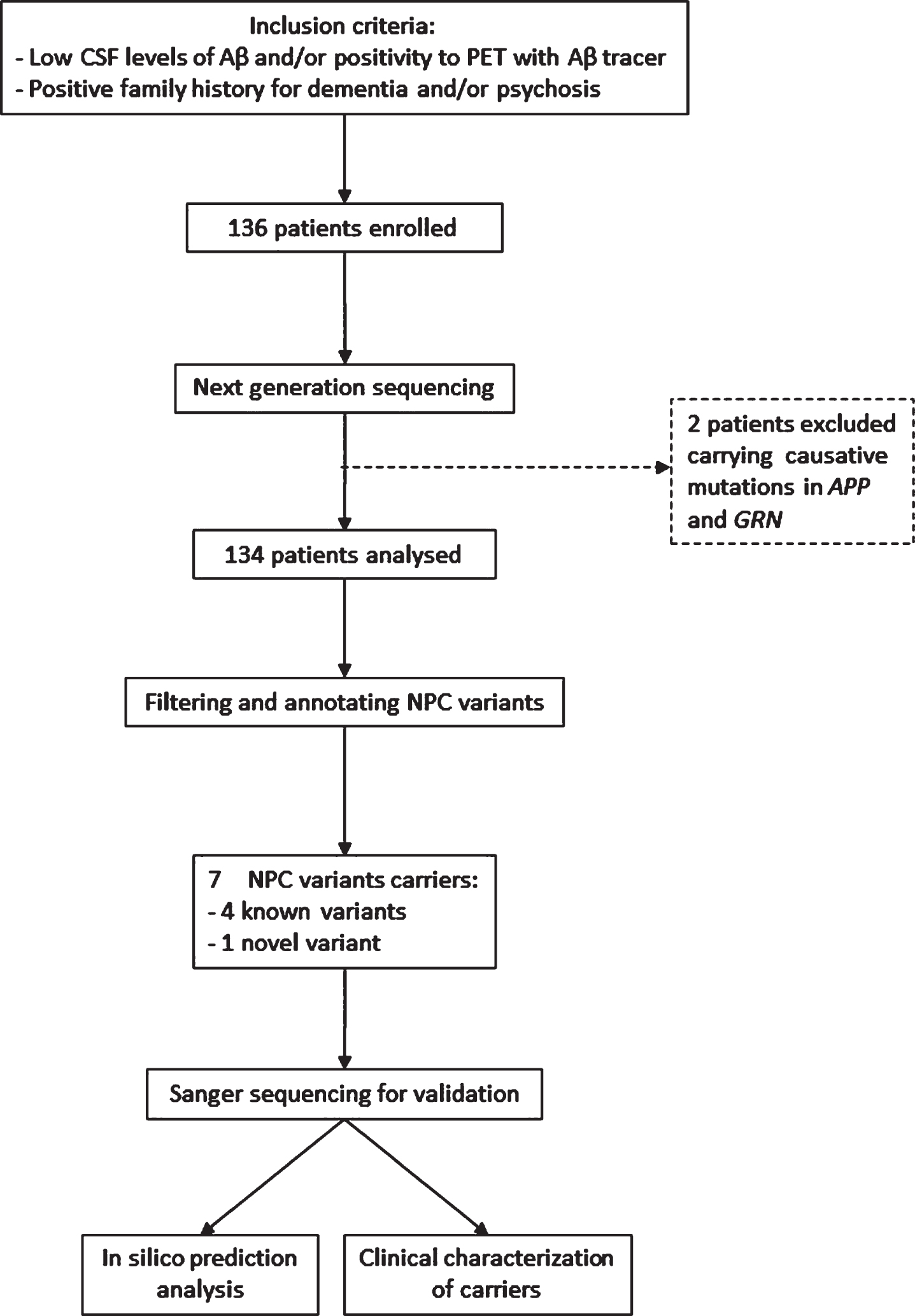
Flowchart of cohort selection.
A cohort of 200 non-demented geriatric subjects from the Geriatric Unit of the same Institution was enrolled for the frequency estimation of any variant found (Table 1). Informed consent was obtained from all participants or their caregivers, and the study was approved by the local Institutional Review Board.
CSF analysis
CSF samples were collected into 15 mL polypropylene tubes by lumbar puncture in the L3/L4 or L4/L5 interspace. Samples were centrifuged at 2000 r/min for 10 min at 4°C. The supernatants were stored at –80°C until use. CSF Aβ, tau and Ptau were measured with ELISA kits (Fujirebio, Ghent, Belgium). Normal values of biomarkers were: Aβ > 600 pg/mL; tau < 400 pg/mL and Ptau < 61 pg/mL [18].
Haloplex target enrichment system
A targeted NGS panel was customized to screen NPC1 and NPC2 genes and the main genes related to autosomal dominant forms of cognitive neurodegenerative diseases: APP, PSEN1, PSEN2 (causing familial AD), GRN and MAPT.
Genomic DNA was extracted from whole blood. Libraries were obtained with HaloPlex HS Target Enrichment System (Agilent Technologies), following manufacturer’s instructions. 50 ng of gDNA were fragmented using 16 restriction enzymes. Fragments were hybridized to probes targeting exons and 25 bp of exon padding to cover flanking regions: HaloPlex HS probes hybridized to both ends of target DNA, directing circularization. During hybridization, each sample was uniquely indexed in order to pool several samples per sequencing lane. Following, the circularized hybrids were closed in a ligation step. The DNA-HaloPlex HS Probe hybrids containing biotin were captured on streptavidin beads and the target libraries were amplified by PCR. After purification, libraries were validated and quantified using the 2100 Bioanalyzer (Agilent Technologies). Further, the sequencing pool was loaded on Illumina Cartridge V2 300 cycles and run on Illumina MiSeq platform according to the instruction of the manufacturer.
Sequence analysis
The fastq NGS data were analyzed using Alissa Align & Call (Agilent Genomics) that aligned the reads to human reference genome and led to vcf analysis files (UCSC hg19, GRCh37, February 2009). The annotation was performed using the web interface to Annotate Variation Software (wANNOVAR), the Sure Call analysis software v 4.1 and the Alissa Interpret software v2.1.1 (Agilent Genomic).
Sanger sequencing and allelic discrimination
Variants were validated by Sanger Sequencing: DNA was amplified using primers designed to cover exons of interest (Eurofins Genomics). Amplicons were sequenced on a 3130 Genetic Analyser (Applied Biosystem).
Using a TaqMan allelic discrimination assay (Thermo Fisher Scientific), performed on QuantStudio 12 K Real-time system (Applied Biosystem), patients were characterized for ApoE genotype and the minor allele frequency (MAF) for each variant was determined.
Chromosome 9 Open Reading Frame 72 (C9ORF72) analysis
All samples were genotyped with AmplideX PCR/CE C9ORF72 Kit (Asuragen, Inc.). Genomic DNA was amplified using a three-primer G4C2-Repeat Primed (RP)-PCR configuration, followed by fragment sizing on a 3100 Genetic Analyzer (Thermo Fisher). ROX 1000 was used for sizing by capillary electrophoresis and the size of the PCR products were converted to the number of G4C2 repeats using size and mobility conversion factor with GeneMapper v 4.1 software (Thermo Fisher).
In silico analysis
The impact of rare variants was assessed by in silico predictions using PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/) which evaluates the effect of missense substitutions on protein sequence and structure, and Mutation Taster (http://www.mutationtaster.org/) which accesses the effect of missense and nonsense substitutions, and intronic alterations, based on the effect on protein sequence.
Statistical analysis
Descriptive statistics, including means and standard deviations, or counts and percentages were calculated. For data measure, analysis of variance (ANOVA) was used. If data were not normally distributed, non-parametric tests were employed. Fisher exact test was performed for allele frequency distribution.
Statistical analyses were carried out using GraphPad Prism version 6.00 (San Diego, CA). Significant threshold was set at p < 0.05.
RESULTS
Genetic analysis
The NGS approach was completed for all the 136 samples investigated in the study and led, unexpectedly, to the identification of two patients who displayed causal mutations: one in APP (p.V717I) in a male patient diagnosed with AD at 55 years, and one in GRN (p.C157fs), predicted to lead to haploinsufficiency and previously associated with phenotypes of FTD spectrum [19], in a woman diagnosed with prodromal AD at 76 years of age. After the diagnosis, the patient carrying the APP p.V717I mutation deteriorated fast (last visit at 57 years of age, MMSE = 9/30). Conversely, the patient carrying the GRN variant (predicted in silico to lead to haploinsufficiency) worsened slowly (last visit at 79 years of age, MMSE = 22/30) and did not show any behavioral disturbance despite the mutation is associated with FTD. She died at 82 years of age. The remaining 134 patients did not carry causal variants for AD and FTD, including the C9ORF72 expansion, and were considered for subsequent NPC1 and NPC2 analysis. The genetic screening revealed that seven patients presented heterozygous variants in NPC1 and NPC2 genes, accounting for 5.2%of our population (Table 2).
NPC mutations carriers
y, years; AD, Alzheimer’s disease; CBS, Corticobasal syndrome. *Frontal variant of AD.
There were no significant differences in terms of age at onset between NPC variant carriers and non-carriers. We did not find any homozygous or compound heterozygous pathogenic variant identifying a genuine NPC case.
Among the seven carriers, four displayed variants previously found in NPC patients (in homozygosity or compound heterozygosity): the near-splicing variant c.441 + 1 G > A [20], rs1401300028, located in the consensus donor splice site of intron 4 of the NPC2 (one with CBS and two with AD) and the missense NPC2 p.V30M [21], rs151220873 (in one AD patient). The former was predicted to be disease causing by Mutation Taster. The latter was predicted to be possibly damaging by PolyPhen-2 software, while Mutation Taster categorized it as tolerated.
Furthermore, we identified two rare known variants: the missense p.K71R in NPC2 and the nonsense p.Q241X in NPC1, each found in patients diagnosed with AD (Table 2). The former, according to Mutation Taster Software, is disease causing since it causes a premature stop signal and therefore leads to an absent or disrupted protein product. The latter was predicted, in silico, to have a disease-causing impact by Poly-Phen2 and Mutation Taster.
Noteworthy, a novel variant of uncertain significance was found in NPC1 (NM_000271: exon11, c.T1708C, p.Y570H) in one patient with AD (Table 2, Fig. 2A). Sanger sequencing confirmed the presence of the variant (Fig. 2B), which leads to the substitution of a Tyrosine with a Histidine residue and is predicted to be disease-causing by in silico analysis performed with Mutation Taster software, while PolyPhen2 categorized it as polymorphism.

Novel variant in NPC1. A) NGS reads alignment. B) Sanger validation of the novel variant. The arrow indicates the variant site.
To test whether the novel variant p.Y570H is a common polymorphism or may have a pathogenic role, the allelic frequency was investigated in a cohort of 200 non-demented geriatric subjects. The variant was absent in controls. The remaining variants were tested as well: three out of 200 controls (1.5%) presented two variants in heterozygosity: two controls displayed the c.441 + 1 G > A (MAF = 0.005), one healthy subject presented the variant p.K71R (MAF = 0.0025) whereas no one showed the p.V30M and p.Q241X variants. There was no significant difference in MAF distribution for the detected variants in patients compared with controls (Table 3).
NPC1/2 variant frequencies
*number of variant carriers, percentage within the cohort.
Apolipoprotein E (APOE) status is shown in Table 2. Four out of 7 NPC variant carriers were also carrier of the ɛ4 AD risk allele.
Phenotypic correlations
Besides cognitive impairment, which was a selection criterion for all patients, five carriers developed behavioral disturbances, language dysfunctions or delusions at onset or during the course of the disease, resembling FTD or psychosis as compared with 58 of 134 in non-carriers (70 versus 43%, p > 0.05). Two carriers developed movement symptoms, leading in one case to the diagnosis of CBS with evidence of Aβ deposition, in consideration of the severity of the extrapyramidal dysfunctions, as compared with 14 of 134 non-carriers (28 versus 10%, p > 0.05).
All carriers underwent morphological neuroimaging study (CT or MRI), which showed frontal atrophy in all cases, associated to temporal atrophy in six carriers and to parietal atrophy in five of them. Five carriers underwent also functional neuroimaging (FDG-PET) which showed temporal hypometabolism in all of them, associated to frontal hypometabolism in two carriers and parietal hypometabolism in four. In carrier #5, neuroimaging revealed marked asymmetry in atrophy and metabolism, characterized by a larger involvement of the left hemisphere (Table 4, Fig. 3), in the absence of language disturbances.
Clinical characteristics of NPC variant carriers
n.r, normality reference, pg/ml (18); n/a, not available; L, left hemisphere; R, right hemisphere.
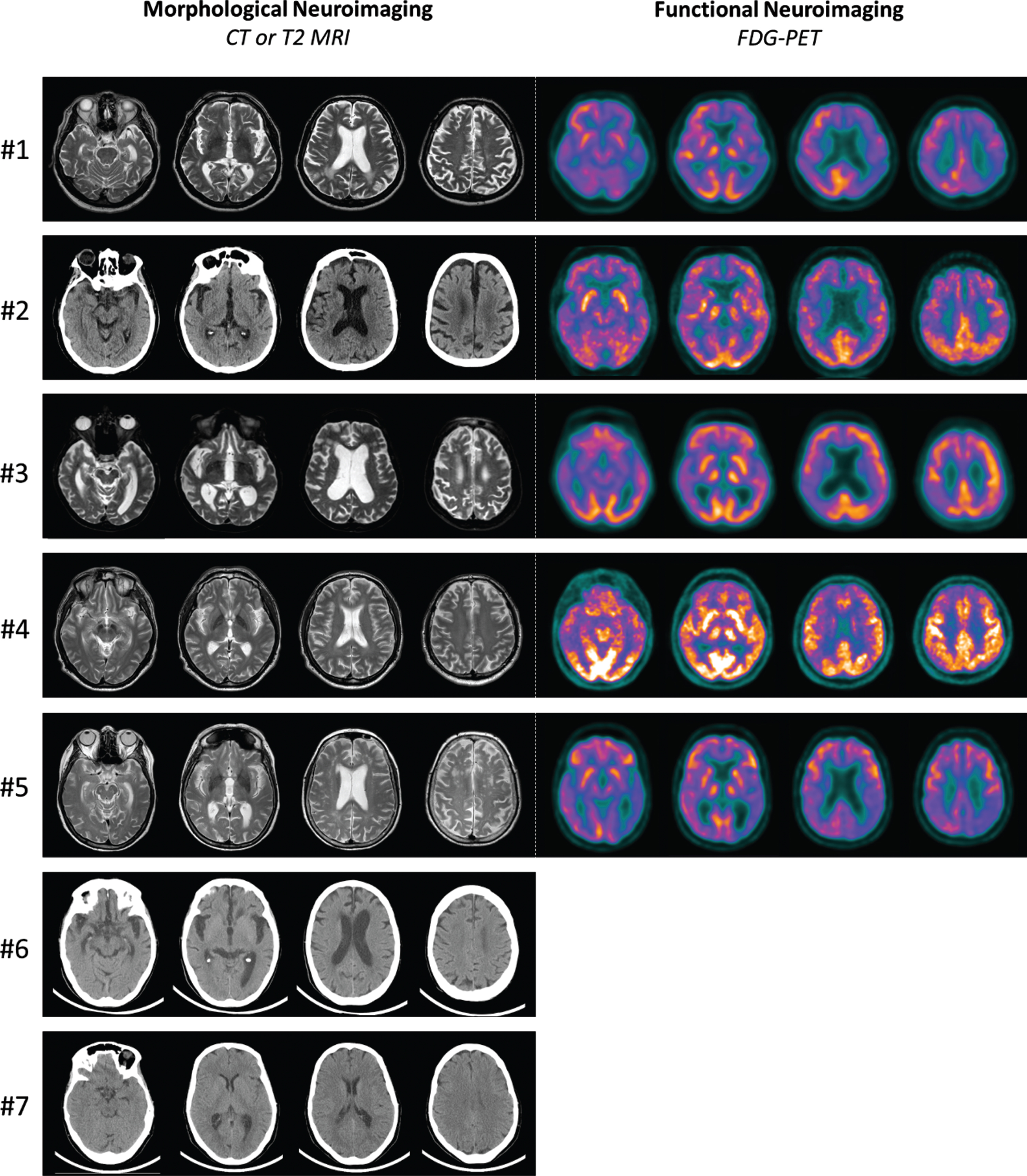
Morphological (CT or T2 MRI scans) and functional (FDG-PET scans) neuroimaging of NPC variant carriers.
DISCUSSION
Herein, we showed that the frequency of genetic variants in NPC1 and NPC2 in patients with dementia and instrumental evidence of Aβ deposition in the brain is higher than in cognitive normal elderly and is associated with the development of behavioral and psychiatric symptoms at onset or during the course of the disease.
Regarding the frequency of variants, 5.2%of our cohort carried a variant as compared with 1.5%in healthy geriatric controls, in accordance with evidence previously reported in the EVP (2%) and the recent paper by Bremova Ertlz et al. (2020), that estimated the occurrence of NPC heterozygosity of 1 : 200 in the general population [22]. This result is in line with previous studies in cohorts of patients with neurodegenerative or psychiatric disorders [10 –12] suggesting that NPC variants may influence the susceptibility to neurodegenerative diseases driven by Aβ deposition.
Moreover, we found a novel variant in NPC1, p.Y570H, absent in 200 non-demented geriatric subjects. These results, together with the in-silico analysis, lead us to raise the possibility that the variant may have a causative role for NPC, although a larger study would be needed to exclude it is a very rare polymorphism. In support of a causative role, the carrier had a remarkable positive family history (five relatives over three generations).
Notably, our cohort was not exclusively defined by the clinical phenotype but based on instrumental analyses suggestive of an ongoing amyloid deposition in the brain. Therefore, we can speculate on a correlation between the presence of NPC1 and NCP2 variants and the ongoing pathogenic amyloid cascade, leading to neurodegeneration. In this regard, to our knowledge, no studies are available assessing the exact role of NPC mutations in different types of dementia characterized with CSF biomarkers and/or amyloid PET to confirm (or exclude) the clinical diagnosis of AD. Genome-wide association studies (GWAS) [23] revealed a significant association between AD and NPC2, but failed to prove a consistent association with NPC1, probably due to the exclusion of SNPs with low allele frequencies (< 5%) [5] and to the lack of biomarkers supporting the clinical AD phenotype. NPC variants are likely too rare for being captured in GWAS studies; thus, we cannot exclude that they might play a role in neurodegenerative diseases.
Regarding the clinical phenotype, beside cognitive impairment, which was one of the main inclusion criteria, five of seven carriers developed psychosis (70%) as compared with non-carriers (43%), mainly delusions, and two had extrapyramidal symptoms (28%, compared with 10%in non-carriers). Notably, patient #1 did DAT-Scan SPECT that was positive for decreased dopamine transporters in putamen and caudate but had no beneficial effects from treatment with levodopa.
As reviewed by Schneider and colleagues [24], several studies have been performed to access clinical, biochemical, and neurophysiological features in heterozygous carriers. They reported a total of 19 patients with neurodegenerative syndromes (i.e., atypical parkinsonism, dystonia, and dementia), drawing the conclusion that NPC heterozygous mutations could be enough to exert a pathogenic effect [25, 26]. Three out of four known variants present in our population were previously found in the heterozygous status in three genetic screening conducted independently in cohorts of patients from different European countries affected by neurodegenerative diseases. In particular, the p.V30M mutation was found in one patient with PD [10] and one with CBS [11]. The c.441 + 1 G > A variant was present in patients diagnosed with PD, FTD [10], and CBS [11] and in a patient with psychosis [12]. The screening in a cohort of geriatric controls showed the presence of the variant in one subject not showing at present any neurological disorder. This evidence could suggest that NPC heterozygosity might promote neurodegeneration acting in combination with other environmental or genetic factors. The p.K71R variant was instead harbored in one patient diagnosed with FTD [10]. It has, however, to be underlined that all the above studies did not include biomarkers for diagnosis, which was merely clinical and some of them were published before the definition of current AD criteria, which include atypical presentations, particularly the frontal variant, resembling, from a clinical point of view, FTD. Therefore, we can speculate that NPC variants are associated with behavioral and psychiatric symptoms and the evidence of amyloid deposition. However, the significance threshold was not reached, possibly due to the small size of the cohort of carriers.
Two patients also displayed movement symptoms. In this regard, a recent study from Ouled et al. [27] widely investigated NPC1 genetic variability in a large cohort of PD patients aiming to find novel genetic association with the disease. However, the study concluded that both common and rare variants in NPC genes were not associated with PD.
As already pointed out, all patients recruited had low CSF Aβ levels or evidence of Aβ deposition at PET [18], thus suggesting amyloid deposition in the brain. Nevertheless, after the complete workup, seven patients were diagnosed with other dementias rather than AD. To explain the observed low amyloid levels in patients diagnosed with other neurodegenerative diseases than AD, different hypotheses may be raised, including: 1) the accuracy of the test is not 100%and the definition of the threshold for defining CSF/PET Aβ levels “normal” is debatable; 2) amyloid deposition may co-occur with other pathologies. For instance, low CSF amyloid levels have been previously reported in carriers of the C9ORF72 expansion [28], associated with TDP-43 deposition in the brain, or in patients with LBD, associated with synuclein deposition.
An unexpected result of the study was the presence of two causal mutations, one for AD (APP) and another for FTD (GRN). Regarding the former, the genetic counselling was not considered at time of diagnosis in light of current literature, suggesting an onset in the fifth decade of life and a complete penetrance [29]. The patient worsened over two years from diagnosis and was then lost at follow up. Regarding the latter, it is known that symptoms at presentation as well as the age at onset are very heterogeneous [30], and presentation with memory disturbances has been reported previously [31]. Despite GRN mutations are associated with behavioral disturbances, the patient never developed such symptoms over time (until the last visit, three years prior to death), never meeting current criteria for FTD [32].
In conclusion, this is the first study of NPC1 and NPC2 variability in a cohort of demented patients with evidence of brain Aβ deposition. We showed that the frequency of NPC1 and NPC2 heterozygous variants in patients with CSF or amyloid-PET evidence of amyloid deposition is higher than in the general population and is associated with behavioral and psychiatric symptoms. Even though NPC is a recessive inherited disorder, growing evidence highlighted the possible pathogenicity of heterozygous mutations. As speculated by Bauer et al., heterozygous mutation in NPC could have a dominant effect with reduced penetrance [12]. The high proportion of neurodegenerative diseases among NPC families further support this hypothesis. Nevertheless, further studies are needed to highlight the connection between NPC, amyloid deposition, and clinical phenotype.