
Abstract
Background:
The pathophysiology of frontotemporal dementia (FTD) is poorly understood but recent studies implicate neuroinflammation as an important factor. However, little is known so far about the role of the resolution pathway, the response to inflammation that allows tissue to return to a homeostatic state.
Objective:
We aimed to measure the concentrations of lipid mediators including specialized proresolving mediators (SPMs) and proinflammatory eicosanoids in the cerebrospinal fluid (CSF) of people with FTD.
Methods:
15 people with genetic FTD (5 with C9orf72 expansions, 5 with GRN mutations, and 5 with MAPT mutations) were recruited to the study along with 15 age- and sex-matched healthy controls. Targeted liquid chromatography-tandem mass spectrometry techniques were used to measure the CSF concentrations of lipid mediators in the docosahexaenoic acid (DHA), n-3 docosapentaenoic acid, eicosapentaenoic acid, and arachidonic acid (AA) metabolomes.
Results:
Only the C9orf72 expansion carriers had higher concentrations of SPMs (DHA-derived maresins and DHA-derived resolvins) compared with controls. In contrast, GRN and MAPT mutation carriers had normal concentrations of SPMs but significantly higher concentrations of the proinflammatory AA-derived leukotrienes and AA-derived thromboxane compared with controls. Additionally, the C9orf72 expansion carriers also had significantly higher concentrations of AA-derived leukotrienes.
Conclusion:
This initial pilot study of lipid mediators provides a window into a novel biological pathway not previously investigated in FTD, showing differential patterns of alterations between those with C9orf72 expansions (where SPMs are higher) and GRN and MAPT mutations (where only proinflammatory eicosanoids are higher).
INTRODUCTION
The term frontotemporal dementia (FTD) encompasses a group of neurodegenerative disorders with a wide spectrum of clinical manifestations and complex disease mechanisms [1]. FTD is a highly heri-table disorder, with the majority of genetic FTD caused by mutations in progranulin (GRN), micro-tubule-associated protein tau (MAPT), and chromosome 9 open reading frame 72 (C9orf72) [2–4]. In common with other neurodegenerative diseases, inflammation appears to play a crucial role in the development of FTD and both molecular and clinical studies over recent years have highlighted its importance (reviewed in Bright et al. [5]). Neuroinflammation is complex and involves multiple stages: once it is triggered there is activation of glial cells with upregulation of several proteins, including cytokines and chemokines, that help guide the process. However, consistent inflammatory activation destroys the healthy structures surrounding the inflammatory core and leads to cellular necrosis or apoptosis. A regulatory mechanism that ameliorates inflammation-related damage is therefore required, and this is the process called ‘resolution’, which emerges after innate and adaptative inflammation has occurred [6]. Resolution involves a number of cellular processes that promote removal of dead cells and debris, restoration of vascular integrity and perfusion, and regeneration of tissue [7]. Key to the process of resolution are the specialized proresolving lipid mediators (SPMs) that are synthesized by a variety of cells including endothelial cells, macrophages and neutrophils [8]. While studies have started to investigate the presence of measures of microglial activation, cytokines, chemokines, and complement proteins in the biofluids of people with FTD (reviewed in Swift et al. [9]), there has been little investigation of SPMs and how the resolution pathway might be altered in FTD [10]. However, recent studies have suggested that impaired resolution may be a contributing mechanism leading to chronic inflammation in dementia [11].
Although the majority of lipid mediators are involved in resolution (Fig. 1), a number of those derived from arachidonic acid (eicosanoids) are in fact proinflammatory [12–14] and while little is currently known about their role in FTD, they have previously been shown to exacerbate pathology in Alzheimer’s disease (AD) [13, 14].

Schematic representation of the role of lipid mediators in modulating neuroinflammation (adapted from Tiberi et al., 2021 [12]).
We therefore set out to measure the lipid mediators underlying the resolution pathway (SPMs), as well as the closely related proinflammatory eicosanoids, in the cerebrospinal fluid (CSF) of a group of people with genetic FTD.
METHODS
Participants
Fifteen people with genetically-confirmed symptomatic FTD (5 with MAPT mutations, 5 with GRN mutations, and 5 with C9orf72 expansions) with available CSF were consecutively recruited from the University College London Genetic FTD Initiative study. The group consisted of 10 men and 5 women, with a mean (standard deviation) age of 63.8 (5.7) years old at sample collection. In the individual genetic groups: MAPT 3 men, 2 women, 63.1 (4.2) years old; C9orf72 4 men, 1 woman, 63.4 (6.9) years old; GRN 3 men, 2 women, 64.8 (6.7) years old. 15 healthy controls were recruited over the same time period: 10 men and 5 women, 64.0 (5.9) years old. No significant differences were seen between groups in terms of age or sex.
The London Queen Square Ethics committee approved the study.
Targeted lipid mediator profiling
All samples were extracted using solid-phase ex-traction columns as previously described [15]. Prior to sample extraction, deuterated internal stan-dards, representing each region in the chromatographic analysis were added to facilitate quantification in 4 volumes of cold methanol. Lipid mediators were then measured via liquid chromatography tandem mass spectrometry using targeted multiple reaction monitoring. Each lipid mediator was identified using established criteria, including matching retention time to synthetic and authentic materials and at least 6 diagnostic ions. Calibration curves were obtained for each using synthetic compound mixtures at 0.78, 1.56, 3.12, 6.25, 12.5, 25, 50, 100, and 200 pg, which gave linear calibration curves with r2 values of 0.98–0.99 [15].
Lipid mediators in the following groups were identified: 1) in the docosahexaenoic acid (DHA) metabolome, maresins, protectins, and resolvins; 2) in the n-3 docosapentaenoic acid (n-3 DPA) meta-bolome, maresins, protectins, and resolvins (D-series and 13-series); 3) in the eicosapentaenoic acid (EPA) metabolome, resolvins; and 4) in the arachidonic acid (AA) metabolome, lipoxins, leukotrienes, prostaglandins, and thromboxane. Concentrations of individual lipid mediators were combined to give a single measure for each of the 12 groups outlined above. This is detailed in Supplementary Table 1 (and previously described in [15]). Of note, the leukotrienes, prostaglandins and thromboxanes are proinflammatory eicosanoids whilst the other measures are all SPMs.
Statistical analysis
All statistical analyses were performed in STATA (v.16). The concentrations of each measure were compared between groups using non-parametric tests: the Wilcoxon Rank Sum test for comparison between the total FTD group and controls and the Kruskal-Wallis test (with post hoc pairwise tests) for comparisons between the individual genetic groups and controls. Spearman correlation coefficients were determined to investigate the relationship between the concentrations of each measure and disease duration.
RESULTS
Concentrations were below the lower limit of detection for the DHA-derived protectins, n-3 DPA-derived protectins and n-3 DPA-derived D-series resolvins. However, differences were seen between groups in four of the other nine measures (Table 1, Fig. 2). Among the SPMs, concentrations were higher in the C9orf72 expansion carriers for the DHA-derived maresins (controls mean 0.6 (standard deviation 1.6) pg/mL, MAPT 4.6 (7.9), C9orf72 7.1 (5.9), GRN 0.0 (0.0)) and the DHA-derived resolvins (controls 1.3 (2.9) pg/mL, MAPT 0.9 (0.7), C9orf72 7.7 (4.7), GRN 0.6 (1.0)) in comparison with controls and the GRN mutation carriers (but not with the MAPT group). No differences were seen in the MAPT or GRN mutation carriers compared with controls, although concentrations of the DHA-derived maresins were positively correlated with disease duration in the MAPT group (rho = 0.89, p = 0.041). No other significant correlations were seen comparing SPMs with disease duration apart from in C9orf72 expansion carriers for n-3 DPA-derived 13-series resolvins levels (rho = –0.89, p = 0.041).
Demographics and mean (standard deviation) concentrations (pg/mL) for the lipid mediators in controls, the total FTD group and each of individual genetic groups (MAPT, C9orf72, and GRN mutations)
Significant differences between disease groups and controls are shown in bold: *p < 0.05, **p < 0.01, ***p < 0.001. The only significant differences between individual genetic groups was between the C9orf72 and GRN groups for DHA-derived maresins and DHA-derived resolvins (both p < 0.05). N/A, not assessed as all values were at the lower limit of detection.
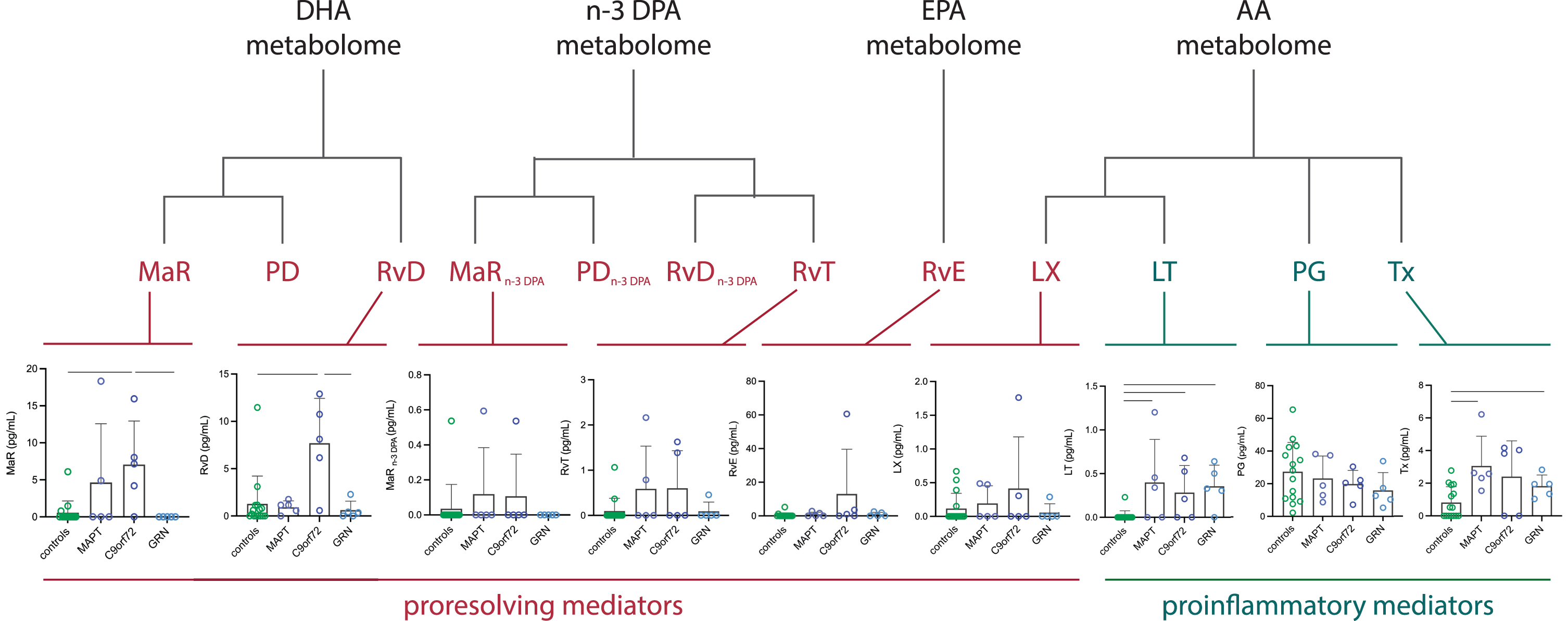
Concentrations of proresolving and proinflammatory lipid mediators. Results expressed in pg/mL in controls and genetic FTD groups (MAPT, C9orf72, and GRN). Significant differences between groups shown by bars. In the docosahexaenoic acid (DHA) metabolome: MaR, maresins; PD, protectins; RvD, resolvins. In the n-3 docosapentaenoic acid (n-3 DPA) metabolome: MaRn - 3DPA, maresins; PDn - 3DPA, protectins; D-series RvDn - 3DPA and 13-series (RvT), resolvins. In the eicosapentaenoic acid (EPA) metabolome: RvE, resolvins. In the arachidonic acid (AA) metabolome: LX, lipoxins; LT, leukotrienes; cysteinyl leukotrienes; PG, prostaglandins; Tx, thromboxane.
In contrast, both the GRN and MAPT mutation carriers had significantly higher concentrations of the AA-derived leukotrienes (controls 0.0 (0.1) pg/mL, MAPT 0.4 (0.5), C9orf72 0.3 (0.3), GRN 0.4 (0.2)), and AA-derived thromboxane (controls 0.8 (1.0) pg/mL, MAPT 3.1 (1.8), C9orf72 2.4 (2.2), GRN 1.8 (0.7)) compared with controls. The AA-derived leukotrienes were also higher than controls in the C9orf72 group. For the AA-derived leukotrienes, concentrations were strongly positively correlated with disease duration in the MAPT group: rho = 0.97, p = 0.005. No other correlations were found in any of the groups comparing proinflammatory eicosanoid levels with disease duration.
DISCUSSION
In this preliminary study, we show that SPMs and the proinflammatory eicosanoids are abnormal in genetic FTD, with a differential pattern in the different forms: the SPMs are higher in concentration in C9orf72 mutation carriers but normal in GRN and MAPT mutation carriers whilst the proinflammatory eicosanoids are higher in GRN and MAPT mutation carriers with only AA-derived leukotrienes higher in C9orf72 mutation carriers.
Only one previous study has investigated SPMs in FTD: Fraga and colleagues studied the levels of the lipoxin LXA4 and found no differences in behavioral variant FTD in either plasma or CSF compared to healthy controls or people with AD [10]. In contrast, studies of AD have shown abnormalities of SPMs both in tissue and CSF, e.g., downregulation of DHA-derived mediators such as MaR1, PD1, and RvD5 as well as LXA4 is seen in the entorhinal cortex and hippocampus [16, 17], while concentrations of LXA4 are lower in the CSF of people with AD [18], suggesting that the impairment of resolution may potentiate chronic inflammation in AD [11]. Intriguingly, we found the opposite pattern in C9orf72-related neurodegeneration with increased levels of SPMs. One prior study of DHA-derived resolvins does in fact show increased levels in the spinal cord tissue of people with amyotrophic lateral sclerosis [19], one of the phenotypes of C9orf72 expansions. The mechanisms of how this affects the pathophysiology of FTD or amyotrophic lateral sclerosis remains unclear, and further studies both in tissue and biofluids will be important to understand this more clearly.
The finding of abnormal proinflammatory eico-sanoids in GRN mutation carriers is consistent with previous studies showing that inflammation plays a major role in this form of FTD [5, 20]. In CSF, abnormalities have previously been shown in microglial activation markers [21–24], chemokines and cytokines [20, 25], and complement proteins [26]. Here, we add to these findings, suggesting a further biomarker that may be useful in disease modifying trials—lowering of the concentrations of the proinflammatory eicosanoids may help to show a therapeutic effect in GRN-related FTD with improvement of chronic neuroinflammation, although more validation work would need to be done in the first instance. An increase in proinflammatory eicosanoids was seen in MAPT-related FTD as well; this group has also been previously associated with abnormal neuroinflammation in prior studies [27]. Only an increase in the AA-derived leukotrienes was seen in the C9orf72 group, likely due to the complex nature of neuroinflammation and the different pathways affected.
Although there has been little work in SPMs in FTD, there is a growing literature on the involvement of lipids in the pathophysiology of the disease [28–30], including biomarker studies in CSF. One study showed an association with survival [30] (with cholesterol levels), whilst a lipidomic study found widespread abnormalities in different lipid species [29]. However, most of the studies so far have focused on clinically-defined populations with the association of abnormal lipid processing in FTD and underlying molecular pathology remaining unclear.
The limitations of the study include the small number of cases investigated here, reducing the power to detect changes. However, even with these small numbers, we found differences within the genetic groups. One further potential limitation is that as this was an exploratory study we did not correct for multiple comparisons [31]. Further studies will be needed to extend this pilot investigation, including a wider group of mutation carriers across the spectrum from the presymptomatic to symptomatic stages. Another limitation was the lack of data collected on concurrent medication including particularly anti-inflammatory drugs; such information will be an important factor to take into account in future studies.
Overall, our results suggest that there are differential alterations in SPMs and proinflammatory eicosanoids in the different forms of genetic FTD and that these changes can be detected and monitored in CSF supporting the use of such lipids as biomarkers to understand underlying disease pathophysiology in FTD, and potentially as markers of neuroinflammation in future therapeutic trials. Further studies will be needed in a larger cohort, including in very early disease, to determine when and how alterations of lipid mediators occur, and how they change over time.
Footnotes
ACKNOWLEDGMENTS
We thank the research participants for their contribution to the study.
The Dementia Research Centre is supported by Alzheimer’s Research UK, Alzheimer’s Society, Brain Research UK, and The Wolfson Foundation. This work was supported by the NIHR UCL/H Biomedical Research Centre, the Leonard Wolfson Experimental Neurology Centre (LWENC) Clinical Research Facility, and the UK Dementia Research Institute, which receives its funding from UK DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK. ASE is supported by the UK Dementia Research Institute which receives its funding from DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK. JDR has received funding from an MRC Clinician Scientist Fellowship (MR/M008525/1) and the NIHR Rare Disease Translational Research Collaboration (BRC149/NS/MH) as well as the Brain Research UK Miriam Marks Fellowship; his work is also supported by the MRC UK GENFI grant (MR/M023664/1), the Bluefield Project and the JPND GENFI-PROX grant (2019-02248). This work was also supported by funding from the Barts Charity (grant no: MGU0343) to JD.).