
Abstract
Background:
Tauopathies are a group of neurodegenerative disorders, including Alzheimer’s disease (AD) and frontotemporal lobar degeneration with tau pathology. Hyperphosphorylation modification promotes tau protein misfolding and aggregation into neurofibrillary tangles, leading to impairments of synaptic plasticity and learning and memory. However, very limited therapeutic strategies are available.
Objective:
In the present study, we wanted to investigate the potential effects of Dihydroartemisinin (DHA) on tauopathies.
Methods:
We constructed adeno-associated virus carrying hTau cDNA (AAVhTau) to establish a mouse model of tauopathy through intrahippocampal microinjection. Using a combination of behavioral test, electrophysiological recording, and western blotting assay, we examined the neuroprotective effects of DHA on learning and memory deficits in mice with tauopathy.
Results:
DHA improved learning and memory and increased hippocampal CA1 long-term potentiation (LTP) in mice overexpressed human tau (hTau) in the hippocampus. More importantly, further study revealed that DHA could induce protein O-GlcNAcylation modification and reduce protein phosphorylation. O-GlcNAc transferase inhibitor alloxan could suppress DHA-induced protein O-GlcNAcylation, and subsequently prevent therapeutic effect of DHA on the deficits of learning and memory as well as synaptic plasticity in hTau mice.
Conclusion:
These results indicate that DHA may exert neuroprotective role in tauopathy through a crosstalk between O-GlcNAcylation and phosphorylation, suggesting a potential therapeutic for learning and memory deficits associated with tau pathology.
Keywords
INTRODUCTION
O-GlcNAcylation is an important post-translational modification [1]. Different from glycation, O-GlcNAcylation is involved in the addition of O-linked-β-N-acetyl- glucosamine (O-GlcNAc) onto serine and threonine residues by O-GlcNAc transferase (OGT), and it could be removed by O-GlcNAcase (OGA). The main function for O-GlcNAcylation appears to be the inhibition of protein aggregation. Several studies have revealed a crosstalk between O-GlcNAcylation and phosphorylation by competition for the same Ser/Thr residues, although not at all sites, which are responsible for neuronal function, learning, and memory [2, 3].
Tau pathogenesis involves both hyperphosphorylation and dominantly N-terminal truncation of tau (without C-terminus a.a. 428–441) in Alzheimer’s disease (AD) [4]. Phosphorylated tau (P-tau) accumulation in interneuron reduces GABAergic signaling, and consequently impairs adult hippocampal neurogenesis and increases astrogliosis [5]. When it comes to tau, not only S400 but also at least another four O-GlcNAc sites (T123, S208, S238, and one of S409/412/413) were identified together, and all of them are sensitive to phosphorylation [6–8]. It has been reported that O-GlcNAcylation may block hyperphosphorylation, so as to hinder subsequent oligomerization of tau by enhancing its solubility without perturbing monomer activity [9]. Genetic knock-out of OGT or inducing hyperphosphorylation with the phosphatase inhibitor okadaic acid would decrease tau O-GlcNAcylation [10], leading to a significant increase in P-tau and Aβ, and a progressive reduction in hippocampal and cortical neurons. On the contrary, chronic inhibition of OGA diminishes P-tau in the brain and total tau in cerebrospinal fluid [11]. Notably, O-GlcNAcylation goes far beyond that, and it can also modify proteins like amyloid-β protein precursor (AβPP) as well [12, 13].
Artemisinin (ART) and its derivatives were popular antimalarial drugs, which is a prodrug waiting to be converted into the active metabolite dihydroartemisinin (DHA) in vivo. ART play an important role in dealing with malarial, tumor, diabetes, inflammation, hyperlipidemia, pulmonary hypertension, and iron overload [14–16]. More importantly, ART is able to exert neuroprotective function from oxidative damage and excitotoxicity via activation AMPK [17] and ERK signal pathways [18, 19], consequently inhibit neuronal death and improve learning and memory [20]. With unique structural features, ART shows high binding affinity of interaction to inhibit and disaggregate the cytotoxicity peptides human islet amyloid polypeptide (hIAPP) and amyloid-β (Aβ) fibrillization to protect neuronal cells [21]. Besides Aβ, ART also can reduce the deposition of P-tau in AD models and improve cognitive function [22]. Thus, it is reasoned that ART/DHA may reduce hTau-induced protein phosphorylation by enhancing O-GlcNAcylation, and subsequently ameliorate synaptic and cognitive functions in tauopathy. In the present study, we investigated this hypothesis using a combination of electrophysiological and behavioral assessments in a mouse model of tauopathy.
MATERIALS AND METHODS
Animals
C57BL/6 male mice, aged 8 weeks, were purchased from Charles River Laboratories and raised at the animal care center of Children’s Hospital affiliated to Chongqing Medical University. Mice were housed in plastic cages under a 12/12-h light/dark cycle (7:00 a.m. to 7:00 p.m.) with free access to food and water in a temperature and humidity-controlled SPF room. All animal experiments were conducted in accordance with the Chongqing Science and Technology Commission guidelines and approved by the Chongqing Medical University Animal Care Committee.
Chemical reagents and antibodies
DHA (#HD155631) was purchased from Chenguang Biotechnology Co., Ltd (Baoji, Shanxi). Complete Protease Inhibitor Cocktail Tablets (#04693116001) was obtained from Roche Applied Science. Alloxan (#A7413) was purchased from Sigma-Aldrich. Pierce TM BCA Protein Assay Kit (#23225) was ordered from Thermo Fisher Scientific.
O-GlcNAc mouse monoclonal antibody (RL2) (#MA1-072) and tau mouse monoclonal antibody (#AHB0042) were obtained from Invitrogen. OGT rabbit polyclonal antibody (#11576-2-AP), MGEA5 (OGA) rabbit polyclonal antibody (#14711-1-AP) were obtained from Proteintech Group, Inc (Wuhan, China), and Anti-Phospho (Ser/Thr)-Phe rabbit polyclonal antibody was obtained from Abcam Inc (Cambridge, MA).
Adeno-associated virus (AAV) and stereotaxic injection
To overexpress human tau (hTau) in vivo, adeno-associated virus-mediated hTau (AAVhTau) and its control virus (AAVmcherry) were constructed by OBiO Technology (Shanghai, China). Titers were 3×1012 TU/ml. Mice were anesthetized with 60 mg/kg pentobarbital sodium, and then fixed in stereotaxic instrument (RWD Life Science Co., Shenzhen, China). Two holes were drilled, and AAV was microinjected into CA1 region of hippocampus (–2.5 mm posterior,±2.0 mm lateral and 2.5 mm ventral relative to bregma) [23]. The syringe attached to a nanoliter infusion pump (Legato130, KD Scientific, USA), and a volume of 0.6μl of AAV was microinjected into each hole at a rate of 0.2μl/min. The needle syringe was left for 5 min before withdrawal. After the skin was sutured and sterilized, mice were sent back to their home cage.
Drugs and treatment
DHA was dissolved in DMSO with a 50 mM concentration of storage solution and diluted with PBS to 5 mM for use. Alloxan was dissolved in PBS with a 12 mg/ml concentration for use. Mice were divided into 6 groups: AAVmcherry, AAVmcherry + DHA, AAVmcherry + DHA + Alloxan; AAVhTau, AAVhTau + DHA, and AAVhTau + DHA + Alloxan. For mice in the AAVmcherry + DHA and AAVhTau + DHA groups, they were treated with DHA (20 mg/kg/day, i.p.) from 3 weeks after AAV microinjection to the beginning of the behavioral test. For mice in the AAVmcherry + DHA + Alloxan and AAVhTau + DHA + Alloxan groups, an additional alloxan (75 mg/kg/day, i.p.) was used for another 9 days (Fig. 1).

Diagram illustrating the experimental protocols. Mice aged 8 weeks received AAVmcherry or AAVhTau stereotaxic injection into the hippocampal CA1 area on day 1. Three weeks after AAV microinjection, mice received daily DHA treatment (i.p.) for 50 days until behavioral tests began. After behavioral tests, mice were sacrificed for electrophysiological recordings and biochemical assay.
Open-field test
Mouse was put into an open box (40×40×60 cm) to test general motor ability and anxiety-like behavior. Each mouse was placed at the central area to explore freely for 10 min, and was recorded by using ANY-maze tracking system (Stoelting, USA)
Barnes maze test
The apparatus consists of a white circular platform 0.75 m in diameter, with 18 holes (5 cm in diameter) around, and an escape box was located underneath one of these holes. A CCD camera was suspended above the maze center to record the latency and count of errors of finding the escape box, and video outputs are digitized by ANY-maze video tracking system (Stoelting, USA). On day 1, all animals were allowed to explore the maze freely for 3 min to adapt. Following 4 days, animals were trained in spatial learning task for 4 trials per day, with a 15-min inter-trial interval. The trial was completed if the animal found and entered into the escape box within 3 min. If the animal failed to find the escape box, or it found the box but did not enter into the box within 3 min, it would be gently guided to the box and held there for 60 s before being returned to the home cage. Twenty-four hours after the final training trial, mice were placed back to the maze, and a 3-min probe test was performed with the escape box blocked.
Electrophysiological recordings
Mice (about 4 months old) were deeply anesthetized with urethane (1.5 g/kg, i.p.) and transcardially perfused with artificial cerebral spinal fluid (ACSF) (in mM: NaCl 124, KCl 2.8, NaH2PO4.H2O 1.25, CaCl2 2.0, MgSO4 1.2, Na-vitamin C 0.4, NaHCO3 26, Na-lactate 2.0, Napyruvate 2.0, and D-glucose 10.0, pH = 7.4) prior to decapitation. Then, hippocampal slices were coronally sectioned (400μm) with a vibratome (VT1200 S, Leica Microsystems, Bannockburn, IL) with 95% O2 and 5% CO2, and then were incubated in ACSF for 2 h at 35°C. A bipolar stimulating electrode was placed at the Schaffer collaterals of dorsal hippocampus CA3 pyramidal neurons, and a recording pipette filled with ASCF was placed at the ipsilateral striatum radiatum of the hippocampal CA1 area. The test excitatory postsynaptic potentials (EPSPs) were evoked at a frequency of 0.05 Hz and at stimulus intensity adjusted to around 50% of the maximal response size. After a 30-min stable baseline, theta burst stimulation (TBS) was given to induce LTP. TBS consisted of 2 trains of stimuli (at 20-s interval), with each train composed of 5 bursts (4 pulses at 100 Hz in each burst) at an inter-burst interval of 200 ms. Data acquisition was performed with the PatchMaster v2.73 software (HEKA Elektronik, Lambrecht/Pfalz, Germany).
Western blotting
After behavioral tests, the hippocampal tissues were collected and lysed in homogeneous buffer in a mortar and pestle on ice. Each protein samples (approximately 60μg) was boiled with 5× loading buffer at 95°C for 5 min. All protein samples were then separated on 10% SDS-PAGE gels for about 90 min, and then transferred onto an immobilon-PTM polyvinylidene fluoride (PVDF) membrane (Bio-Rad, Hercules, CA, USA). To block non-specific background, the membranes were incubated with 5% fat-free milk in TBS contains 0.1% Tween-20 for 90 min at room temperature or 37°C for 60 min. The target proteins were immunoblotted with primary antibodies overnight at 4°C.
The primary antibodies were re-collected and the membrane was washed with TBS contains 0.1% Tween-20. After then, corresponding horseradish peroxidase (HRP)-conjugated secondary antibody (1:5000, Perkin Elmer) was incubated at room temperature for 90 min, the blots were visualized in the Bio-Rad Imager using ECL Western blotting substrate (Pierce). GAPDH was set as the internal reference. The band intensity of each protein was quantified by the Bio-Rad Quantity One software as described previously [24].
Statistical analysis
All data were presented as mean±SEM. Two-group comparisons were performed by two-tailed Student’s t-test. The differences of spatial learning and memory in Barnes maze test among different groups were analyzed by two-way AVOVA with group as the between-subjects factor and training trials as the within-subjects factor. The data of other behavioral tests were analyzed by one-way ANOVA followed by Tukey’s post hoc test. Statistical significance was set as at *p < 0.05, **p < 0.01, ***p < 0.001.
RESULTS
DHA rescues learning and memory deficits in a mouse model of tauopathy
DHA is believed to alleviate cognitive impairment in AD. However, it is unclear whether DHA exerts beneficial effects on cognitive function and neuropathology in tauopathy. Given hTau overexpression in the hippocampus was sufficient to induce tau hyperphosphorylation [5, 25], we constructed adeno-associated virus carrying hTau cDNA (AAVhTau) or its control (AAVmcherry) to establish a mouse model of tauopathy through intrahippocampal microinjection. The results showed that neither hTau nor DHA affected spontaneous activity, as reflected by no alterations of total distance traveled were observed among these groups during open-field test (Fig. 2A). However, hTau overexpression in the hippocampus dramatically impaired spatial learning during the Barnes test, because the time for finding the escape box in hTau mice (AAVhTau, n = 16, p = 0.028 versus AAVmcherry; Fig. 2D) was much longer than those in control mice (AAVmcherry, n = 19). DHA treatment significantly shortened time for searching for the escape box (AAVhTau + DHA, n = 13, p = 0.913 versus AAVmcherry, p = 0.002 versus AAVhTau; Fig. 2D). Notably, pharmacologically blocking OGT by alloxan prevented the beneficial effects of DHA on spatial learning (AAVhTau + DHA + Alloxan, n = 16, p = 0.066 versus AAVmcherry, p = 0.051 versus AAVhTau,p = 0.011 versus AAVhTau + DHA; Fig. 2D). A long-term spatial memory probe test was performed 24 h after the last training trial with the escape box blocked. The results showed that memory retrieval was impaired in hTau mice since the escape latency to find the escape box was significantly longer compared to control ((AAVhTau, n = 16, p = 0.028 versus AAVmcherry; Fig. 2D), although the number of errors found escape box remained unchanged (p = 0.314 versus AAVmcherry; Fig. 2C). DHA treatment shortened the time to find the escape box (AAVhTau + DHA, n = 12, p = 0.322 versus AAVmcherry, p < 0.001 versus AAVhTau; Fig. 2D), whereas the beneficial effects of DHA on memory retrieval was partially blocked by alloxan (AAVhTau + DHA + Alloxan, n = 16, p = 0.027 versus AAVmcherry, p = 0.051 versus AAVhTau, p = 0.022 versus AAVhTau + DHA; Fig. 2B). Together, these results indicate that DHA plays a neuroprotective role in hTau-induced spatial learning and memory impairment in an OGT-dependent manner.
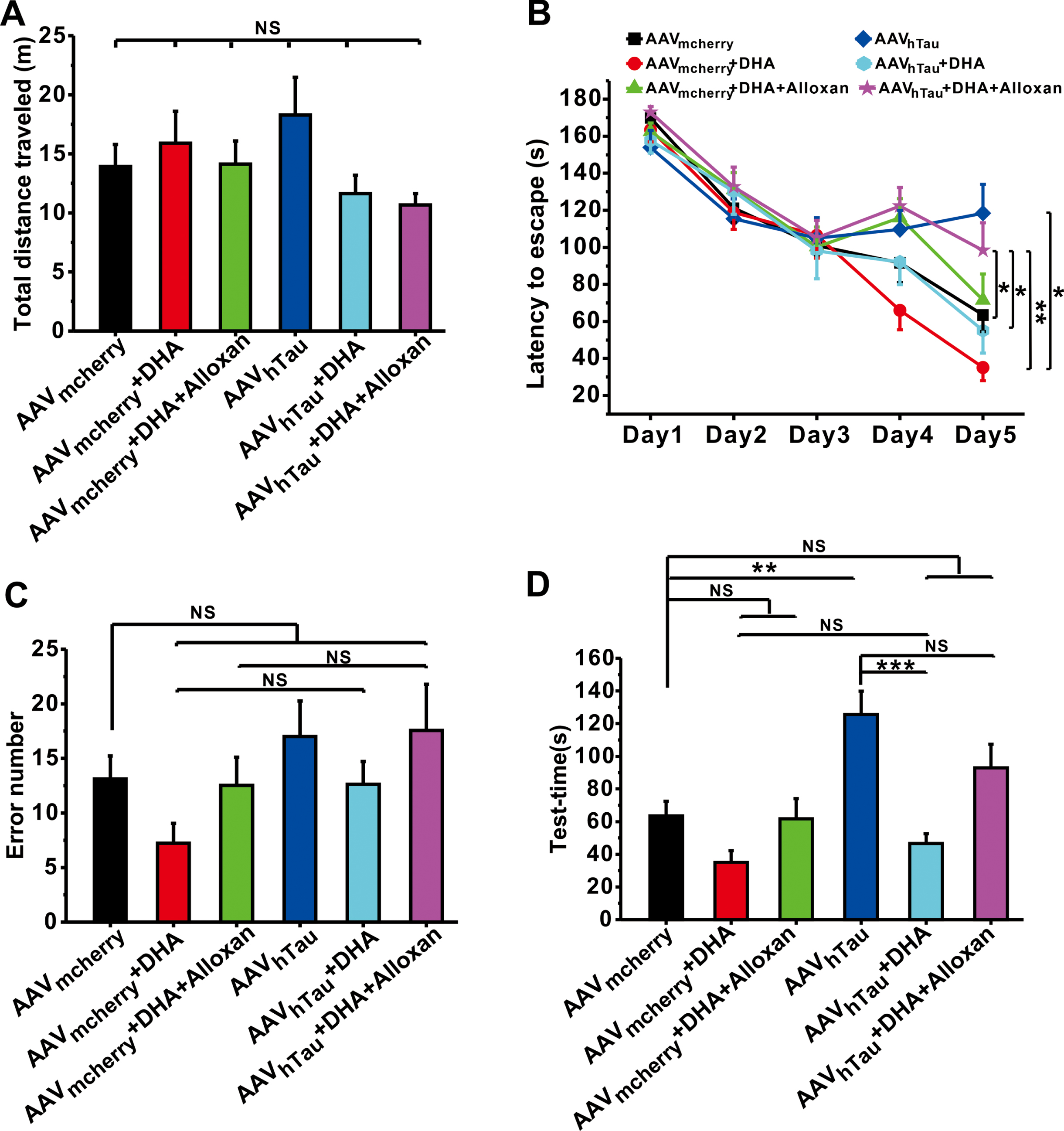
DHA rescues cognitive and memory deficits in hTau mice. A) Total distance traveled during Open-field test (n = 12–14). B) The latency to the escape box during the Barnes maze training (n = 13–19). C, D) The number of errors before finding the target hole (C), and the latency to find the target hole (D) on the test day in the Barnes maze test (n = 13–19). Data are expressed as mean±SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
DHA rescues hippocampal CA1 LTP in a mouse model of tauopathy
Hippocampal synaptic plasticity, especially LTP, is considered to be a major cellular mechanism underlying spatial learning and memory. Nevertheless, hTau accumulation induces the impairment of synaptic plasticity [26–28]. Aforementioned behavioral results show that DHA can ameliorate the impairments of spatial learning and memory in hTau mice. We therefore next wanted to determine whether DHA is able to rescue the impairment of synaptic plasticity in hTau mice. Electrophysiological data showed that a reliable hippocampal CA1 LTP was induced in control mice (AAVmcherry, n = 10 slices from 5 mice; Fig. 3). However, hTau overexpression significantly impaired LTP (AAVhTau, n = 8 slices from 3 mice, p < 0.001 versus AAVmcherry; Fig. 3). As expected, DHA treatment completely restored hippocampal CA1 LTP induction (AAVhTau + DHA, n = 8 slices from 4 mice, p = 0.055 versus AAVmcherry, p = 0.019 versus AAVhTau; Fig. 3), whereas alloxan treatment partially prevented the effect of DHA on hippocampal CA1 LTP (AAVhTau + DHA + Alloxan, n = 7 slices from 4 mice p = 0.002 versus AAVmcherry, p = 0.321 versus AAVhTau, p = 0.170 versus AAVhTau + DHA; Fig. 3).
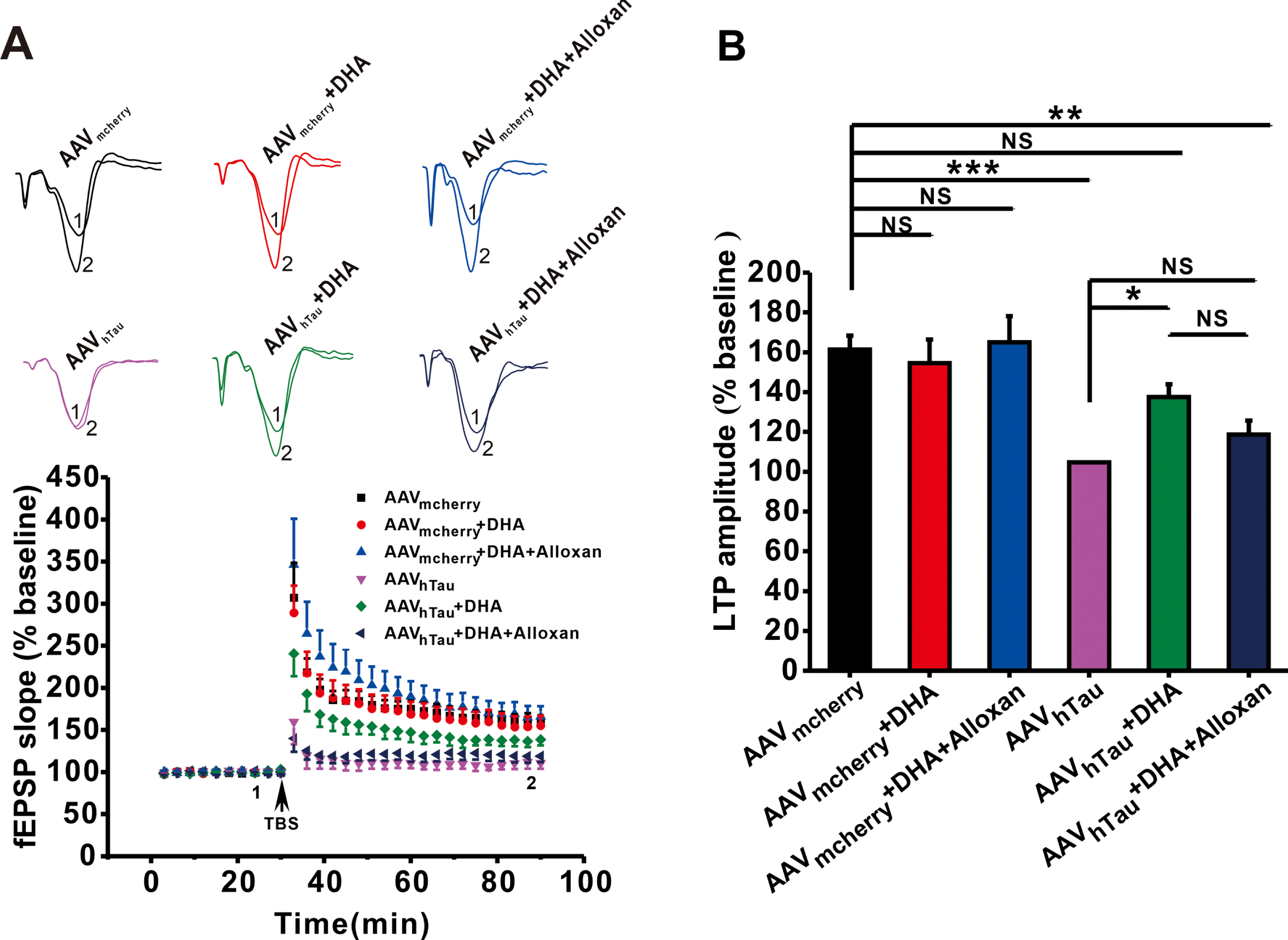
DHA restores hippocampal CA1 LTP in hTau mice. A) Representative traces of fEPSP, and the plots of the normalized slopes of the fEPSP 5 min before and 55 min after TBS delivery. B) Bar graphs of the average percentage changes in the fEPSP slope 55-60 min after TBS delivery (n = 7–10 slices from 3–5 mice in each group). Data are expressed as means±SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
DHA induces O-GlcNAcylation in the hippocampus
Behavioral and electrophysiological results show that OGT inhibitor alloxan is able to prevent the neuroprotective effects of DHA on learning and memory and hippocampal synaptic plasticity in a mouse model of tauopathy. Therefore, we next wanted to directly examine the effects of DHA on protein O-GlcNAcylation and phosphorylation level in the hippocampus. The results showed that AAVhTau mediated hTau overexpression (p = 0.048, AAVhTau versus AAVmcherry; Fig. 4A, B). Although DHA treatment did not affect Tau expression (p = 0.978, AAVhTau versus AAVmcherry; Fig. 4A, B), it reduced hTau-induced protein phosphorylation, leading to an increase in the ratio of O-GlcNAc/Phospho in hTau-overexpressed mice (p < 0.001, AAVhTau + DHA versus AAVhTau; Fig. 4A, C), which was probably attributed to the upregulation of the ratio of OGT and OGA after DHA treatment (p = 0.012, AAVhTau + DHA versus AAVhTau; Fig. 4A, D).
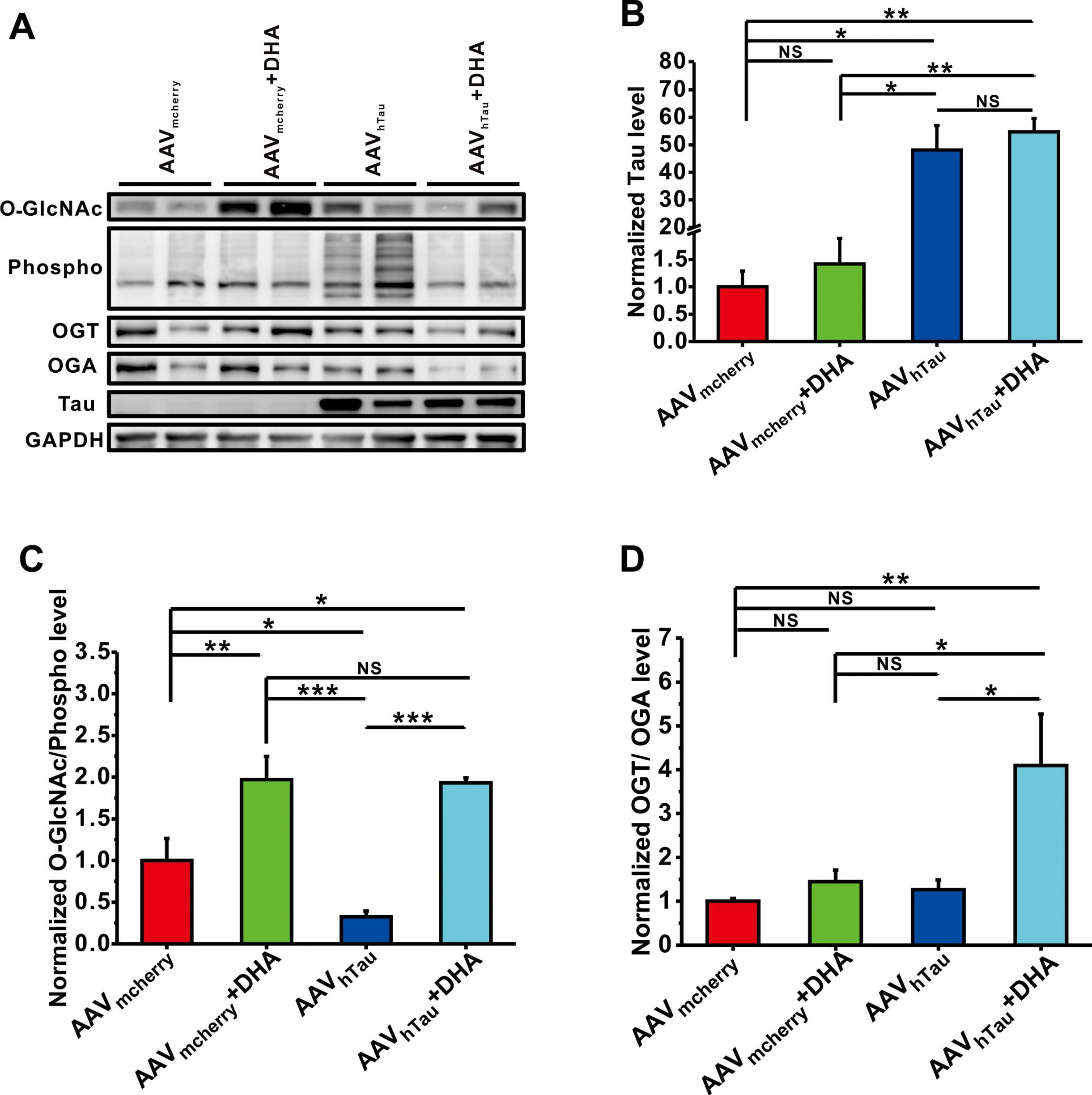
DHA suppresses phosphorylation by inducing O-GlcNAcylation in the hippocampus. The relative protein levels of total tau protein (B), O-GlcNAc/Phospho (C), and OGT/OGA (D) are normalized by control mice. Data are expressed as means±SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
Tau protein plays critical roles in maintaining the stability of microtubule structure under physiological condition. However, hyperphosphorylated tau protein causes misfolding and aggregation to form neurofibrillary tangles, leading to AD and many other neurodegenerative diseases, which are called tau tauopathies. Therefore, clearance of hyperphosphorylation of tau is able to prevent cognitive decline [29]. It has been documented that hTau overexpression could produce tau accumulation and mimic neurofibrillary tangles formation in APP transgenic mice [30]. Tau accumulation induces the impairment of synaptic plasticity and memory by inhibiting NMDA receptor expression [26], nuclear CaMKIV/CREB signaling [27], as well as PKA/CREB/BDNF/TrkB and PKA/GluA1 signals. Consistent with these reports, we have found that hTau overexpression mediated by AAVhTau intrahippocampal microinjection increased total protein phosphorylation level in the hippocampus, and consequently induces apparent impairments of synaptic plasticity (Fig. 3) and learning and memory (Fig. 2).
OGT and OGA promote O-GlcNAc cycling, in which OGT enhance O-GlcNAcylation while OGA do the opposite. Approximately total 4,000 proteins were predicted to have the potential of O-GlcNAcylation that could prevent protein aggregation. Previous studies have suggested a critical role of protein O-GlcNAcylation in neurodegenerative diseases [31–33]. For example, a reduction in the global O-GlcNAcylation levels was observed in the cortex and hippocampus of AD [3, 34]. O-GlcNAcylation of RIPK3 (receptor-interacting serine/threonine protein kinase 3) suppresses phosphorylation of RIPK3 and its interaction with RIPK1, thus inhibit neuron necroptosis and ameliorated AD pathology [35]. Consistently, we here reported that hTau microinjection leaded to a significant decrease in the ratio of O-GlcNAc/Phospho in the hippocampus (Fig. 4), which may induce the impairments of synaptic plasticity and learning and memory.
Previous reports have revealed that ART is able to rescue the deficits of learning and memory via reducing the deposition of P-tau in AD models [22]. Furthermore, ART can significantly suppress the reactive oxygen species production and phosphorylation of p38, ERK, mTOR, and ULK1 in a dose- and time-dependent manner [22, 36–39]. Based on these findings, we speculate that DHA, the active metabolite ART, may reduce phosphorylation by enhancing O-GlcNAcylation, and subsequently improve cognitive functions in hTau mice. It has been documented that splicing of both OGT and OGA results in intron retention, which is regulated by the changes of O-GlcNAc levels. High O-GlcNAc levels increase, while low O-GlcNAc levels decrease OGT retention in the nucleus, thereby decreasing or increasing OGT protein. Conversely, low O-GlcNAc levels increase nuclear retention of OGA [40, 41]. Meanwhile, there is a mutual regulation between OGT and OGA. Overexpression of OGA increases OGT transcription, whereas knockdown OGA significantly decreases OGT levels [41, 42]. Therefore, OGT/OGA would be the best way to reflect the pharmacological effects of DHA. Indeed, we here found that DHA treatment significantly increased the ratios of OGT/OGA and O-GlcNAc/Phospho in the hippocampus (Fig. 4), and may thereby alleviate the impairments of hippocampal LTP (Fig. 3) and learning and memory (Fig. 2), which is supported by previous finding that neuronal O-GlcNAcylation improves cognitive function in the aged mouse brain [3]. These findings suggest that DHA may participate in the crosstalk between O-GlcNAcylation and phosphorylation following the OGT/OGA signal axis. In addition to O-GlcNAcylation, DHA may promote learning and memory through other different ways. For instance, DHA ameliorates LPS-induced neuroinflammation in the hippocampus by inhibiting PI3K/AKT pathway [43], and improves learning and memory through promoting autophagic clearance for Aβ [44]. ART has ever been reported to strengthen GABAA receptor signaling pathway by stabilizing the interaction gephyrin-GABAA receptor in vitro [16]. Moreover, OGT is able to maintain the AMPA receptor expression including GluA2 and GluA3 subunits, thereby promoting synapse maturity in the hippocampus [45, 46].
Notably, although DHA exposures dramatically increase the protein O-GlcNAcylation in both hTau and control mice (Fig. 4), it can only improve the spatial learning and memory in hTau mice, but not in control mice (Fig. 2). One possibility is that O-GlcNAcylation is dysregulated in tauopathy, but it keeps relative higher level in normal conditions. Therefore, increased O-GlcNAcylation by DHA may not further enhance the stability or solubility of tau protein, thereby no beneficial or harmful to control mice. However, we here only measure the total phosphorylation and O-GlcNAcylation of protein rather than tau in the brain. Thus, future experiments directly examining the phosphorylation and O-GlcNAcylation of tau after DHA treatment will help determine whether DHA’s therapeutic effect in tauopathy can be attributed to Tau O-GlcNAcylation. In addition, we here studied the effects of DHA on synaptic plasticity and learning and memory in mice overexpressed exogenous tau by AAV microinjection. The effect of DHA on endogenous tau overexpression in hTau transgenic mice needs to be further explored in the future.
In summary, the present study demonstrates that DHA may induce O-GlcNAcylation and reduce phosphorylation, and thereby rescue hTau overexpression-induced synaptic and cognitive impairments, suggesting a potential therapeutic for learning and memory deficits associated with tau pathology.
Footnotes
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (NSFC) 82071395, 82001158 and 91749116, the National Natural Science Foundation of Chongqing cstc2020jcyj-zdxmX0004 and cstc2019jcyj-bshX0016, the Science and Technology Research Program of Chongqing Municipal Education Commission KJZD-K201900403 and Innovation Research Group at Institutions of Higher Education in Chongqing CXQTP19034. We are also grateful to other members in the Dong laboratory for the technical support and helpful suggestion.