
Abstract
Alzheimer’s disease (AD) is the most common form of dementia. Although AD is one of the most socioeconomically devastating diseases confronting humanity, no “curative” disease modifying drug has been identified. Recent decades have witnessed repeated failures of drug trials and have called into question the utility of the amyloid hypothesis approach to AD therapeutics design. Accordingly, new neurochemical processes are being evaluated and explored as sources of alternative druggable targets. Among these newly identified targets, neuroinflammation is emerging as a front-runner, and within the realm of neuroinflammation, the inflammasome, particularly the NLRP3 complex, is garnering focussed attention. This review summarizes current data and approaches to understanding the role of the NLRP3 inflammasome in neuroinflammation and AD, and systematically identifies and evaluates multiple targets within the NLRP3 inflammasome cascade as putative drug targets.
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia; estimates suggest that the incidence of sporadic AD will exceed 15 million by 2050 in North America alone [1]. Due to the aging demographic of the global population, AD is not only an immense socioeconomic burden [2], but also a major unmet global medical need [3]. Regrettably, the pathological alterations of AD start more than 20 years prior to the onset of symptoms [4], rendering the process of rationally designing targeted therapeutics into a formidable challenge; consequently, no disease modifying therapeutics are currently available to prevent or delay AD’s onset and progression.
Currently approved AD treatments include cholinesterase inhibitors such as donepezil, galantamine, and rivastigmine, as well as the N-Methyl-D-Aspartate receptor antagonist memantine [5, 6]. However, these treatments offer only temporary symptomatic improvement in mild-to-moderate AD, and do not alter the disease progression [7] or prevent neuronal death [8]. Numerous Phase III therapeutic clinical trials of disease modifying agents for AD, including therapeutics that target the amyloid hypothesis, have failed to demonstrate significant benefits—failures arising in part from the underlying complexity and heterogeneity of the pathobiology and pathophysiology involved in AD [9]. The development of disease-modifying drugs for AD is a neuropharmacological priority.
Recent research has shown neuroinflammation to play a major role in AD [10]. Hyperactivation of microglial cells causes activation of the nucleotide-binding oligomerization domain-like receptor (NLR) leading to chronic inflammation that drives AD pathogenesis [11]. A multiprotein complex, known as the NLRP3 inflammasome, is part of the innate immune system [12] responsible for induction of the inflammatory responses and pyroptosis [13], further contributing to neurodegeneration. Activated NLRP3 proteolytically activates caspase-1 to generate pro-inflammatory cytokines, including IL-1β and IL-18 [14].
Neuroinflammation caused by aberrant signaling of the NLRP3 inflammasome and subsequent production of excessive cytokines drives the development and progression of AD [15]. Hence, regulation of aberrantly activated NLRP3 inflammasome signaling may constitute a promising therapeutic approach to AD. Pharmacological inhibition of the inflammasome in preclinical rodent-based studies has shown significant therapeutic effects [16], highlighting the druggable potential of the NLRP3 inflammasome. Herein we review the NLRP3 inflammasome and its role in AD, from the mechanisms of NLRP3 activation to downstream inflammatory effectors. Finally, a review of the NLRP3 inflammasome as a therapeutic target for the treatment of AD is discussed.
ALZHEIMER’S DISEASE AND NEUROINFLAMMATION
Clinically, AD is categorized by age of onset [17]. Late onset AD (LOAD; people aged ≥65 years), accounts for 95%of all cases; early onset AD (EOAD, < 65 years of age) is substantially less common [18]. This skewed aged distribution is significant given the increase in inflammation and inflammatory-related disorders with age. Furthermore, a number of established risk factors for AD include traumatic brain injury (TBI), hypertension, physical inactivity, diabetes, cardiovascular risk factors, obesity [19], old age [20], and genetics [21]. Some of the modifiable risk factors function through overlapping mechanisms of neuroinflammation to increase the propensity for AD [22].
The core pathological hallmarks of AD are the accumulation of extracellular amyloid plaques composed of amyloid-β (Aβ) proteins and the intraneuronal accumulation of neurofibrillary tangles (NFTs) of aggregated and hyperphosphorylated tau. Although oligomeric Aβ accumulation in the brain is an early pathological marker of AD, it occurs up to 20 years prior to the onset of clinical symptoms [4]. Numerous therapeutic approaches targeting Aβ, including γ-secretase inhibitors, BACE inhibitors, and anti-Aβ monoclonal antibodies have failed in the clinic, raising doubts about the relevance of Aβ as a sole therapeutic target [23, 24]. Although the Aβ-targeting therapy aducanumab, has recently been given approval by the FDA [25], emerging research suggests that the accumulation of Aβ is multi-variable [3], and that the degree of cognitive decline does not correlate strictly with the magnitude of amyloid deposit [26].
Likewise, formation of hyperphosphorylated tau, NFTs, and synaptic and neuronal loss are directly linked to cognitive decline [27], and for many years were assumed to be responsible for immediate symptom-producing neuronal toxicity [28, 29]. However, recent animal studies observed neurodegeneration even in the absence of NFTs and overexpression of tau [30, 31], suggesting that the exact mechanism of tau-mediated neurotoxicity has yet to be fully elucidated [32]. Moreover, recently a Phase II trial of a tau-targeting antibody, semorinemab, was halted when it failed to meet its primary efficacy endpoint of reducing dementia. The failures of these clinical trials further highlight the inadequacies of currently accepted theories of AD.
Over the past decade an increasing appreciation of the role of chronic inflammation as one of the core pathological hallmarks of AD has emerged [33]. Triggers of neuroinflammation in AD are multiple, but include Aβ aggregates and NFTs, both of which activate inflammatory astrocytes and microglia, leading to increased levels of pro-inflammatory cytokines and chemokines [10]. Microglia, a sub-type of glial cells, play a central role in regulating the brain’s immune system and information transmission in the brain via synapses [34, 35]. Microglial dysfunction is an established contributor to neurodegeneration [36], with constant stimulation of Aβ deposits in AD leading to sustained continuous activation of microglial cells [37]. In response to minor injuries, activated microglia release anti-inflammatory cytokines and neuroprotective growth factors [38]. However, when microglia sense danger molecules secreted from damaged or dying cells (e.g., protein misfolding byproducts, Aβ aggregates, α-synuclein, prion protein, superoxide dismutase, extracellular ATP, complement proteins) [39], they become activated to release toxins and pro-inflammatory cytokines [38]. Due to this double-edged sword nature of microglial cells in AD, its neuroinflammatory response is beneficial only if it does not induce damage of neighboring areas [40]. In early AD, microglial response serves as a protective factor to mediate Aβ clearance and induce synaptic growth. As Aβ, pro-inflammatory cytokines, and toxins accumulate over time, microglial cells assume a pro-inflammatory phenotype, leading to chronic neuroinflammation [41]. Aβ-mediated activation of the NLRP3 inflammasome in microglia has also been implicated as a key player in the neuroinflammatory pathways associated with AD [42]. Therefore, targeting the NLRP3-induced inflammatory signaling pathways early in the initial stages of AD may be a rational neuroprotective strategy.
THE NLRP3 INFLAMMASOME IN NEUROINFLAMMATION
As the first line of defense against pathogenic threat, the innate immune response initiates downstream inflammatory responses through pattern recognition receptors (PRRs) mediated by recognition of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) [43]. The NOD-like receptor containing a pyrin domain 3 (NLRP3) inflammasome is an integral component of the cytoplasmic [44] multi-protein complex involved in the innate immune response to pathogenic threat [12]. This complex consists of a sensor NLRP3, ASC adaptor protein (adaptor molecule apoptosis-associated speck-like protein containing a CARD) and caspase-1 effector protein (Fig. 1) [45, 46]. The inflammasome regulates the activation of caspase-1 through ASC to induce production of IL-18 and IL-1β pro-inflammatory cytokines [14], eventually leading to pyroptosis via cleavage of gasdermin D (GSDMD) [13].

Structure of the NLRP3 inflammasome complex.
Enhanced expression of caspase-1 through aberrant activation of NLRP3 has been shown to play a role in the neuroinflammation of AD [46, 47]. Accumulating evidence has recognized activated microglia to express increased levels of interleukin 1β (IL-1β) in the extracellular deposits of Aβ [48]. IL-1β is a pro-inflammatory cytokine implicated in neurodegeneration [49]. In cerebrospinal fluid samples taken from AD patients, elevated IL-1β levels were observed [50], suggesting involvement of the downstream immune response mediators in AD progression. Furthermore, overexpression of the NLRP3 inflammasome has been identified in postmortem analysis of human AD brains [15] and has been shown to be involved in various inflammatory disorders including Parkinson’s disease [51], type 1 diabetes [52], and multiple sclerosis [53]. As the NLRP3 inflammasome appears to play a crucial role in inflammatory disorders, targeting this pathway may contribute to the development of novel treatment strategies for AD (Fig. 2).
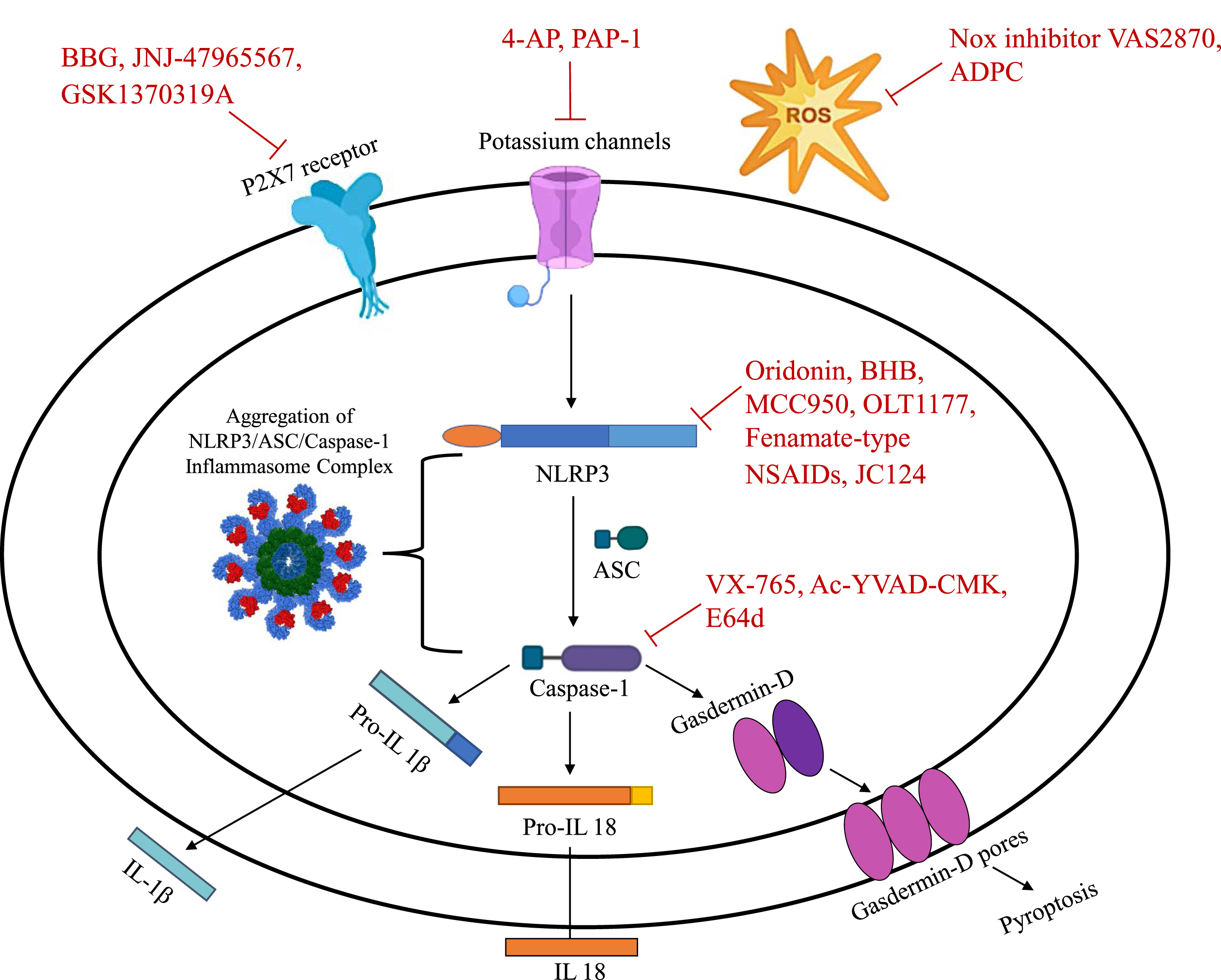
Pharmacological targets of the NLRP3 inflammasome.
MECHANISMS OF NLRP3 ACTIVATION
In AD, NLRP3 is activated in microglia when it senses dysregulation such as protein misfolding, Aβ aggregates, α-synuclein, prion protein, superoxide dismutase, extracellular ATP, or complement proteins [39, 54]. Canonically, activation of the NLRP3 inflammasome requires an initial priming signal followed by a second activation step. Exposure of macrophages to priming stimuli such as ligands for toll-like receptor (TLR), nod-like receptors (NLRs), and cytokine receptors activate nuclear factor kappa-B (NF-κB) which then constitutes a transcriptional role by increasing the expression of NLRP3, pro-IL-1β, and pro-IL-18 [46]. Binding of TLR4 ligand lipopolysaccharide to its receptor triggers the first signal, causing increased expression of inflammasome components NLRP3, procaspase-1, and pro-IL-1β. The priming signal is then mediated by adaptor molecules MyD88, IRAK1, and IRAK4 [55]. Via a second activation signal, NLRP can respond to various stimuli including extracellular ATP, bacterial toxins, potassium ionophores, and crystalline or particulate substances to become assembled and activated. The three main signaling events induced in response to various stimuli to active the NLRP3 inflammasome are K+ efflux, mitochondrial dysfunction causing reactive oxygen species (ROS) production, and lysosomal damage [46]. A subsequent activation signal results in generation of active caspase-1 that cleaves the pore-forming GSDMD and induces pyroptosis inflammatory cell death [55].
Several regulatory mechanisms are involved in the activation of the NLRP3 inflammasome. Although Muñoz-Planillo et al. reported that K+ efflux can activate NLRP3 [56], the recent identification of GB111-NH2, imiquimod, and CL097 as independent NLRP3 inflammasome activators suggest that K+ efflux is downstream from existing activation pathways [57, 58]. Ca2+ mobilization, Na+ influx, and Cl efflux were likewise initially implicated in inflammasome activation; however, alternative mechanisms have been proposed that suggest otherwise [46]. Thus, specific molecular based mechanisms for NLRP3 inflammasome activation are not fully understood and further elucidation is required.
EVIDENCE OF NLRP3 IN AD
Accumulating evidence has indicated the role of NLRP3 inflammasome in AD pathogenesis (Fig. 3). During disease progression, accumulation of Aβ aggregates associated with microglia triggers activation of the NLRP3 inflammasome [59]. Constitutive activation of the NLRP3 inflammasome by microglia in severe AD impairs microglial clearance of Aβ and NFT [60]. Oligomers and protofibrils of Aβ in primary microglia have recently been shown to trigger activation of the NLRP3 inflammasome, producing chronic levels of pro-inflammatory mediators such as IL-1β and IL-18 [61]. This chronic inflammation can further exacerbate the negative effects of senile plaques and thereby contribute to the progression of AD pathology [62]. Venegas et al. recently demonstrated that clusters of Aβ surrounding ASC specks in the extracellular matrix enhanced activation of NLRP3, IL-1β release, and generation of Aβ fibrils [63]. Ojala et al. reported the association of tau phosphorylation with Aβ-related inflammatory reaction and the induction of IL-18 production by Aβ [64]. Levels of IL-18 were also increased in mild AD patients compared to control [65], suggesting that this downstream pro-inflammatory molecule of the NLRP3 inflammasome pathway possibly plays a role in AD progression.
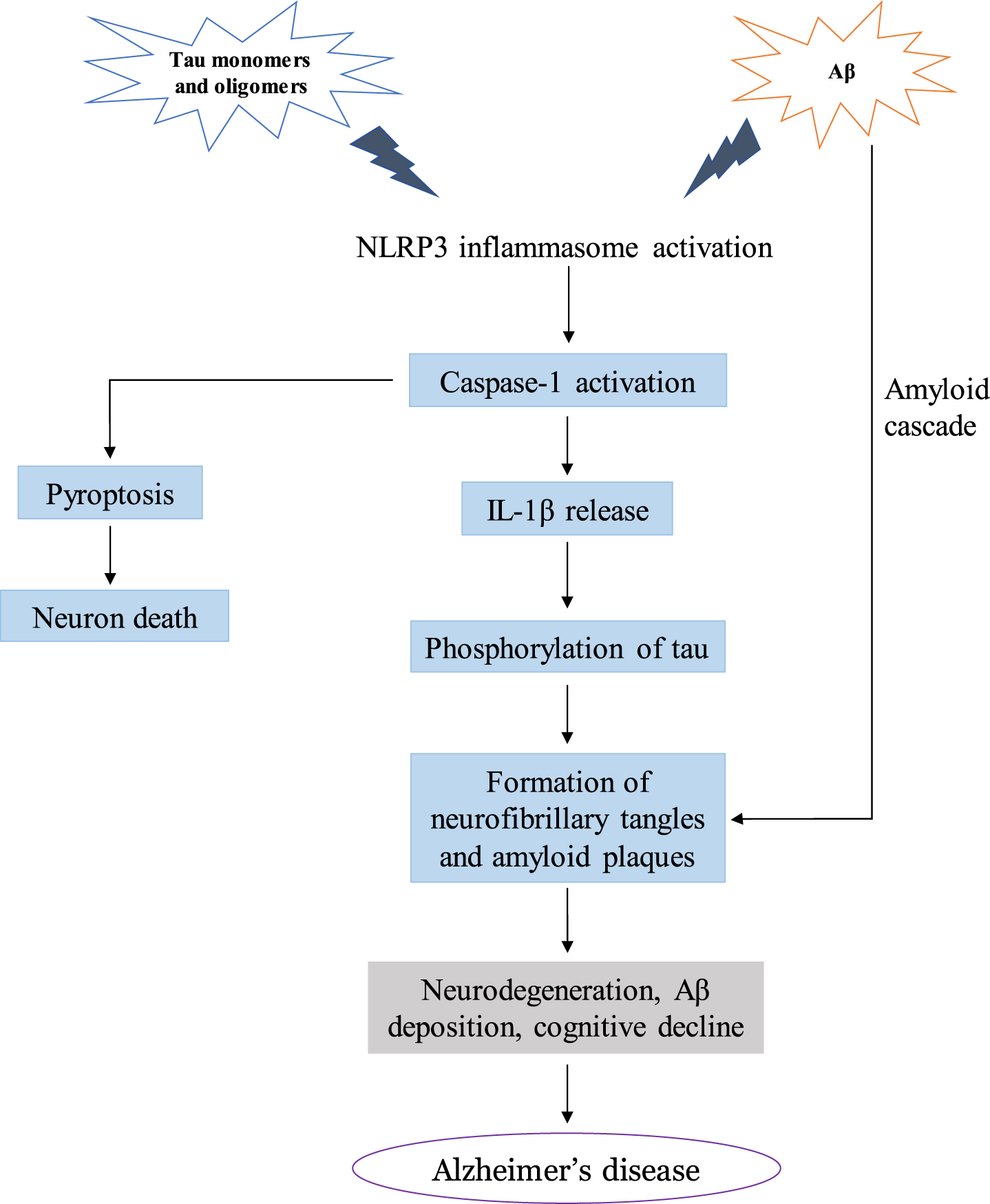
Mechanisms of NLRP3 inflammasome-mediated AD pathology.
Tau aggregates have also been observed to activate NLRP3 in primary microglia [66], implicating tau in exacerbation of AD neurodegeneration through chronic inflammation. In tau mice with a deficiency in either the NLRP3 gene (Nlrp3) or ASC gene (Asc), there was reduced activity of tau kinase and increased tau phosphatase, leading to attenuated tau pathophysiology and decreased progression of cognitive impairments [66, 67]. Transcriptome analysis studies in a tau-induced rat model demonstrated an increase in pro-inflammatory markers such as IL-1β [68]. Moreover, tau gene transfer caused microgliosis and neurodegeneration [69]. As indicated by these studies, chronic inflammation through aberrant NLRP3 activation may therefore enhance the neurodegenerative effects of tau.
Ising et al. found that patients with dementia-related proteinopathies such as frontotemporal dementia have elevated levels of cleaved caspase-1, ASC, and IL-1β [67]. Increased caspase-1 expression has been associated with risk of AD-related dementias [70], and inhibition of caspase-1 has been shown to reduce cognitive impairment and AD brain pathologies in an AD mouse model [71]. In a separate study, components of the inflammasome (NLRP3, PYCARD, caspase-1) and downstream cytokines (IL-1β and IL-18) were upregulated in both severe and mild AD [47], suggesting the involvement of activated NLRP3 inflammasome in AD pathogenesis.
In a study by Heneka et al., deletion of NLRP3 in a mouse model of AD diminished neuroinflammation and Aβ accumulation and prevented cognitive dysfunction [15]. Ablation of the NLRP3 inflammasome in mice conferred increased lifespan and attenuated age-related neurodegeneration [54], providing further support for the hypothesis that the NLRP3 inflammasome plays a key role in the progression of AD-related pathologies [15].
THERAPEUTIC APPROACHES TOWARD NLRP3
In AD, activated NLRP3 is responsible for the maturation of pro-caspase-1 [72]. Activated caspase-1 induces the innate immune response by cleaving and activating inflammatory cytokine precursors, pro-IL-1β and pro-IL-18 [73]. Additionally, caspase-1 triggers cleavage of GSDMD to induce pyroptosis which further releases pro-inflammatory molecules. These pro-inflammatory mediators result in the activation of downstream signaling pathways, causing chronic neuroinflammation and neuronal cell death [74, 75].
Despite a lack of drugs directly binding to and inhibiting the NLRP3 inflammasome, the presence of multiple activation points and involvement of several downstream signaling pathways provide promising pharmacological targets for AD (Fig. 2). Current therapeutic strategies targeting the NLRP3 axis consist of biologicals such as anakinra, an IL-1β antagonist, and canakinumab, a neutralizing monoclonal antibody [76]. However, the use of these drugs is limited due to high costs and low brain bioavailability [76]. Recently, new targets have been identified that play a prominent role in modulation of the NLRP3 inflammasome, representing new potential avenues for the development of disease-modulating AD therapeutics. As NLRP3 activation promotes neuroinflammation through release of proinflammatory molecules, downstream signaling pathways and mediators may also be valuable drug targets.
Considering the downstream effects of caspase-1, inhibiting its activation may diminish inflammation and pyroptosis. Likewise, preventing secretion of pro-inflammatory mediators downstream of NLRP3 and caspase-1 activation may be another approach by which chronic inflammation can be modulated in AD. Additionally, by building upon current data regarding mechanisms of NLRP3 as well as components of NLRP3, related studies can be used to translate the clinical efficacies of current drug trials and approved drugs for use in AD therapeutics.
Although exact mechanisms of NLRP3 activation and regulation are not fully elucidated, further research could allow for exploitation and modulation of NLRP3-associated pathways. We will review the role of potential inhibitors of the inflammasome, summarize current therapeutic findings, and suggest possible indications in AD pharmacotherapies (Table 1).
List of Compounds Targeting the NLRP3 Inflammasome Pathways
UPSTREAM TARGETS OF THE NLRP3 INFLAMMASOME ACTIVATION ION CHANNELS
P2X7 receptor
Postmortem analysis of AD patients identified enhanced expression of purinergic receptor P2X7 (P2X7R), an ATP-gated ion channel in both astrocytes and microglial cells [108]. Activation of P2X7R is triggered in response to extracellular particles including Aβ [109] and ATP released by impaired neurons, astrocytes, and microglia [110, 111]. ATP subsequently induces secretion of pro-inflammatory cytokines and chemokines such as IL-1β [112], tumor necrosis factor-α (TNF-α) [113], and chemokine ligand 3 (CCL3) [114]. Activation of P2X7R through prolonged ATP exposure triggers inflammasome activation, causing an excessive production of microglial IL-1β [114]. The downstream effect of P2X7R activation through cytokine release plays a critical role as a secondary step in AD pathogenesis. Although tonic activation of P2X7R has been associated with microglial proliferation [115], constitutive and aberrant activation of P2X7R results in pathologies, most prominently necrotic cell death through microglial membrane pore formation [116]. In the Tg2576 mouse model of AD, evidence of P2X7R overexpression was reported in astrocytes and microglia neighboring Aβ plaques [117]. Another study investigating APP/PS1 mice found Aβ-stimulated microglia to mediate excessive production of ROS through P2X7R activation, leading to neuronal damage [118]. Further in vivo evidence of the role of P2X7R in AD was demonstrated in a mice model of tauopathy in which co-localization of PX27R with astrocytes was determined [119]. In AD patient brains, there was enhanced expression of P2X7R in microglia, primarily in association with co-localized Aβ plaques [120]. Consistent with the aforementioned studies, in AD mice deficient in P2X7R, Martin et al. demonstrated reduced Aβ lesions as well as improved cognition and synaptic plasticity [108]. Deficiency in P2X7R also led to decreased levels of CCL3, the chemokine correlated with recruitment of CD8 + T-cells [108]. A study assessing transgenic mice reported that accumulation of T-cells due to CCL3 upregulation had detrimental cognitive impact [121]; therefore, reducing levels of CCL3 through P2X7R activity may have neuroprotective effects.
P2X7R antagonism
In early research on P2X7R pharmacological inhibition, Ryu et al. reported that administration of a P2X7R antagonist, brilliant blue G (BBG), in an animal model of AD reduced inflammatory reactivity and conferred neuroprotection [122]. Likewise, in a rat brain injected with Aβ, subsequent BBG administration led to neuroprotection and inhibition of neuroinflammation even in the presence of a P2X7R agonist [122]. In a study with APP transgenic mice, inhibiting P2X7R with BBG led to a decrease in Aβ in the hippocampus [123]. BBG was also shown to have neuroprotective and neuroregenerative effects in animal models of Parkinson’s disease [124]. Consequently, through preclinical studies highlighting the role of P2X7R in inflammasome activation, inhibition of the P2X7 signaling pathway may evolve to be a viable therapeutic intervention in the treatment of AD.
The potent CNS-permeant P2XR7-selective antagonist, JNJ-47965567, was partially successful in mitigating progression of ALS in female mice [79]. Another specific P2X7 antagonist, GSK1370319A, had neuroprotective effects through inhibition of IL-1β release from ATP-induced microglia [80]. Through the inhibitory effects of GSK1370319A on the effect of NLRP3 inflammasome assembly in microglia on IL-1β release, this mechanism could be used to modulate neurodegeneration progression. In a separate study, using GSK1370319A as a comparison, pyroglutamate-based P2X7R antagonism led to decreased production of IL-1β and ROS [125]. Further clinical trials of CNS-permeable P2X7R antagonists will be required to evaluate the feasibility of these therapeutics for the treatment of AD.
Potassium channels
The voltage gated K+ channel, Kv1.3, is involved in regulating microglial function [97]. In vitro data from activated microglial cells reveal increased expression of Kv1.3 for mediation of microglial priming by Aβ and production of ROS [126]. Immunohistochemical analysis of human brain cortices displayed higher expression of Kv1.3 channels in cortical microglia in AD patients compared to non-AD controls [126]. Likewise, increased Kv1.3 channels were also observed in transgenic mice microglia [127]. Moreover, microglia expressing Kv1.3 channels were found to be in close proximity to Aβ plaques, thereby suggesting an association between Kv1.3 and AD pathogenesis [126].
Franciosi et al. demonstrated that a broad-spectrum voltage gated K+ channel inhibitor, 4-aminopyridine (4-AP), suppressed microglial activation and subsequent neuronal death [128]. As 4-AP can also be used to inhibit Kv1.3 channels to suppress microglial priming, this supports Kv1.3 as a viable target for AD therapeutics design [129]. Maezawa et al. used a Kv1.3 inhibitor 5-(4-phenoxybutoxy)psoralen (PAP-1) to demonstrate involvement of Kv1.3 in Aβ-induced neuroinflammatory microglial responses [127]. Extrapolating from these studies, it can be hypothesized that targeting the Kv1.3 channel will have potential for reducing AD pathologies caused by pro-inflammatory and Aβ-induced neurotoxic microglial responses [127].
The stimulatory effect of cytosolic K+ efflux has been identified as a modulator of NLRP3 inflammasome activation [56]. Stimuli such as extracellular ATP [130] and factors causing phagosomal destabilization reduce levels of cytosolic K+ [131]. Reduced extracellular ATP concentrations activate P2X7R to cause influx of Ca2+ and Na+ and efflux of K+ [132]. This is one of the various conditions that deplete cytosolic K+ to initiate NLRP3 signaling. Greaney et al. reported bacterial endotoxins also elicit K+ efflux to trigger NLRP3 activation, while toxin-deficient bacteria are unable to activate the inflammasome [133]. Investigating the molecular mechanisms of K+ depletion as a trigger for NLRP3 inflammasome activation may allow for the development of novel AD treatments.
Considering the widespread role of voltage gated K+ channels in neuronal and cardiac muscle excitability (and other non-excitable tissues), drugs targeting Kv1.3 channels may be susceptible to unwanted off-target effects. Further investigation is needed to establish the specific NLRP3 inflammasome pathways downstream of K+ channels to determine the exact role of K+ channels in NLRP3 driven AD pathogenesis.
Madry et al. recently identified another K+ channel, THIK-1, a two-pore domain channel expressed in microglia cells, thought to play a role in microglial IL-1β release [134]. However, further studies are required to assess the regulatory mechanisms of THIK-1 in neuroinflammation to decipher its role in AD.
REACTIVE OXYGEN SPECIES
Evidence has been put forth on the contributions of Aβ-induced oxidative stress-induced neurotoxicity in AD pathogenesis [135]. Oxidative stress, a process associated with aging brain, arises from an imbalanced redox state caused by impaired antioxidant defense or increased oxidants [135, 136]. Generation of free radicals in AD is affected by various factors [137] including damaged mitochondria [138], chronic and neurotoxic microglial activation [139], and Aβ plaques [140]. Excess free radical production is cycled through a toxic cascade of Aβ-induced oxidative mitochondrial damage resulting in DNA damage and impaired antioxidant defense mechanisms [141]. Lysosomal destruction, ROS production, and K+ efflux are some of the various signals which trigger activation of the NLRP3 inflammasome [12].
Assembly of the NLRP3 inflammasome components is mediated through an increase in the cytoplasmic concentration of Ca2+ [142]. ROS-dependent Ca2+ influx through the TRPM2 channel triggers activation of the NLRP3 inflammasome, thereby causing secretion of IL-1β [143]. As demonstrated by Aminzaeh et al., activation of the NLRP3 inflammasome is mediated by ROS-dependent Ca2+ influx via the TRPM2 [143]. It was also demonstrated that inhibition of Ca2+ influx consequently decreased production of IL-1β through blockage of Aβ-induced NLRP3 inflammasome activation [143]. They also reported an increase in NLRP3 inflammasome activation and IL-1β production in response to ROS [143].
By targeting sources of ROS production, such as NADPH oxidase [144], specific inhibitors may be used to prevent chronic activation of NLRP3. Inhibition of NADPH oxidase by ROS inhibitors including the novel Nox inhibitor VAS2870 [145] and ammonium pyrrolidinedithiocarbamate (APDC) [146] were demonstrated to reduce levels of IL-1β, possibly via inhibition of NLRP3 activation [143]. Deficiency in the TRMP2 channel was shown to prevent maturation of pro-caspase-1 [143], and significantly decrease levels of NLRP3 inflammasome activation and IL-1β production [147]. As NLRP3 activation depends partly on TRMP2 channels activated through ROS and NADPH oxidase [147], targeting TRMP2 may be an effective therapeutic strategy for the treatment of NLRP3-associated inflammatory diseases including AD. Although TRPM2 is highly expressed in the brain, it is also found in various tissues including bone marrow, heart, spleen, and liver. Modulation of this channel may therefore have off-target effects. Further research on precise mechanisms of TRPM2 in AD are needed to justify safe targeting of this pathway.
Despite unclear mechanisms of oxidative stress induced NLRP3 activation, studies have identified association of cathepsin B (CTSB) with oxidative stress. Bai et al. demonstrated the involvement of CTSB in mediation of H2O2 induced inflammasome activation [148]. It was also found that pharmacological inhibition of CTSB hindered caspase-1 maturation and thus, decreased IL-1β release even in the presence of H2O2 [148]. There have also been studies reporting accumulation of CTSB in areas of increased oxidative stress with microglia mediated Aβ plaques [149]. In a mouse model of AD, it was found that inhibition of CTSB improved memory deficits and attenuated Aβ plaques [150, 152]. A cathepsin B inhibitor, E64d, was orally administered in a mouse model of AD, and was observed to reduce Aβ deposition and enhance memory, suggesting it may have potential value for therapeutic use in AD [152]. H2O2 induces IL-1β secretion through CTSB and NLRP3 signaling [148]; thus, preventing release of CTSB may halt NLRP3 inflammasome mediated neuroinflammation in AD.
NEK7
NIMA-related kinase 7 (NEK7) is a crucial mediator situated upstream in the NLRP3 inflammasome activation pathway [153]. Although recent evidence has considered NEK7 unlikely to be the sole activator of the NLRP3 inflammasome [154], it is thought to be involved in modulation of upstream NLRP3 signals, or in the facilitation of NLRP3 and ASC interactions [155]. Several studies have recently identified NEK7 as a mediator of K+ efflux in NLRP3 inflammasome assembly [155]. Chen et al. reported the role of NEK7 as a modulator of NLRP3 assembly and in conducting neuroinflammatory signals triggered by K+ efflux through mediation of NEK7-NLRP3 assembly, caspase-1 maturation and pyroptosis in damaged neurons [153]. It was also demonstrated that in response to NLRP3-triggering stimuli, cells lacking NEK7 had reduced caspase-1 activation and IL-1β production [156], thus supporting the role of NEK7 as a mediator in NLRP3 activation. In vivo downregulation of NEK7 in a mouse model of TBI improved neurological deficits, decreased NLRP3 and caspase-1 activation, and attenuated neuronal damage [153]. Knockdown of NEK7 inhibited NLRP3 inflammasome activation and caspase-1 activation to prevent pyroptosis [153]. Increased interactions of NEK7 and NLRP3 may therefore explain aberrant NLRP3 inflammasome signaling [153]. Furthermore, Song et al. reported phosphorylation of a serine residue by NEK7 to be a crucial priming event for the signal-2 induced assembly and activation of the NLRP3 inflammasome [157]. Considering the enhanced pro-inflammatory pathogenesis occurring downstream of NLRP3 signaling events, targeting NLRP3 phosphorylation by NEK7 may be a promising therapeutic intervention for AD [157].
NEK7 inhibition
Oridonin is an active diterpenoid [158] derived from the traditional Chinese medicine, Rabdosia rubescens [159]. It has recently gathered attraction as a neuroprotective agent for its potent anti-inflammatory and neurological effects. He et al. reported oridonin’s mechanism of action to be through covalent inhibition of the NLRP3 inflammasome [159]. Oridonin treatment suppressed interaction of NLRP3 and NEK7, a crucial step for inflammasome assembly and for subsequent NLRP3 oligomerization and ASC recruitment [159]. In vivo oridonin treatment in the presence of an NLRP3 agonist, monosodium urate crystals (MSU), alleviated IL-1β production and inhibited the NLRP3-induced inflammatory response [159]. In contrast to previously discussed inhibitors of the inflammasome which targeted the NLRP3 ATPase activity, oridonin interacts directly with NLRP3 to suppress its activation without affecting upstream signaling pathways, such as K+ leakage or mitochondrial dysfunction [159]. In an AD mouse model, oridonin inhibited microglial activation and subsequent inflammatory cytokine release, thereby alleviating cognitive impairment [160] and preventing synaptic loss [161]. It was also found to enhance expression of nerve nuclear growth factor (NGF), critical for neuronal differentiation and survival, suggesting a potential neuroprotective effect of oridonin [158]. Despite the associated risk of idiosyncratic toxicity and hypersensitivity associated with covalently binding drugs, many have previously been approved for human administration [162]. Furthermore, oridonin’s mechanisms of action as a covalent drug may potentially be pharmacologically beneficial as a long-term therapy for AD due to its increased duration of action and potency [162, 163].
DIRECT INHIBITORS OF THE NLRP3 INFLAMMASOME ACTIVATION
MCC950
MCC950 is a small molecule inhibitor of the NLRP3 inflammasome, which functions by binding to NLRP3, blocking its ability to hydrolyze ATP, and thus preventing it from maintaining its energy-dependent active structural conformation, thereby inhibiting NLRP3-induced ASC oligomerization and reducing cleavage of caspase-1 [164]. MCC950 displayed neuroprotective effects in models of TBI by reducing levels of caspase-1 and IL-1β [164]. Following TBI, daily intraperitoneal administration of MCC950 reduced microglial NLRP3 inflammasome activation, decreased IL-1β release, and improved cognitive function [165]. Stancu et al. demonstrated that MCC950 inhibited tau aggregates from inducing activation of IL-1β, thereby preventing tau-mediated pathology [66]. Hull et al. observed in a mouse model of frontotemporal dementia that MCC950 reversed inflammatory and frontotemporal dementia [166]. Dempsey et al. found that MCC950 attenuated Aβ accumulation [167], while Qi et al. concluded that in a rat model of AD, MCC950 improved impairment in synaptic plasticity [168]. Based on preclinical studies on MCC950, a related brain penetrant NLRP3 inhibitor, inzomelid, recently completed a Phase I clinical trial. Although inzomelid was evaluated against Cryopyrin-associated periodic syndrome [169], its successful pharmacokinetic and safety profile may accelerate future trials of similar drugs for use in AD.
OLT1177
OLT1177 is an active β-sulfonyl nitrile small molecule that prevents the non-canonical and canonical activation of the NLRP3 inflammasome by directly binding and inhibiting NLRP3 ATPase activity [16]. In vitro analysis observed that nanomolar concentrations of OLT1177 directly reduced IL-1β and IL-18 release [170]. In a mouse model of AD, OLT1177 was also shown to reverse cognitive dysfunction and to reduce microglia activation and cortical plaque deposition [97]. The pharmacokinetic and safety profile of OLT1177 has already been confirmed in a Phase I trial [171], suggesting it may be a possible therapeutic option for AD.
Fenamate-type non-steroidal anti-inflammatory drugs (NSAIDs)
Fenamate-type NSAIDs were found to selectively inhibit the NLRP3 inflammasome [99]. In a mouse model of AD, fenamate NSAIDs inhibited cognitive impairment and demonstrated both protective and therapeutic efficacies [171]. Diclofenac, a structurally related NSAID, was similarly shown to reduce levels of IL-1β [172]. Based on the clinical availability of fenamate NSAIDs, repurposing them as NLRP3 inhibitors may be a rapid therapeutic AD treatment strategy [99].
JC124
JC124, a specific small molecule inhibitor of the NLRP3 inflammasome [101], was recently observed to selectively suppress inflammasome formation by inhibiting ASC aggregation and caspase-1 activation thereby decreasing in vitro and in vivo levels of IL-1β. [173] In a transgenic mouse mode of AD, JC124 treatment exhibited neuroprotective effects by decreasing levels of Aβ, increasing synaptophysin expression, and attenuating oxidative stress [174]. Furthermore, JC124 treatment following traumatic brain injury provided neuroprotection by decreasing expression levels of NLRP3, ASC, IL-1β, and caspase-1 to reduce neuroinflammation [101].
DOWNSTREAM TARGETS OF THE NLRP3 INFLAMMATORY CASCADE
ASC
Activation of inflammatory sensors triggers ASC polymerization and accumulation of ASC specks [175]. ASC specks recruit and cause activation of pro-caspase-1 and subsequent IL-1β maturation and pyroptosis [175]. Venegas et al. reported that supernatants from mutant ASC macrophages did not support Aβ aggregation [63]. Moreover, hippocampal administration of ASC in transgenic mice enhances Aβ accumulation and aggregation, an indication of ASC cross-seeding in Aβ aggregation [63]. In a mouse model of AD, ASC-Aβ complexation elevated with age whereas this interaction was absent in control mice. Likewise, analysis of human brain samples exhibited similar results showing increased ASC-bound Aβ with age, confirming the association between seeding and spreading of Aβ [63]. While exogenous ASC administration in primary mouse microglia mutants was reported to induce activation of the NLRP3 inflammasome, mice mutant in ASC showed enhanced cognition and neuroprotection. Additionally, ASC-bound Aβ amplified NLRP3 inflammasome activation and led to pyroptosis compared to either ASC or Aβ alone [176]. Moreover, microglial Aβ clearance was also dysfunctional in the presence of extracellular ASC, therefore contributing to microglial toxicity [176]. Based on these findings, it may be possible to pursue NLRP3 inflammasome activation through ASC-speck release as a druggable target for sporadic AD [63]. Further studies to identify precise pathophysiological associations of the inflammasome and ASC are required to advance the development of effective therapeutic strategies for AD.
Modulation of caspase-1
Upon inflammasome activation, enzymatic action of caspase-1 triggers cleavage of IL-1β and IL-18 precursors to their active forms and induces pyroptosis [177]. Excessive pyroptosis, inflammatory cell death, characterized by cell lysis and IL-1β release, leads to aberrant inflammatory responses involved in neurodegeneration [178]. Mice deficient in caspase-1 and impaired IL-1β release were resistant to endotoxic shock [179], suggesting the potential value of caspase-1 inhibitors in treatment of inflammatory pathologies.
Interesting results have emerged from a small molecule prodrug, VX-765, which works by inhibiting caspase-1. In transgenic mice, VX-765 was shown to reverse episodic and spatial memory deficits, prevent Aβ accumulation, and reduce neuroinflammation [71] As clinical safety of VX-765 was confirmed in Phase II trials of epilepsy, this suggests it may be an effective therapeutic intervention in mild-cognitively impaired AD patients [71] Follow-up experiments assessing VX-765 concluded its benefit in both AD treatment and prevention [180].
A selective irreversible inhibitor of caspase-1, Ac-YVAD-CMK was shown to attenuate release of IL-1β and IL-18 [181]. In Parkinson’s disease rat models, Ac-YVAD-CMK inhibited expression of the NLRP3 inflammasome, thus indicating that downstream inhibition of the NLRP3/caspase-1/IL-1β axis may be a possible anti-inflammatory treatment of neurodegenerative diseases [182]. Highlighting the contribution of inflammation in age-related memory impairments, administration of Ac-YVAD-CMK in rats increased age-related hippocampal neurogenesis [183]. Infusion of Ac-YVAD-CMK in aged rats was also shown to decrease hippocampal IL-1β and improve memory [184].
OTHER TARGETS
Wendeln et al. found that a single administration of lipopolysaccharide (LPS) in an AD mouse model prior to Aβ accumulation led to priming of microglia and increased Aβ aggregated deposition six months later. However, when LPS was administered at four separate time-points, microglial tolerance was developed and Aβ deposition was attenuated [185]. Administration of TLR ligands in AD mouse models reduced Aβ neurotoxicity and NFTs and improved cognition [186, 188]. Therefore, phagocytosis mediated by the microglial TLR response can be exploited for targeting in the AD therapeutics design.
β-Hydroxybutyrate (BHB) is one of the primary ketone bodies that inhibit the NLRP3 inflammasome. Recently, in a mouse model of AD, Shippy et al. found that exogenous BHB decreased overall AD pathology by attenuating the formation of Aβ plaques, microgliosis, ASC formation, and activation of caspase-1 [189]. In addition, they observed that BHB administered in water effectively crossed the blood-brain barrier, thereby increasing its therapeutic potential as a treatment strategy for AD [189].
CONCLUSION, CHALLENGES, AND FUTURE THERAPEUTIC\\ STRATEGIES
Despite the major medical burden of AD, there are currently no effective disease modifying therapies. Historically, the main explanation for the neurodegeneration of AD was the aggregation and accumulation of Aβ and tau. However, the failure of more than 200 drug candidates as therapies for AD shines a spotlight on the need for new approaches and new therapies.
There is mounting evidence regarding the role of neuroinflammation in the pathogenesis of AD with NLRP3 inflammasome being a potentially druggable target rich zone within the neuroimmune-neuroinflammation cascade. From the initial discovery of the inflammasome in the early 2000 s to our current knowledge, its precise molecular workings remain incompletely elucidated. Greater investigation is required to identify microglial signaling cascades and to determine modulatory mechanisms as potential therapeutic approaches in AD; further research on the NLRP3 inflammasome and associated regulatory mechanisms are needed to explore the utility of this target for AD.
Although numerous compounds have been successful in targeting NLRP3 in vitro and in vivo, their specific clinical efficacies and applications to AD remain far from established. A considerable body of evidence supports the role of inflammasome over-activation in AD pathology, but relatively little is known about the beneficial or detrimental clinical outcomes of targeting the inflammasome. There remains a significant knowledge gap pertaining to the characterization of signaling pathways and the implications of directly targeting these pathways. Additional investigation is required to control for non-NLRP3 inflammatory signaling and to evaluate the applications of molecular targets of NLRP3 in the treatment of AD.
One of the greatest challenges in developing NLRP3-targeted therapeutics is and shall continue to be the lack of blood-brain barrier permeability. To accelerate pharmacokinetic profiling, repurposing small molecule drugs may be a potential option. Recently, novel therapeutic strategies are being explored, such as nanotechnology, which mitigate the brain permeability limitation. However, recent work has demonstrated that the nanomaterial properties are themselves capable of inducing NLRP3 inflammasome activation [190]. Further research is therefore required to test various nanoengineered nanomaterials for drug delivery optimization.
To enhance future drug development research on NLRP3 inflammasome targets, there needs to be greater knowledge on the molecular targets and their interactions with innate and adaptive immune mediated responses. As the NLRP3 inflammasome is involved in various diverse peripheral cellular signaling events, lack of specificity is a major limitation in AD drug development. Understanding the detailed mechanisms of the NLRP3 inflammasome in AD is crucial in the assessing implications of different molecular agents on inflammatory signaling pathways and to enable the prediction of off-target effects. Additionally, early intervention in AD using modulators of the inflammasome will require further studies regarding long-term health implications. A greater understanding on the exact regulatory mechanisms of NLRP3 in AD is pivotal for paving the road towards successful development of targeted pharmacotherapies in AD.