
Abstract
Background:
A significant subset of patients with Alzheimer’s disease (AD) exhibit low bone mineral density and are therefore more fracture-prone, relative to their similarly aged neurotypical counterparts. In addition to chronic immune hyperactivity, behavioral dysregulation of effector peripheral sympathetic neurons—which densely innervate bone and potently modulate bone remodeling—is implicated in this pathological bone reformation.
Objective:
Thus, there exists a pressing need for a robust in vitro model which allows interrogation of the paracrine interactions between the putative mediators of AD-related osteopenia: sympathetic neurons (SNs) and mesenchymal stem cells (MSCs).
Methods:
Toward this end, activated SN-like PC12 cells and bone marrow derived MSCs were cultured in poly(ethylene glycol) diacrylate (PEGDA) hydrogels in the presence or absence of the AD-relevant inflammatory cytokine tumor necrosis factor alpha (TNF-α) under mono- and co-culture conditions.
Results:
PC12s and MSCs exposed separately to TNF-α displayed increased expression of pro-inflammatory mediators and decreased osteopontin (OPN), respectively. These data indicate that TNF-α was capable of inducing a dysregulated state in both cell types consistent with AD. Co-culture of TNF-α-activated PC12s and MSCs further exacerbated pathological behaviors in both cell types. Specifically, PC12s displayed increased secretion of interleukin 6 relative to TNF-α stimulated monoculture controls. Similarly, MSCs demonstrated a further reduction in osteogenic capacity relative to TNF-α stimulated monoculture controls, as illustrated by a significant decrease in OPN and collagen type I alpha I chain.
Conclusion:
Taken together, these data may indicate that dysregulated sympathetic activity may contribute to AD-related bone loss.
INTRODUCTION
Alzheimer’s disease (AD) is a terminal neurodegenerative disorder characterized by progressive memory loss, confusion, and in developed cases, loss of executive function and mental faculties [1]. Osteoporosis, stemming from deviations in osteo-homeostatic processes toward bone reformation, results in a significant loss in bone mineral density (BMD) and increased fracture risk [2]. While these two age-related disorders initially appear pathologically distinct, multiple preclinical and retrospective clinical studies have suggested their notable and, potentially, mutually antagonistic relationship [3–6]. AD patients present with, on average, significantly diminished BMD relative to their sex- and age-matched neurotypical counterparts and are over twice as likely to sustain debilitating hip fractures [7]. Select proinflammatory cytokines known to be systemically upregulated in AD patients; indeed, tumor necrosis factor alpha (TNF-α) levels in the cerebrospinal fluid (CSF) and serum of AD patients are significantly upregulated and correlated with disease progression [8], directly potentiate bone reformation in part through synergistic interactions with receptor activator of nuclear factor kappa-B ligand (RANKL), upregulating osteoclast differentiation [9]. TNF-α has also been shown to indirectly suppress osteoblast production and proliferation by inhibiting insulin-like growth factor-1, osterix (OSX), Wnt, and runt-related transcription factor 2 (RUNX2) signaling [10–12].
In addition, BMD loss in AD also appears to be due to a complex, dysregulated, and incompletely understood interplay between systemic inflammation and local neural inflammatory activity [13, 14]. Indeed, local chronic immune hyperactivation and behavioral dysregulation of effector peripheral sympathetic neurons (SNs), which densely innervate bone and potently regulate bone remodeling, have been implicated in exacerbating this pathology [15]. Available in vivo data suggest that chronically activated SNs may prompt pathological increases in bone reformation largely via paracrine interactions with osteoblasts and their precursors [16, 17]. Elevated noradrenergic signaling prompts differentiation of osteoclasts via upregulation of RANKL by stimulating osteoblast β-adrenergic receptor β2AR [18]. Increased expression of dopamine β-hydroxylase, a rate-limiting enzyme in the biosynthesis of catecholamines, likewise induces bone reformation, and its genetic ablation in mice was found to markedly increase osteoblast proliferation and activity, and subsequently, bone mass [19]. Central infusion of neuropeptide Y (NPY) in mice was found to induce bone reformation like that seen in leptin-deficient cohorts, and conversely, selective knockout of NPY receptor Y2 stimulates osteoblast activity, resulting in increased cortical and trabecular bone mass [20].
Under normal physiological conditions, bone marrow derived human mesenchymal stem cells (hMSCs) directly mediate osteogenesis during the regenerative phase of the bone reformation cycle. In response to a complex milieu of mechanical and biomolecular stimuli, including transforming growth factor-beta (TGF-β), RUNX2, and peroxisome proliferator-activated receptor-γ signaling [21], these osteo-progenitors migrate, proliferate, and subsequently differentiate into osteoblasts [22]. However, multiple studies have shown that chronic exposure to pro-inflammatory microenvironments, like those in AD patients, inhibits mesenchymal stem cell (MSC) osteogenesis and impedes their ability to mediate inflammation remission [23, 24]. This, combined with an aging-driven increased propensity toward adipogenic lineage commitment [25], may tip the osteo-regulatory scale toward pathological bone reformation.
Despite the abundance of research regarding possible mechanisms underlying AD-related BMD loss, comparatively little is known about how these mechanisms may be exploited for designing effective therapeutic interventions. The current clinical “gold standard” for stimulating functional regeneration of damaged bone tissue in this patient segment involves the implementation of harvested autografts. Positive outcomes are limited due to tissue availability, donor site morbidity, complex grafting procedures, and high engraftment failure rates [26]. Moreover, in AD, fracture healing is slow relative to age-matched controls, and often, incomplete [27]. Thus, there exists a pressing need for a robust in vitro model which allows evaluation of the interactions between putative key cellular mediators of AD-induced BMD loss: SNs and MSCs.
In developing this model system, we therefore implemented the SN-like rat neuroendocrine pheochromocytoma-12 (PC12) cell line in 3D transwell coculture with hMSCs. In addition to their ubiquity and ease-of-use relative to primary human SNs, PC12s secrete critical peripheral neurotransmitters and neuropeptides including noradrenaline and NPY [28, 29]. Importantly, PC12s secrete little to no epinephrine—a neurotransmitter found in cortical, but not peripheral neurons [30]. SN-like PC12s and hMSCs were separately encapsulated in poly(ethylene glycol) diacrylate (PEGDA) hydrogels and subsequently cocultured using semi-permeable transwells to allow for focus on paracrine cell interactions. In this study, following 14 days of coculture in an AD-analogous inflammatory environment (TNF-α), PC12 activation and hMSC osteogenesis were assessed at the protein level relative to monoculture and unstimulated controls.
METHODS
Fabrication of PEGDA hydrogels
PEGDA was synthesized per previous protocols [31]. Adhesion peptide arginine-glycine-aspartate-serine was reacted with acrylate-derivatized PEG-NHS at a 1:3 molar ratio, purified, and added (3.9 mg/mL) to the polymer precursor solution (10%w/v in DPBS; Life Technologies). Immediately prior to cell encapsulation, 1%v/v photoinitiator (262 mg/mL Irgacure 2959 in 70%EtOH) was added to the polymer precursor solution, which was then filtered (0.22μm membrane) and protected from light until use.
PC12 activation and cell encapsulation
To induce pathological activation of SN-like PC12s (ATCC), cells were pre-exposed to 50 ng/mL recombinant murine NGF (PeproTech) for 72 h. Activated PC12s and hMSCs (P6; Texas A&M Institute for Regenerative Medicine) were then harvested and resuspended in the PEGDA precursor solution at 5×10–6 cells/mL. Concurrently, 200μL aliquots of the resulting suspension were distributed in the wells of a 48-well plate (Falcon) and cured via longwave UV exposure (10 mW/cm2) for 6 min.
Cell culture
Cell-laden hydrogels were cultured in osteogenic medium composed of high-glucose Dulbecco’s Modified Eagle Medium (DMEM-HG; Corning), 10%fetal bovine serum (FBS Premium; Gibco), 1%antibiotic solution (P/S: 10,000 U/mL penicillin, 10,000μg/mL streptomycin; Life Technologies), and 50 ng/mL recombinant human bone morphogenic protein 2 (rhBMP-2; PeproTech) in the presence or absence of 10 ng/mL TNF-α (PeproTech). Blank PEGDA hydrogels were implemented in coculture with hMSC or PC12 laden hydrogels for hMSC- and PC12-only control groups, respectively. Full experimental medium changes were performed every 2–3 days. This experimental design is summarized in Fig. 1.
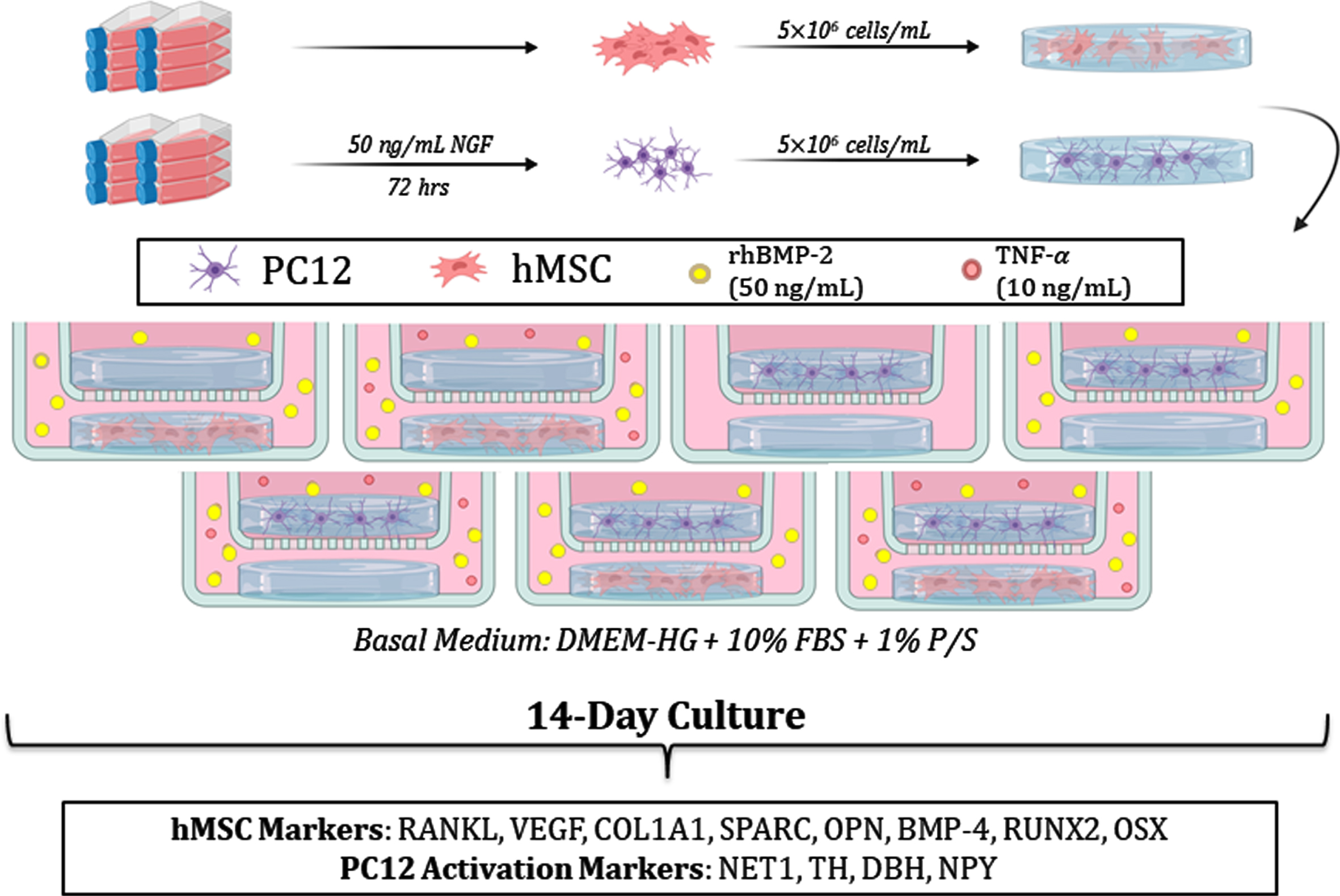
Schematic of the experimental design. hMSCs and PC12s were encapsulated in PEGDA hydrogels and subsequently cultured for 14 days in the presence or absence of TNF-α, after which the cell-laden hydrogels were lysed and analyzed for the expression of markers associated with osteogenesis (hMSCs) and neural activation (PC12s).
Cell lysis and protein extraction from hydrogels
Following 14 days of coculture, cell culture supernatant was collected and immediately stored at –80°C in 1.7 mL microfuge tubes (VWR) for subsequent analysis. Hydrogels were halved, flash-frozen in liquid nitrogen, and likewise stored in 1.7 mL microfuge tubes. After addition of 200μL lysis buffer (100 mM Tris, 500 mM LiCl, 10 mM EDTA, 1%LiDS, 5 mM dithiothreitol, pH ∼7.6) containing Halt™ Protease and Phosphatase Inhibitor (Thermo Fisher), flash-frozen hydrogels were homogenized using plastic DNAse- and RNAse-free pestles (VWR). The resulting homogenate was then vortexed vigorously (20 min), centrifuged (16,000×g; 20 min), and the supernatant extracted and stored at –80°C for subsequent proteomic analysis. All protein extraction procedures were performed at 4°C to reduce protease activity.
Western blot
Cell lysates were analyzed for the expression of markers associated with osteogenesis and bone reformation (hMSCs) and neural activation (PC12s) as described previously [32, 33]. Briefly, DNA levels in each sample were quantified using the Quant-It PicoGreen dsDNA Assay kit (Life Technologies). Sample volumes corresponding to 1500 ng and 1000 ng DNA for hMSCs and PC12s, respectively, were concentrated using 3 kDa Amicon Ultra-0.5 mL filter units (Millipore) and subsequently exposed to sodium dodecyl sulfate (Sigma-Aldrich) and heat treatment (95°C; 10 min) for protein denaturation. Each sample was then loaded into separate wells of 6–15%acrylamide gradient gels and separated via electrophoresis. Separated proteins were then transferred to nitrocellulose membranes and placed in a blocking solution containing TBST/NaN3 (25 mM Tris-HCl, pH 7.5, 137 mM NaCl, 0.1%Tween 20, 0.05%NaN3) and 5%bovine serum albumin (BSA; Fisher Scientific) for 1 h at room temperature. Membranes were sectioned to isolate molecular weight ranges of target proteins.
Primary antibodies corresponding to hMSC markers for bone reformation and osteogenesis, including RANKL (1:1000; sc-9073; Santa Cruz Biotechnology), RUNX2 (1:1000; sc-12488; Santa Cruz Biotechnology), and OSX (1:500; sc-22538-R; Santa Cruz Biotechnology), as well as those corresponding to SN activation—TNF-α (1:500; sc-12744; Santa Cruz Biotechnology) cyclooxygenase-2 (COX-2; 1:500; sc-166475; Santa Cruz Biotechnology) endothelial nitric oxide synthase (NOS-3; 1:500; sc-654; Santa Cruz Biotechnology), IL-6 (1:1000; ab6672; Abcam), noradrenaline transporter 1 (NET1; 1:500; ab41559; Abcam) and tyrosine hydroxylase (TH; 1:500; sc-25269; Santa Cruz Biotechnology)—were diluted in TBST/NaN3 + 5%BSA, applied to designated nitrocellulose membrane sections, and incubated at 4°C overnight with constant agitation. Following multiple TBST washes, bound primary antibody was detected by incubating appropriate horse radish peroxidase- and alkaline phosphatase-conjugated secondary antibodies (1:1000; Jackson ImmunoResearch) for 1 h at room temperature. Following incubation with Luminol (Santa Cruz Biotechnology) or Novex chemiluminescent (Life Technologies) substrates, the membrane sections were imaged using the ChemiDoc™ XRS+ Imager using Image Lab™ software (Bio-Rad), applying controlled exposure periods to prevent image saturation. The integrated optical density of each band was then assessed via semi-quantitative image analysis using Adobe Photoshop.
MAGPIX multiplex immunoassay
Culture supernatants were subjected to MAGPIX multiplex immunoassay as described previously [34] and assessed for the expression of markers associated with hMSC osteogenesis, in accordance with the manufacturer’s protocol (R&D Systems). Target proteins included vascular endothelial growth factor (VEGF), secreted protein acidic and rich in cysteine (SPARC), bone morphogenic protein 4 (BMP-4), osteopontin (OPN), and COL1A1. Sample median fluorescence intensities were assessed using a corresponding standard curve to calculate sample concentrations of the proteins of interest. Results were normalized to the total DNA content measured from each sample.
NPY ELISA
Culture supernatants were applied toward characterizing PC12 secretion of NPY via an ELISA kit, in accordance with the manufacturer’s protocol (Millipore). The dose response curve was generated by applying a sigmoidal five-parameter logistic regression to the included standards. NPY release was calculated from the absorbance of each sample at 450 nm, corrected by the absorbance at 590 nm. Results were normalized to total DNA content measured from each sample via Quant-It™ PicoGreen™ dsDNA Assay Kit (Thermo Fisher) according to the manufacturer’s instructions.
Statistical analysis
All data are reported as mean±standard error of the mean (SEM). Statistical comparisons between the experimental groups were performed in SPSS using a standard alpha level of 0.05. Homogeneity of variance was validated using Levene’s test (α= 0.05). Two-way ANOVAs were conducted to evaluate the individual effects of the categorical independent variables (MSC/PC12 coculture; TNF-α stimulation) as well as to assess their interactions with respect to each marker studied. Pairwise sample means were compared using Tukey’s post-hoc tests. In accordance with the chosen alpha level, all comparisons with p < 0.05 were considered statistically significant.
RESULTS
In developing an in vitro model system that can accurately reflect the pathological effects of aberrant chronic inflammation in AD, we utilized TNF-α to stimulate an AD-analogous inflammatory environment. After confirming the effect of TNF-α exposure on PC12s and hMSCs in monoculture, co-culture studies in the presence of TNF-α were conducted to allow assessment of paracrine interactions.
To confirm the influence of TNF-α stimulation on PC12 activation state, markers for inflammation-related activation and catecholamine synthesis in TNF-α stimulated PC12s were compared to unstimulated PC12 controls. In response to longitudinal TNF-α exposure, PC12s produced significantly greater levels of proinflammatory enzyme COX-2 (∼2.5-fold, p = 0.02) and proinflammatory cytokine TNF-α (∼2.5-fold, p = 0.0006). Furthermore, NOS-3, an enzyme involved in nitric oxide synthesis, was increased ∼3.1-fold (p = 0.002) relative to unstimulated negative controls (Fig. 2A). While TNF-α stimulated PC12s appeared to secrete more IL-6 relative to unstimulated controls, this effect did not attain statistical significance. Although there were no observable changes in catecholamine synthesis (Fig. 2B), TNF-α stimulated PC12s also demonstrated a significant downregulation of NPY (∼1.4-fold, p = 0.004) relative to unstimulated controls.
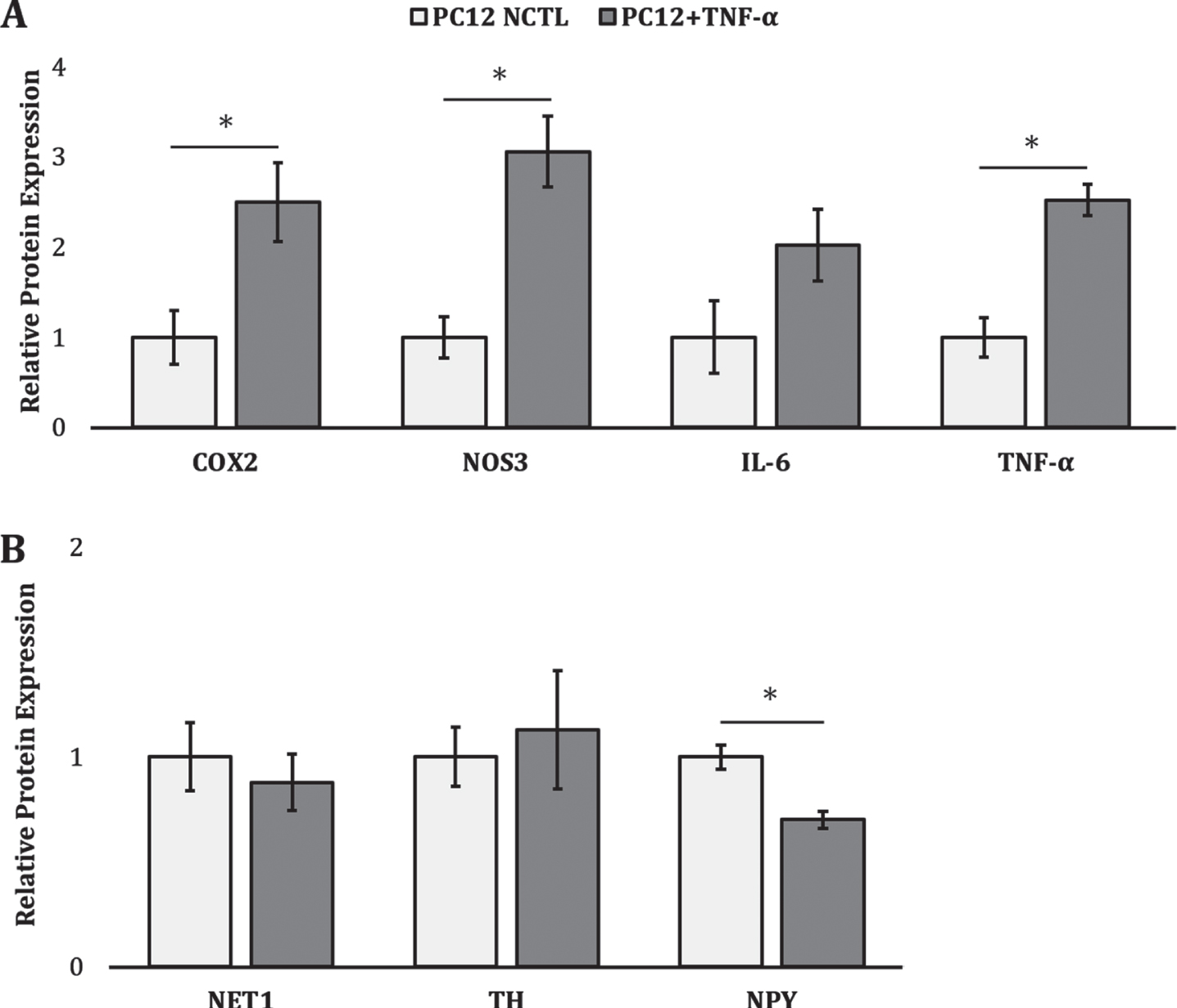
PC12 activation following TNF-α stimulation. Relative protein expression of markers associated with (A) inflammatory activation and (B) catecholamine synthesis. * denotes a significant difference between TNF-α stimulated PC12s (dark grey) and unstimulated controls (white) (p < 0.05).
Similarly, exposure of hMSCs to 10 ng/mL TNF-α drove a significant decrease in angiogenic factor VEGF (∼1.8-fold, p = 0.0003) relative to unstimulated controls (Fig. 3). Although COL1A1, OSX, and RUNX2 levels were unaffected, OPN was significantly reduced (∼2.3-fold, p = 0.03) following prolonged TNF-α treatment. Conversely, expression of hMSC RANKL, a canonical marker of osteoclastogenesis and bone reformation [35], increased following TNF-α stimulation (∼8.7-fold, p = 1.4×10–6). Altogether, these data may support the notion that prolonged exposure to an AD-analogous inflammatory environment induces abnormal activation of SNs and hMSCs.

Inhibition of hMSC osteogenesis following TNF-α stimulation. Relative protein expression of osteo-regulatory markers shown. * denotes a significant difference between TNF-α stimulated hMSCs (light green) and unstimulated controls (white) (p < 0.05).
Activated PC12s strongly inhibit the osteogenic capacity of hMSCs
We then evaluated the interplay between the TNF-α activated PC12s and TNF-α activated hMSCs over a 14-day coculture period relative to monoculture controls. As can been seen from the osteo-regulatory markers investigated (Fig. 4), hMSCs cocultured with PC12s in the presence of TNF-α demonstrated significantly reduced expression of OPN (∼7.7-fold, p = 3.4×10–5) and COL1A1 (∼2.1-fold, p = 0.03) compared to TNF-α stimulated hMSCs in monoculture. While hMSCs cocultured with PC12s appeared to also demonstrate a reduction in SPARC, RANKL, and OSX, and an increase in RUNX2, these effects did not reach statistical significance. Given the largely unchanged levels of RUNX2 and OSX, which are both key transcription factors expressed during the early stages of hMSC osteoblastic differentiation [21], this may indicate that PC12s exert their osteo-inhibitory effects at later differentiation timepoints related to ECM deposition, mineralization, and osteocyte commitment.
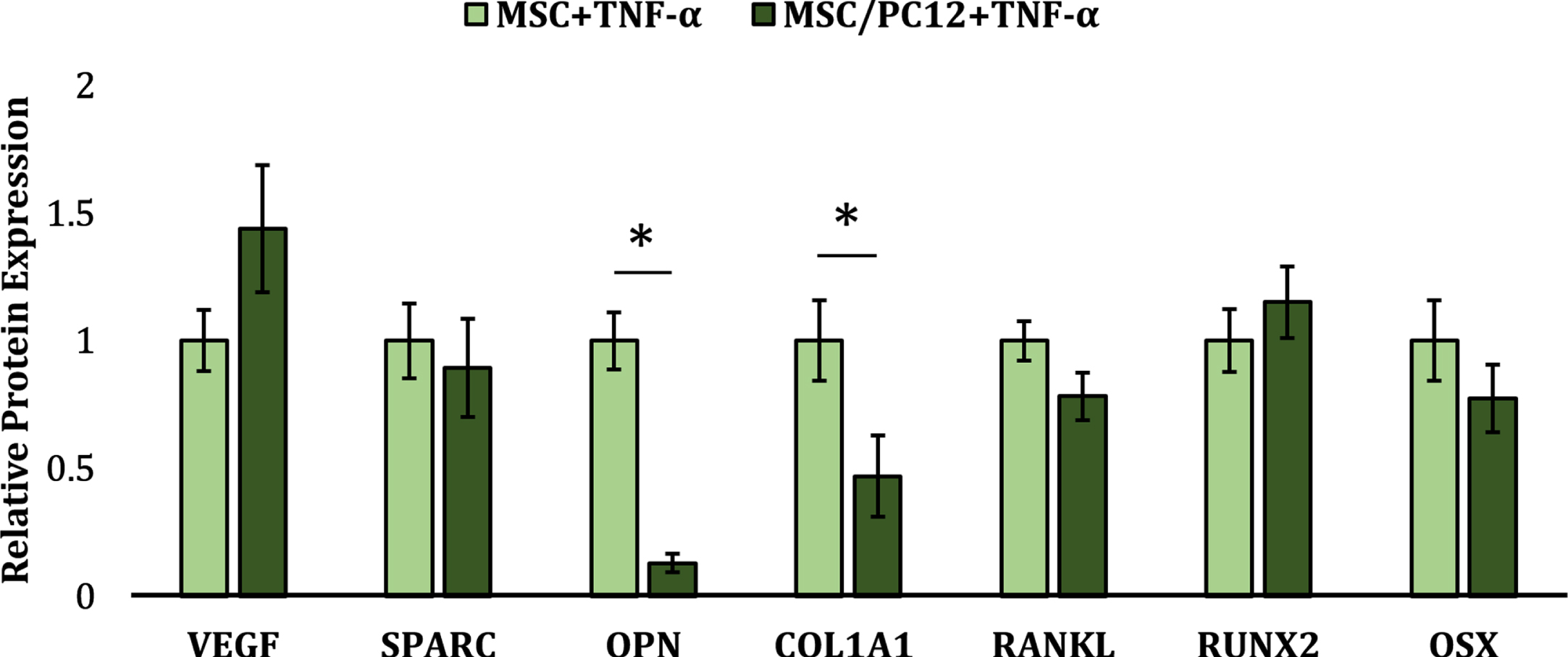
Relative protein expression of osteo-regulatory markers in hMSCs stimulated with TNF-α in the presence or absence of PC12s. * denotes a significant difference between MSC/PC12 cocultures (dark green) and monoculture controls (light green) (p < 0.05).
Activated PC12s secrete pro-inflammatory cytokines in response to hMSC coculture
As shown in Fig. 5, we likewise investigated the response of chronically activated PC12s to hMSC coculture in the presence of TNF-α to further characterize the complex paracrine interaction between these cells. Interestingly, PC12s cocultured with hMSCs exhibited a clear increase in the expression of pleiotropic inflammatory cytokine IL-6 (∼3.9-fold, p = 1.49×10–5) relative to TNF-α stimulated PC12 monoculture controls. While there appears to be a trend toward greater PC12 expression of inflammatory markers COX-2 and TNF-α, as well as upregulated expression of NET1, an indicator of norepinephrine release, under co-culture conditions these differences were not statistically significant. IL-6 is involved in a wide gamut of immunomodulatory, developmental, and hematopoietic processes and is known to induce osteoclastogenesis and subsequent bone reformation [36]. Thus, these data may indicate a pathological shift of this osteo-homeostatic paracrine interaction toward osteoblastic inhibition and further bone reformation.
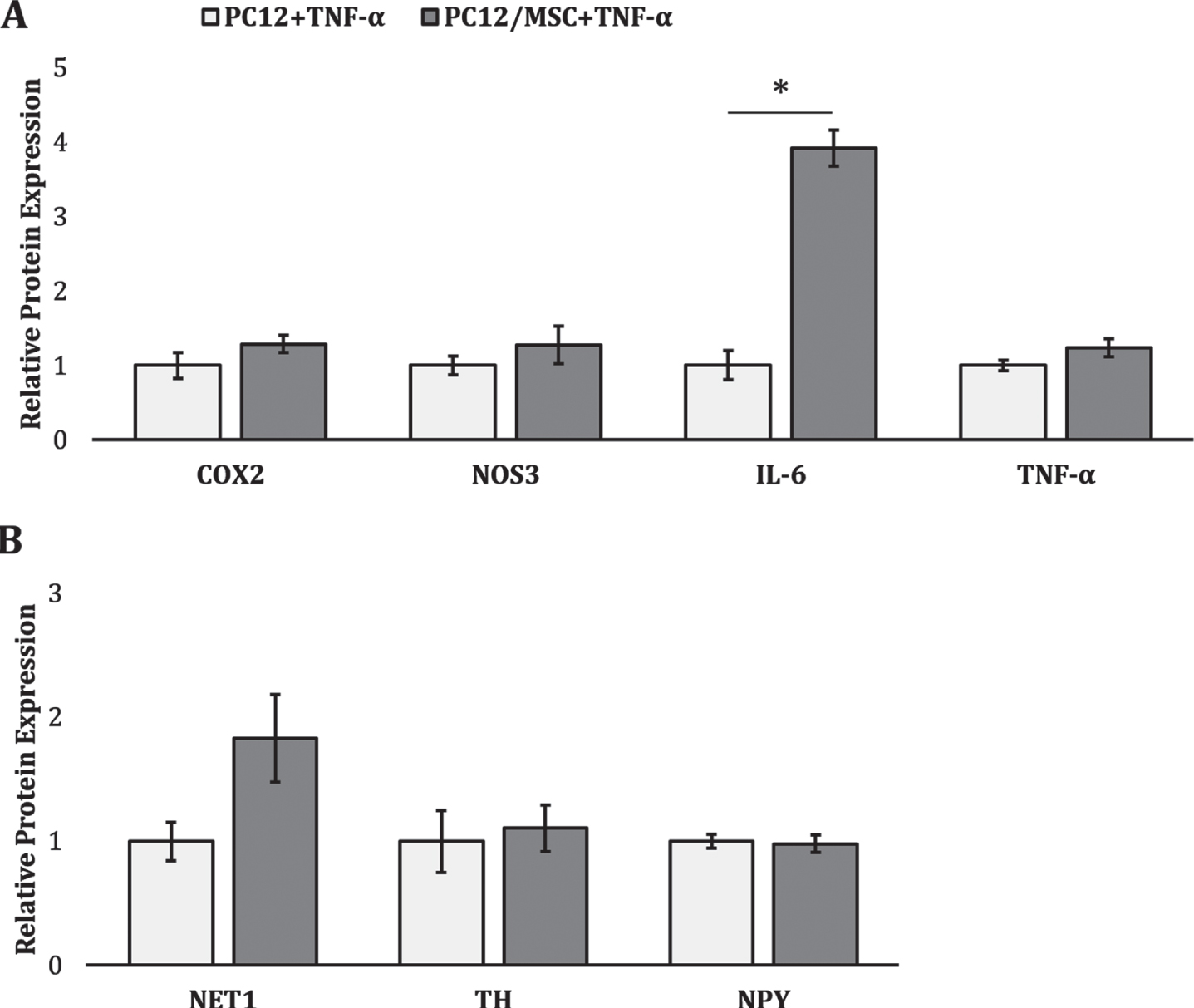
Effect of MSC coculture on PC12 activation state in the presence of TNF-α. Relative protein expression of markers associated with (A) inflammatory activation and (B) catecholamine synthesis. * denotes a significant difference between PC12/MSC cocultures (dark grey) and monoculture controls (white) (p < 0.05).
DISCUSSION
The goal of the present investigation is two-fold: 1) to characterize the paracrine interactions between PC12 cells and hMSCs in an AD-analogous inflammatory environment and 2) to determine whether activated SN-like cells can influence the osteogenic lineage progression of osteoblast precursors. Toward this goal, we encapsulated both cell types in separate PEGDA hydrogels and cocultured them using transwell inserts in osteogenic medium (BMP-2) in the presence or absence of TNF-α—a highly-elevated and well-conserved proinflammatory cytokine implicated in the pathogenesis and progression of neurodegeneration in AD [37]. The dosage of TNF-α implemented in this investigation, while higher than the chronic low-grade inflammation observed in the CSF and serum of patients with AD [38, 39] is in line with recent in vitro literature separately evaluating the effects of this proinflammatory cytokine on hMSC osteogenesis [40–42] and PC12 activation [43–45]. After 14 days of culture, the cell-laden hydrogels were homogenized, and the lysate subjected to proteomic analyses to identify changes in PC12 inflammatory activation and catecholamine synthesis and hMSC osteogenic capacity.
To confirm pathological PC12 activation following TNF-α stimulation, we first analyzed PC12 lysates for the expression of proteins associated with inflammatory activation and catecholamine synthesis. Our data demonstrate a significant upregulation in inflammatory markers NOS3, COX-2, endogenous TNF-α, and a significant downregulation in sympathetic signaling marker NPY in TNF-α stimulated PC12s relative to unstimulated controls. Under normal physiological conditions, NOS3 expression and subsequent nitric oxide (NO) secretion is overall neuroprotective [46, 47] and osteoprotective [48, 49]; however, the high levels of localized NO present in chronic inflammation are associated with neurodegeneration, bone reformation, and tissue damage [50–52]. Indeed, chronic excess NO has been shown to exacerbate AD pathology via nitrotyrosination and subsequent increased aggregation of Aβ fragments [53], impairment of γ-secretase activity leading to increased Aβ42/40 ratio [54], and mitochondrial fragmentation [55].
The significant increases in COX-2 and endogenous TNF-α observed in monocultures of TNF-α stimulated PC12s are in line with prevailing literature attributing the upregulation of these factors to neuroinflammation and sympathetic hyperactivity [56–58]. The proinflammatory enzyme COX-2 is upregulated in PC12 cells following activation through a variety of mechanisms, including lipopolysaccharide stimulation [59, 60], hypoxia [61], and exogenous Aβ administration [62]. COX-2 is known to be upregulated in the brains and CSF of patients with AD [63], and peripheral activation of the sympathetic nervous system induces preferential increases in COX-2 expression [64, 65]. TNF-α has been identified as a prominent pro-inflammatory cytokine involved in the pathogenesis and maintenance of neuroinflammation in the AD brain [66, 67] and its expression in peripheral neurons has been shown to be upregulated following their activation [68]. That endogenous TNF-α expression is upregulated following exogenous TNF-α stimulation may indicate that inflammatory activation in chronic disease states is perpetuated in a positive feedback loop manner. Interestingly, PC12 endogenous TNF-α production under this pro-inflammatory condition was abrogated with hMSC coculture (Fig. 5), indicating the presence of immunomodulatory crosstalk. The anti-inflammatory capacity of immunologically licensed hMSCs has been thoroughly studied in recent years: following exposure to similar dosages of pro-inflammatory cytokines TNF-α and interferon gamma, hMSCs have been found to secrete anti-inflammatory factors, including IL-10, TGF-β, prostaglandin E2, indoleamine 2,3-dioxygenase, and hepatocyte growth factor [69, 70]. While not evaluated in the present study, the secretion of these immunomodulatory proteins may have contributed to the marked decrease in PC12 TNF-α production shown here.
NPY, one of the most abundant neuropeptides in both the central and peripheral nervous systems [71], is secreted by SNs in response to stress [15, 72]. Under normal physiological conditions, NPY preferentially inhibits bone formation via stimulation of Y1 and Y2 receptors on osteoblasts and neurons, respectively [15]. Contrarily, under conditions of chronic stress, NPY secretion may be osteoprotective: genetic ablation of NPY exacerbated BMD loss compared to wild-type controls in a murine model of chronic stress. Additionally, Y2R-deficient mice were found to be more sensitive to stress-induced bone loss than unmodified controls [73]. The significant reduction in NPY expression in TNF-α stimulated PC12s seen herein may therefore indicate a contribution of the NPY/Y1/Y2 axis to sympathetic neuron mediated bone reformation.
We next evaluated the influence of TNF-α stimulation on the osteogenic lineage progression of hMSCs in monoculture and found a significant downregulation of VEGF and OPN, and concurrent upregulation in RANKL, in TNF-α stimulated hMSCs relative to unstimulated controls. VEGF is a principal growth factor involved in a plurality of inflammatory [74], tissue regenerative [75], wound healing [76], and developmental [77] processes. VEGF expression necessarily precedes bone formation and regeneration [78]. Thus, its reduction herein may reflect a reduction in osteogenic capacity in this context. OPN is preferentially expressed at interfacial mineralized sites in calcified bone and is an integral component of the bone ECM [79]. Its expression is positively correlated with bone formation, mineralization, and turnover as well as hMSC osteogenesis [32, 80]. Indeed, a recent study demonstrated that OPN also plays a critical role in hMSC lineage determination: genetic ablation of OPN in hMSCs was shown to significantly decrease osteogenesis [81]. The downregulation in OPN observed herein following longitudinal TNF-α stimulation is in line with investigations reporting a reduction in hMSC osteogenic capacity in chronic inflammatory environments [37]. RANKL is a type II transmembrane protein predominantly expressed by hMSCs, osteoblasts, and T-leukocytes [35]. In response to RANKL stimulation, mononuclear precursors terminally differentiate into preosteoclasts, resulting in enhanced bone reformation [82]. The synthesis of RANKL by hMSCs following inflammatory cytokine stimulation, especially via TNF-α, is a fundamental characteristic of bone reformation [83]. The upregulation of RANKL shown here is consistent with the pathologically elevated levels of RANKL in patients with chronic inflammatory disorders, including osteoporosis [84].
To our knowledge, this investigation represents the first attempt to characterize the potential contributions of sympathetic hyperactivity to the pathological bone reformation observed in patients with AD. Under these inflammatory coculture conditions, hMSCs demonstrated a significant reduction in OPN and COL1A1 expression, and PC12s showed a significant increase in IL-6, relative to monoculture inflammatory controls. Type I collagen is the most abundant protein found in bone, constituting over 90%of the organic matrix [85]. Given that a primary function of osteoblasts is the synthesis and deposition of type I collagen [86], reduction in COL1A1 suggests a reduction in hMSC osteogenesis [87–89]. Similarly, multiple in vitro studies have found that suppression or blockage of OPN signaling induces MSC adipogenic rather than osteogenic differentiation [81, 90]. A pathological shift toward hMSC adipogenesis has been associated with multiple inflammatory age-related disorders manifesting in bone loss, including AD and osteoporosis, designated “inflammaging” [37, 92]. Our observation that paracrine signaling from chronically activated PC12s induces a significant decrease in hMSC OPN expression seems to be in line with this paradigm.
IL-6 is a pleiotropic cytokine whose signaling is involved in a host of major physiological systems including immune response, inflammation, tissue regeneration, and synaptic plasticity [93]. Increased neuronal IL-6 secretion is a canonical response to injury and inflammation [94]; unsurprisingly, IL-6 has been shown to be heavily upregulated in the brains and CSF of AD patients [95, 96]. Clinical studies have also found heightened serum levels of IL-6 in patients with elevated sympathetic tone [97]. The significant increase in PC12 IL-6 expression shown here is in line with data from previous investigations demonstrating increased PC12 IL-6 secretion following TNF-α stimulation (Fig. 5) [43], as well as with an investigation reporting a three-fold increased IL-6 expression in APPsw-PC12/MSC coculture supernatants relative to those taken from monoculture controls [98]. In the present study, TNF-α stimulation and hMSC coculture appear to act in concert; that a similar upregulation in PC12 IL-6 expression is not seen with TNF-α stimulation alone (Fig. 2A) merits further investigation. Importantly, exogenous IL-6 treatment has been shown to upregulate hMSC proliferation, inhibit osteogenic, adipogenic, and chondrogenic differentiation, and augment in vitro wound healing [99]. This may indicate that paracrine signaling from chronically activated SN-like cells inhibits hMSC osteogenesis partly via an IL-6 dependent mechanism.
Lineage-specific differentiation capacities of hMSCs are potently modulated by a wide gamut of mechanical and biomolecular cues [100]; under certain conditions, hMSCs have demonstrated the ability to efficiently differentiate into neuronal precursor cells, mature neurons, and glial cells [101]. A shift toward neuronal lineage commitment due to the paracrine factors released by SN-like PC12 cells may have contributed to the reduction in hMSC osteogenic capacity shown here. A recent study found that coculturing hMSCs with neural stem cells (NSCs) induced a significant reduction in osteogenic and adipogenic potential, and enhanced chondrogenic differentiation relative to monoculture controls [102]. The researchers speculated that differences in hMSC cell density and NSC TGF-β signaling were responsible for these differences, but changes in the expression of early neuronal markers were not evaluated. In contrast, another study found that culturing MSCs in Schwann cell conditioned medium enhanced osteogenesis and pre-vascularization in a tissue-engineered bone graft [103]. These phenomena merit further investigation, as they carry major implications for the usage of hMSCs as a therapeutic agent in neurodegenerative disease.
Taken together, our data indicate that paracrine signaling from SN-like PC12 cells subjected to longitudinal stimulation by TNF-α—one of the prominent proinflammatory cytokines upregulated in AD patients—potently inhibits the osteogenic capacity of hMSCs. While these findings add credence to the recent hypothesis that BMD loss in AD patients is due, at least in part, to elevated SN activity [15], further investigation incorporating longer timepoints (>21 days), a more comprehensive marker panel, 3D scaffold architectures optimized for each cell type, and a cytokine cocktail more faithfully recapitulating the complex inflammatory milieu in AD are required to further establish causality in this relationship. In addition, future studies should incorporate human AD-derived sympathetic neurons by implementing induced pluripotent stem cells (iPSCs) to more faithfully recapitulate the AD environment and to avoid the crosstalk issues associated with the use of cells derived from different host species. Finally, as further information accumulates regarding the local expression of TNF-α within tissues of interest, TNF-α dosages and treatment periods can be further refined. Incorporating these variables into future experiments will facilitate the discovery of novel tissue engineering approaches for modeling this complex pathology, enabling the generation of effective therapeutic interventions.