
Abstract
Background:
A complex network of aging-related homeostatic pathways that are sensitive to further deterioration in the presence of genetic, systemic, and environmental risk factors, and lifestyle, is implicated in the pathogenesis of progressive neurodegenerative diseases, such as sporadic (late-onset) Alzheimer’s disease (sAD).
Objective:
Since sAD pathology and neurotoxicity share microRNAs (miRs) regulating common as well as overlapping pathological processes, environmental neurotoxic compounds are hypothesized to exert a risk for sAD initiation and progression.
Methods:
Literature search for miRs associated with human sAD and environmental neurotoxic compounds was conducted. Functional miR analysis using PathDip was performed to create miR-target interaction networks.
Results:
The identified miRs were successfully linked to the hypothetical starting point and key events of the earlier proposed tau-driven adverse outcome pathway toward memory loss. Functional miR analysis confirmed most of the findings retrieved from literature and revealed some interesting findings. The analysis identified 40 miRs involved in both sAD and neurotoxicity that dysregulated processes governing the plausible adverse outcome pathway for memory loss.
Conclusion:
Creating miR-target interaction networks related to pathological processes involved in sAD initiation and progression, and environmental chemical-induced neurotoxicity, respectively, provided overlapping miR-target interaction networks. This overlap offered an opportunity to create an alternative picture of the mechanisms underlying sAD initiation and early progression. Looking at initiation and progression of sAD from this new angle may open for new biomarkers and novel drug targets for sAD before the appearance of the first clinical symptoms.
INTRODUCTION
Alzheimer’s disease (AD) is the most common type of dementia, representing 60–80% of all dementia cases worldwide [1]. Late-onset or sporadic AD (sAD), which accounts for more than 95% of AD cases, is a neurodegenerative disease affecting adults over 65 years of age and results in a gradual decline in memory, cognitive function, and eventually irreversible brain damage. It is anticipated that AD cases will be increased up to 107 million worldwide by 2050 [2]. Based on the AD’s progressive cognitive decline, four AD-stages (preclinical, mild, moderate, and late) have been described [2]. Pathological alterations are associated with damages in different brain regions, affecting initially the entorhinal cortex and hippocampus in preclinical stage, and spreading gradually to the cerebral cortex resulting in mild, moderate, and severe AD-stage. The preclinical AD-stage can last more than 10 years before the onset of cognitive decline and is often asymptomatic [3, 4]. Since no significant irreversible brain damage has occurred at this stage, it provides a window of opportunity for identification of biomarkers capable of detecting early pathological processes manifesting in this stage, as well as for development of drugs preventing progression into symptomatic stages.
sAD is a multifactorial disease resulting from an age-related deterioration of the maintenance of homeostatic processes that is amplified by genetic, systemic, and environmental risk factors, and lifestyle [5–7]. Although several dysregulated cellular and biological processes have been reported by genetic, biochemical, or neuropathological studies to be implicated in the pathogenesis of sAD [8], the accumulation of extracellular amyloid plaques (amyloidopathy) and intracellular neurofibrillary tangles (tauopathy) seem to be the most extensively studied throughout literature. Developed biomarkers for the early AD diagnosis, based on these two pathological mechanisms, remarkably fail due to their low accuracy [9, 10]. Thus, these two pathologies may not be the primary causes of the AD development, but other more elusive processes may trigger the initiation and progression of sAD.
In an attempt to collect and structure mechanistic data on sAD pathogenesis, a tau-driven adverse outcome pathway (AOP) toward memory loss has been previously constructed [11]. This AOP suggests a series of potential early molecular and cellular key events (KEs), including glucose and cholesterol dysmetabolism (hypothetical starting point), both assumed to contribute to sAD pathology due to their tight interplay, mitochondrial dysfunction (KE1), oxidative stress (KE2), hyperphosphorylation of tau (KE3), impaired autophagy (KE4), accumulation of cytosolic toxic tau (KE5), dysfunctional axonal transport (KE6), synaptic dysfunction (KE7), neuroinflammation (KE8), and neuronal dysfunction (KE9), to lead to the adverse outcome (AO) of memory loss. These proposed memory loss-related pathological processes may be dysregulated already at the early preclinical stage of AD, long before the appearance of the clinical symptoms, preceding the onset of clinically evident cognitive impairment.
The contribution of environmental factors to the risk for acquiring sAD is substantiated by evidence from epidemiological and experimental studies [5, 139]. Neurotoxicity is the consequence of any adverse effect caused by exposure of nervous system to chemical, biological, or physical agents. In particular, use of alcohol [12], nicotine [13], or drugs [14] has been reported to lead to altered neuronal function and confer neurotoxicity. Human exposure to metals such as aluminum, arsenic, lead, manganese, mercury, or trimethyltin, an organotin compound, has been associated with metal-induced neurotoxicity [15]. Moreover, pesticides are known for their neurotoxic potencies and their human exposure is often linked to neurodegenerative diseases, including cognitive impairment [16] and dementia [17]. In addition, neurotoxic effects of air pollution exposure have been associated with increased risk of cognitive decline and AD [18]. Several neurotoxicity-related cellular and biological processes, including oxidative stress [19], Ca2 + dyshomeostasis [20], mitochondrial dysfunction [21], dysfunctional autophagy [22], neuroinflammation [23], neurotransmitter dysfunction [24], insulin resistance [25, 26], cholesterol dysmetabolism [27], hyperphosphorylation of tau and synaptic dysfunction [28], are dysregulated in sAD, as well. Hence, it can be hypothesized that neurotoxicity and sAD share common pathological processes, which opens new opportunities for investigating disease initiation and progression.
microRNAs (miRs) have gained significant attention from researchers due to their important role in a wide range of cellular and biological processes, and their capacity to regulate several processes from development to disease. miRs are small molecules (19–22 nucleotides) known as master regulators of gene expression acting at posttranscriptional level, while controlling more than 60% of human genome. To date, approximately 2,650 human mature miRs have been identified [29], with 70% known to be localized in the brain [30], while 50% are either brain-specific or brain enriched [31]. Many miRs have been suggested as promising molecular biomarkers for numerous diseases, including AD [32]. In addition to AD development [33–41], several miRs have been associated with neurotoxicity [42], probably regulating pathophysiological processes shared by both environmental neurotoxicity and sAD.
In this study, existing data on AD- and neurotoxicity-related miRs were collected, and their gene interactions-networks, linked to the proposed events of the tau-driven AOP for memory loss [11], were identified with functional miR analysis. The resulting miR-target network may provide new mechanistic insight with benefit for AD clinical research, by providing a better understanding of sAD initiation and progression, and for the discovery of reliable biomarkers and new drugs targets capable of addressing the preclinical stage of the disease.
METHODS
Strategy for the selection of the candidate miRs implicated in sAD and/or neurotoxicity
A comprehensive search for publicly available articles referring to “microRNA”, “miRNA”, “miR” in combination with “Alzheimer’s disease”, “AD”, and “environmental neurotoxicity”, “chemical-induced neurotoxicity” in PubMed and ScienceDirect databases was conducted. First, the miR search covered implication of human miRs, detected in biofluids, such as blood, serum, plasma, cerebrospinal fluid, and in brain tissues, from patients with clinical sAD. The retrieved miR profiles of sAD patients were compared to those of healthy controls. Classification of sAD stage was based on several clinical diagnostic criteria, including Mini-Mental State Examination scoring (MMSE), Braak staging, or Clinical Dementia Rating (CDR), established by National Institute of Neurological Disorders or Stroke in collaboration with the Alzheimer’s Association (NINDS-AA), National Institute of Neurologic and Communicative Disorders and Stroke/Alzheimer’s Disease and Related Disorders Association (NINCDS–ADRDA), or US National Institute on Aging–Alzheimer’s Association (NIA-AA). Additionally, miRs related to environmental neurotoxicity were retrieved from available epidemiological and in vivo human and animal studies, relevant to the nervous system and investigating brain tissue, brain-cells and/or blood, following exposure to neurotoxicants such as industrial chemicals, drugs, pollutants, or metals. All retrieved miRs were quantitatively detected, by using either highly sensitive and specific methods such as quantitative polymerase chain reaction (qPCR), or high-throughput technologies (e.g., microarrays, next generation sequence). Next, the available molecular and cellular information related to the potential role of the miRs in AD or neurotoxicity was collected and placed in the context of the proposed tau-driven AOP toward memory loss. Search priority was given to human studies, but animal studies were considered to fill knowledge gaps when human studies were not available. Only articles written in English were included, which were published from 1999 till 2021.
Functional miR analysis
To understand the role of miRs, in silico-based functional analysis was performed. This approach typically consists of target prediction following functional analysis of identified miR targets. Human miR–target interactions validated at experimental level were retrieved by using the MiRTarBase [43] for all identified miRs implicated in sAD and neurotoxicity. The analysis resulted in a list of genes, whose expression is modulated by the investigated miRs, according to the database. The number and the reliability of hits accounting for each miR-target interaction were registered. The identified genes were organized in predictive sets, including genes that are potential targets of the miRs related to the hypothetical starting point and to the KEs defined by the AOP. In order to functionally characterize each predictive set, the list of pathways where target genes are involved was obtained by using PathDip 4, an annotated database of human signaling cascades, comprising core pathways from 24 major curated pathways databases [44]. For each event of interest, text-mining analysis superimposed specific semantic parameters to filter pertinent pathways and to select among the targets only genes involved in these pathways. The list of miRs that, according to functional analysis, potentially affect these pathways was reversed engineered and compared to the literature outputs. The results of functional analysis were visualized by using the software flourish.studio and R 3.6.0 (packages ggplot2, igraph and alluvial).
RESULTS
miRs in AD and neurotoxicity
All selected miRs and their detailed information related to sAD and chemical-induced neurotoxicity are provided in the Supplementary Table 1. Based on the selection criteria, 235 human (mature) miRs with sufficient evidence for their involvement in human AD pathology, were identified in cerebrospinal fluid (N = 100), serum (N = 83), plasma (N = 57), whole blood (N = 40), peripheral blood mononuclear cells (N = 16), and brain tissues (N = 89). Additionally, 115 and 87 miRs linked to human and animal chemical-induced neurotoxicity, respectively. Not all the selected neurotoxicity-related miRs were implicated in the human sAD. The numbers of miRs overlapping between sAD (human) and neurotoxicity (human and/or animal) are illustrated, while Table 1 shows common miRs in detail.
miRs shared by AD (human) and environmental neurotoxicity (NT) (human and/or animal)
Among the miRs potentially in sAD and human chemical-induced neurotoxicity, let-7b, let-7f, let-7g, miR-10b, miR-21, miR-27a/b, miR-28, miR-30b/c/d, miR-103a, miR-107, miR-142, miR-144, miR-146a, miR-150, miR-151a, miR-181a, miR-191, miR-199a, miR-222, miR-320a/b/c, miR-378c, miR-409, miR-423, and miR-425 were shown by epidemiological studies to be dysregulated in either human plasma or whole blood after exposure to air pollution [45, 46]. In another human study, an aberrant expression of let-7g, miR-10a, miR-23a, miR-30c, miR-34c, miR-98, miR-124, miR-145, miR-195, miR-376a, miR-498, miR-532, miR-550a, and miR-647 was observed after exposure of human neural progenitor cells to paraquat [47]. Changes in miR-9, miR-125b, miR-128, miR-146a, and miR-322 expression by human brain cells after exposure to neurotoxic metal sulfates such as aluminum sulfate [48–50] or aluminum-maltolate [51], have been reported. Human neuroblastoma cell line SH-SY5Y revealed a modified miR-214 expression following treatment with isoflurane, a commonly used anesthetic [52]. Acyclovir, an antiviral drug against herpes simplex virus-1, upregulated miR-146a in mixed cultures of primary human neuronal and glial cells [53]. Moreover, dysregulation of miR-21 has been reported following treatment of human glioma cells with chemotherapeutic drugs, temozolomide and taxol [54, 55].
With regard to miRs implicated in both human AD and animal neurotoxicity, ethanol-induced neurotoxicity involved changes in let-7b expression in rat [56], in miR-10a/b in mouse brain [57], in miR-10b, miR-29c, miR-30a/e, miR-145, miR-200a, and miR-339 in fetal mouse brain [58], and in miR-9, miR-21, miR-153, and miR-335 in fetal mouse cerebral-cortex neuronal cells [59]. Short-term nicotine exposure was associated with alterations in the expression of miR-10b, miR-21, miR-26a, miR-29a, miR-29b, miR-29c, miR-30a-5p, miR-30c, miR-93, miR-98, miR-125b, miR-137, miR-152, miR-181b, miR-186, miR-188, miR-210, miR-301a, miR-335, and miR-374 in rat or mouse neuronal cultures [60, 61]. Exposure of olfactory system of zebrafish to Cu dysregulated the expression of let-7a, let-7e, let-7f, let-7g, let-7i, miR-23a, miR-27b, miR-30c, miR-31, miR-126, miR-128, miR-138, miR-146a, miR-150, miR-193b, miR-214, and miR-219 [62]. Also, exposure of mice to hexahydro-1,3,5-trinitro-1,3,5-triazine, a common environmental contaminant, induced upregulation of miR-30a-5p, miR-195, and miR-206 [63]. Exposure of mouse serotonergic cells and rat hippocampal neurons to antipsychotic or antidepressant drugs, such as lithium/valproate or fluoxetine, was associated with decreased expression of miR-34a and increased expression of miR-16, respectively [64, 65]. In adult zebrafish, exposure to fipronil or triazophos, or their mixture, induced changes in miR-30b, miR-31, miR-128, and miR-455 levels [66].
Coupling retrieved miRs from literature to the hypothetical starting point and key events of the tau-driven AOP blueprint toward memory loss
The miRs identified in the literature were linked to the hypothetical starting point and KEs described in the proposed AOP for memory loss [11]. Notably, no miRs were found to affect cytotoxic tau oligomers (KE5), dysfunctional axonal transport (KE6), and neuronal dysfunction (KE9). All retrieved miRs, which have been detected in brain (cells/tissues) or blood, with evidence for their links to the starting point and KEs of the AOP are provided below. As aforementioned, priority was given to human studies, however, when they were not available animal studies relevant to the described events were included. In addition, starting point- and KEs-related miRs found in non-brain cells are also discussed.
Glucose and cholesterol dysmetabolism (hypothetical starting point)
Glucose dysmetabolism
While several miRs have been reported to regulate insulin resistance, little is known about miRs implicated in brain insulin resistance [67].
In brain of AD patients, miR-7 was found to target insulin receptor (INSR), insulin receptor substrate 2 (IRS-2), and insulin degrading enzyme (IDE), which are key regulators of insulin signaling in the central nervous system (CNS) and involved in AD [68]. miR-29b levels inversely correlated with insulin growth factor 1 (IGF1) expression in human cortical tissue [69]. In human glioma cells, miR-34a targeted rapamycin-insensitive companion of mechanistic target of rapamycin complex 1 (mTOR) (RICTOR), a component of the mTORC2, which phosphorylates AKT at Ser 473 and promotes cell growth. Overexpression of miR-34a decreased levels of p-AKT (Ser473) and increased glycogen synthase kinase-3 beta (GSK-3β) levels, which are implicated in insulin resistance [70]. Brain enriched miR-128 has been associated with insulin resistance in patients with type 2 diabetes [71, 72]. miR-302 has been indirectly linked to defective insulin signaling via inhibiting AKT signaling and reducing Nanog and miR-302-encoding La-related protein 7 (LARP7) gene expression in blood cells of AD patients [73]. miR-3666 expression in serum has been negatively correlated with blood glucose levels of type 2 diabetes patients [74]. Available evidence suggests that in human AD brain miR-148a can disturb insulin signaling by affecting the insulin growth factor I receptor (IGF1R) and insulin receptor substrate 1 (IRS-1), which are upstream of protein kinase B and mitogen-activated protein kinase (MAPK)/extracellular regulated protein kinases (ERK) signaling pathways [75–77].
In mouse neuroblastoma Neuro2a (N2a)/APP cells, upregulation of miR-98 downregulated IGF1 protein level, regulating insulin signaling [78]. In animal studies, miR-200b/c were demonstrated to modulate expression of ribosomal protein S6 kinase B1 (S6K1), a downstream effector of mTOR, which phosphorylates the IRS-1 protein at serine residues to suppress insulin signaling [79].
In non-brain-related cells, miR-29a and miR-29c have been shown to be increased in primary human skeletal muscle cells regulating glucose uptake and insulin-stimulated glucose metabolism [80]. miR-126 has been also reported to promote insulin signaling via inhibiting IRS-1 in human hepatocellular carcinoma cells [81].
Cholesterol dysmetabolism
With respect to brain cholesterol dysmetabolism, brain enriched miR-128, uniquely expressed in neurons [82], has been negatively associated with circulatory high-density lipoprotein (HDL) cholesterol levels [71, 72]. This miR has been implicated in cholesterol metabolism by targeting sterol regulatory element binding proteins (SREBPs), adenosine triphosphate (ATP)-binding cassette transporters, ABCA1 and ABCG1, and retinoid X receptor alpha (RXRα) proteins [72]. In human brain, miR-1229 directly controls sortilin-related receptor L 1 (SORL1) which binds to lipoprotein including apolipoprotein E (APOE) [83, 84].
In animal studies addressing events of relevance for human pathology, miR-33 has been implicated in the expression of the cholesterol transporter ABCA1 and the inhibition of the systemic HDL cholesterol levels, and the regulation of sterol regulatory element binding transcription factor 1 and 2 (SREBF1 and SREBF2), both implicated in cholesterol synthesis and uptake in brain [85]. miR-106a and miR-106b have been identified as regulators of APOE production and cholesterol metabolism via inhibition of ABCA1 in mouse neurons [86, 87]. A neuron-enriched miR, miR-335, has been shown to regulate cholesterol metabolism via targeting 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) and 3-hydroxy-3-methylglutaryl-CoA synthase-1 (HMGCS1), which are both step-limiting enzymes in cholesterol synthesis pathway, leading to reduced cholesterol and impaired brain memory, in aged cultured astrocytes and aged hippocampal brains of mice [88]. miR-545 regulates LDL receptor-related protein 1 (LRP1), a plasma membrane receptor of APOE, facilitating the import into neurons as shown in mouse brain [89, 90]. miR-137 and miR-181c have been found to regulate brain ceramide synthesis via targeting serine palmitoyl transferase (SPT), in primary rat astrocytes [91].
miR-1 has been reported to regulate B56a, a regulatory subunit of protein phosphatase 2A (PP2A), in rat cardiac myocytes [92]. miR-27a was implicated in cholesterol biosynthesis via targeting the 3-hydroxy-3-methylglutaryl coenzyme A reductase gene (Hmgcr) 3’ untranslated region (3’-UTR) in human hepatocytes [93]. miR-27b has been associated with inhibition of both low-density lipoprotein receptor (LDLR) activity and ABCA1 protein expression in human hepatic cells [94]. Hepatic miR-30c strongly reduced hypercholesterolemia in ApoE–/–mice [95]. miR-148a has been suggested as a negative regulator of LDLR activity and expression, ABCA1 expression and circulating HDL-C levels, in human hepatic cells [96]. miR-206 and miR-613 were shown to control liver X receptor α in human hepatic cells and human macrophages [97, 98].
Mitochondrial dysfunction (KE1)
Several miRs are known to regulate mitochondrial proteins involved in mitochondrial oxidative phosphorylation (OXPHOS) and electron transport chain (ETC) Complexes I-VI, affecting ATP production, mitochondrial metabolism and eventually mitochondrial function [99], in both humans and animals.
miR-7 regulated mitochondrial permeability transition pore (MPTP) via targeting voltage-dependent anion channel 1 (VDAC1) in human neuroblastoma SH-SY5Y cells [100]. miR-34b/c modulated mitochondrial function in differentiated human SH-SY5Y dopaminergic neuronal cells [101]. Increased expression of miR-27a and miR-103 in human SH-SY5Y cells treated with tumor necrosis factor a (TNFα) suppressed the expression of functional units of Complex I, resulting in mitochondrial dysfunction [102].
Several animal studies have produced data likely to be relevant for human pathology. miR-34a negatively affected the integrity of the blood–brain barrier through reduction of mitochondrial oxidative phosphorylation and ATP production, and cytochrome c levels in murine cerebrovascular endothelial cells [103]. An in vivo study revealed that mice lacking miR-29 showed upregulation of iron responsive element binding protein 2 (Ireb2) [104], which has been implicated in intracellular iron delivery in neurons and mitochondrial damage [105]. miR-124 affected mitochondrial localization and function by targeting the intermediate filament vimentin in mouse motor neurons [106]. miR-127 targeted the mitochondrial membrane protein mitoNEET in rat primary cultured spinal neurons [107]. miR-330 has been suggested to alleviate mitochondrial dysfunction in AD by targeting vav guanine nucleotide exchange factor 1 (VAV1) via the MAPK signaling pathway in neuron cells of mice [108]. The brain-specific miR-338 regulated OXPHOS and mitochondrial metabolism via cytochrome c oxidase IV (COXIV) in neurons, involved in ETC in mitochondria of rat neuronal cells [109, 110].
miR-15b, miR-16, miR-195 and miR-424, all sharing the same “seed” sequence, were implicated in ATP production [111], while miR-23a and miR-23b targeted glutaminase, an important player in mitochondrial metabolism, and particularly in ATP production and glutathione synthesis, in different tissues or cells of human or mice [112]. Also, miR-210 was implicated in mitochondrial respiration via targeting mitochondrial iron sulphur cluster homologue in many cell types [109]. miR-15a targeted uncoupling protein 2 (in mouse β-cells), which regulates oxygen consumption and ATP production [109]. Moreover, miR-143 is involved in mitochondrial function via targeting the ERK5 pathway in human colorectal cancer DLD-1 cells [113]. In cancer cells, miR-183 targets isocitrate dehydrogenase 2, an important member of the Krebs cycle, thereby modulating mitochondrial function, as well [114].
Oxidative stress (KE2)
The miR-mediated regulation of oxidative stress has been described in respect to its involvement in several pathological conditions, including neurological disorders [115]. In human or animal studies, miR-9 [116, 117], miR-125b [116], miR-34c [118], miR-142 [119], miR-29, miR-124, miR-128 [117], and miR-210 [120] have been linked to oxidative stress triggered by reactive oxygen species (ROS) accumulation, in brain cells.
Circulating miR-124a, miR-125b, miR-142, and miR-483 have been associated with cellular oxidative stress in mild cognitive impairment (MCI) patients [121]. In serum of Parkinson’s disease patients and in human SH-SY5Y cells, miR-153 has been reported to promote oxidative stress via targeting nuclear factor erythroid 2-related factor 2 (Nrf2) [122].
Moreover, let-7a altered the levels of ROS in murine microglia under inflammatory conditions [123]. miR-132 inhibited the expression of inducible nitric oxide synthase and oxidative stress by inhibiting MAPK1 expression in hippocampus tissue of AD rats, improving their cognitive function [124]. Expression of miR-146a inversely correlated with expression of oxidative stress indicators, including malondialdehyde and p22phox, in the brain of chronic type 2 diabetes rats [125]. The miR-200 family is implicated in oxidative stress through ROS production in rat hippocampal neurons [126]. miR-486 represses neuronal differentiation 6 (NEUROD6) in mouse motor neurons due to progressive neurodegeneration mediated by ROS [127]. miR-330 suppressed oxidative stress in neuronal cells of AD mice by targeting VAV1 via the MAPK signaling pathway [74].
Other oxidative stress-related miRs, including let-7f, miR-9, miR-16, miR-21, miR-22, miR-29b, miR-99a, miR-125b, miR-128, miR-143, miR-144, miR-155, and miR-200c were reported in human cultured cells when treated with H2O2 [117]. Lastly, miR-23b was found to regulate ROS production in cancer cells [128], and to inhibit oxidative stress in mice with diabetic neuropathy via controlling MAPK signaling [129].
Hyperphosphorylation of tau protein (KE3)
Abnormal phosphorylation of tau protein by activation of kinases (e.g., ERKs, MAPKs, cyclin-dependent kinase 6 (CDK6), and GSK-3β), and deactivation of phosphatases (e.g., PP2A), is also mediated by miRs.
Brain miRs, including miR-9, miR-124, miR-132, and miR-137, regulated 4R:3 R tau ratios in sporadic progressive supranuclear palsy patients brain [130], contributing to abnormal tau phosphorylation. miR-219 mediated a decreased tau-tubulin kinase 1 protein and GSK-3β by directly binding to their 3’-UTR, inducing tau phosphorylation in human SH-SY5Y cells [131]. Expression of let-7b in cerebrospinal fluid in human CD4+ T lymphocytes positively correlated with total tau and phosphorylated tau [132]. miR-106b inhibited phosphorylation of tau at Tyr18 by targeting Fyn gene expression in human SH-SY5Y cells stably expressing tau [133]. miR-93 activated phosphoinositide 3-kinase (PI3K)/Akt pathway via directly inhibition of phosphatase and tensin homolog (PTEN), besides PH domain leucine rich repeat protein phosphatases (PHLPP) and forkhead box O3 (FOXO3), in human glioma cells and tissues [134], of which tumor-suppressor PTEN affected tau phosphorylation and aggregation through binding to microtubules in AD patient brains [135]. Also, miR-146a induced abnormal neuronal tau phosphorylation by targeting Rho associated coiled-coil containing protein kinase 1 (ROCK1)/ PTEN pathway in human SH-SY5Y neural cells [136]. In blood of AD patients, miR-103a and miR-107 were found to target cyclin dependent kinase 5 regulatory subunit 1 (CDK5R1), among other targets, and to contribute to tau hyperphosphorylation [137].
In human relevant animal studies, among the miR-16 family, miR-15(a/b) emerged as potent regulator of ERK1/2 signaling pathway in mouse or rat neuronal cells. This pathway is involved in tau phosphorylation [138, 139]. miR-15a, miR-195, and miR-497 were demonstrated to directly interact with the 3’-UTR of ERK1 mRNA in mouse N2a cells [138]. miR-124 inhibited abnormal tau hyperphosphorylation by targeting caveolin-1- PI3K/Akt/GSK3β pathway in mouse neuroblastoma cell lines [140]. miR-148a regulated protein kinase B and MAPK/ERK signaling pathways in rat hippocampal neurons [75], known to contribute to tau hyperphosphorylation. miR-125b induced tau hyperphosphorylation and upregulation of p35, cdk5, and p44/42-MAPK signaling in rat primary neurons [141]. miR-101b targeted 5’ adenosine monophosphate (AMP)- activated protein kinase (AMPK) blocking the histone deacetylase 2 (HDAC2), suggested to be involved in tau pathology, besides dendritic abnormalities, and memory deficits in AD mice [142]. miR-98 increased tau phosphorylation in mouse N2a/WT cells [78]. miR-26a directly regulated GSK-3β gene expression in human airway smooth muscle cells [143], and miR-26b activated CDK6 in rat primary cortical neurons [144], both targeting brain-derived neurotrophic factor (BDNF) [145] and implicated in tau hyperphosphorylation in the hippocampus of a mouse model for Down syndrome during aging. miR-34a and miR-34c regulated the tau expression in human neuroblastoma cell lines [146], with the latter having a negative effect on memory formation via suppression of sirtuin 1 (SIRT1) levels in mouse hippocampus [147]. In transfected primary neurons of rat hippocampus or cortex, miR-128a affected tau hyperphosphorylation via targeting co-chaperone BAG2 pathway [148]. In cultured rat cortical neurons, miR-200c repressed PTEN expression [149] and induced tau hyperphosphorylation in human SH-SY5Y cells [150]. miR-326 decreased hyperphosphorylation of tau by inhibiting c-Jun N-terminal kinase pathway and by targeting VAV1 in neurons of AD mice [151].
Overexpression of brain-enriched miR-138 increased tau phosphorylation in human HEK293/tau cells via targeting the retinoic acid receptor alpha/GSK-3β pathway [152]. Lastly, miR-21 has been shown to regulate matrix metalloproteinase-2 via PTEN pathway in murine cardiac fibroblasts [153].
Dysfunctional autophagy (KE4)
Many miRs have been implicated in the regulation of autophagy by targeting autophagy-related proteins (i.e., autophagy genes (ATG), mTOR) or proteins related to lysosome system [117].
miR-17 regulated the ATG7 and autophagic processes in human glioblastoma cells [154]. miR-100 targeted ATG10 in 1-methyl-4-phenyl-pyridinium ion (MPP+)-intoxicated SH-SY5Y cells [155]. miR-137 reduced starvation-induced autophagy via targeting ATG7 in human glioma cell line [156]. Inhibition of miR-132 in human SH-SY5Y cells suppressed mRNA and protein expression levels of autophagy-related protein light chain 3 (LC3) and Beclin 1 [157]. miR-27a and miR-27b regulated PTEN-induced kinase 1 expression and autophagic clearance of damaged mitochondria in human dopaminergic-like M17 cells [158]. miR-124 regulated autophagy via targeting AMPK/mTOR pathway in human dopaminergic neurons [159]. miR-128 targeted key members in mTOR signaling, including mTOR, RICTOR, IGF1, and phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1), in human glioma cells [160]. miR-181b and miR-590 controlled autophagy by targeting the PTEN/Akt/mTOR signaling pathway, in PC12 cells and human glioma cells, respectively [161, 162]. Also, miR-496 has been reported to reduce the expression of mTOR in peripheral blood mononuclear cells of aged subjects [163].
Several animal studies have addressed this key event of human AD pathology. miR-23b was correlated with cognitive impairment after traumatic brain injury through inhibition of ATG12-mediated neuronal autophagic activity in rat model of traumatic brain injury [164]. miR-214 decreased autophagy of hippocampal neurons via targeting Atg12 expression in SAMP8 mouse model of sAD [165]. In murine neurons, miR-299 suppressed Atg5 and antagonized caspase-dependent apoptosis, inhibiting autophagy [166]. miR-9 was implicated in autophagy in rat bone marrow mesenchymal stem cells differentiating into neurons by regulating the ratio of autophagy-related protein LC3 and the expression of neuron specific enolase and microtubule-associated protein 2 (MAP2) [167]. miR-193b directly targeted tuberous sclerosis 1 to regulate mTORC1 activity, promoting protective autophagy in mouse neuroblastoma NSC-34 cells [168]. miR-34a activated SIRT1/mTOR signaling pathway inducing impaired autophagy in brain aging mice after administration of urolithin A supplementation [169]. miR-199a controlled methyl-CpG binding protein 2 (MeCP2) and mTOR signaling in the brain of mice [170]. miR-106b has been involved in autophagy by targeting to sequestosome 1 (SQSTM1)/p62 in mouse hematopoietic myeloid progenitors [171].
In other cell types, miR-20a and miR-106b regulated leucine deprivation-induced autophagy via direct targeting of the Unc51-like kinase 1 (ULK1) protein in myoblasts [172]. There is evidence supporting the involvement of miR-93 in autophagy by regulating core autophagy kinase, ULK1 expression in mouse embryonic fibroblasts or Chinese hamster ovary cells [173], and of miR-30d and miR-101 through inhibition of Beclin1 and Atg4d expression in human breast cancer cells or hepatocellular carcinoma cells [174]. miR-200c upregulated the endoplasmic reticulum stress signaling via LC3-II expression in human prostate adenocarcinoma PC-3 cells [175], while miR-103/miR-107 regulated end-stage autophagy in mouse epidermis via phospholipase D (PLD)1/2–protein kinase C–dynamin pathway [176]. miR-101 has been suggested as key regulator of autophagy via targeting stathmin 1 (STMN1) in human breast cancer cells. miR-183 has been shown to regulate autophagy via targeting ultraviolet radiation resistance-associated gene, an autophagy regulator promoting autophagosome formation and maturation, in colorectal cancer cells [177]. Finally, miR-15a, miR-20a/b, miR-23a, miR-29a, miR-106a/b, miR-195, miR-590 [117], and miR-101 [178] have been reported as autophagy-related miRs, but mainly in cancer.
Synaptic dysfunction (KE7)
The role of miRs in synaptic transmission and plasticity has been described by others [179].
Let-7 family members regulated synaptic function [180] and modulated synaptic plasticity by controlling BDNF translation [181]. In particular, let-7c was implicated in synaptic function in human neurons [182]. miR-26a/b and miR-374 contributed to the regulation of BDNF expression in human peripheral nuclear blood cells [183]. In human neurons, miR-132 and miR-182 decreased the BDNF protein levels [184]. miR-34 regulated synaptogenesis [185] and mediated the regulation of post-synaptic cytoskeletal SH3 and multiple ankyrin repeat domains 3 (SHANK3) in superior-temporal lobe of sAD patients [186]. miR-130b targeted MECP2 in human brain neocortex [187]. In brains of AD patients, miR-30b regulated ephrin type-B receptor 2, SIRT1, and glutamate ionotropic receptor AMPA type subunit 2 (GluA2), which are all important targets for maintaining synaptic integrity [188]. In human plasma, miR-181c and miR-210 targeted neuronal pentraxin 1 (NPTX1) and neuronal pentraxin receptor (NPTXR), both implicated in activation and clustering of AMPA receptors in postsynaptic terminals, and affecting glutamatergic synapses in the hippocampus and synaptic plasticity [189]. miR-572 targeted neural cell adhesion molecule 1 (NCAM1) expression promoting progressive decrease in myelination in human oligodendroglial cell line [190, 191]. miR-485 was found to regulate synaptic genes, PSD95, surface GluR2, and the miniature excitatory post-synaptic current frequency as shown in human hippocampal neurons [192].
Animal studies addressing this key event of human pathology have identified several miRs with potential human relevance. Among brain-specific miRs, miR-9(-3p) has been associated with hippocampal long-term potentiation (LTP), and learning and memory deficits, via targeting LTP-genes, dystrophin (Dmd) and synapse-associated protein 97 (SAP97) in mouse hippocampus [193]. Also, miR-132, miR-134, and miR-138 regulated the neuronal dendritic spines, excitatory synaptic transmission, and synaptic function [193]. miR-26a and miR-384 were downregulated during LTP, and both targeted ribosomal S6 kinase 3 (RSK3), regulating LTP maintenance and long-lasting spine enlargement in hippocampal slices from mice [194]. Inhibition of miR-34b/c in postischemic hippocampal CA1 pyramidal neurons (mice) rescued impaired synaptic plasticity and diminished memory deficits [195]. miR-124 was implicated in long-term plasticity of synapses (rat PC12 cells) [196] and dendritic actin cytoskeleton via regulating the activity of small Rho GTPases (mouse primary cortical neurons) [197]. In a study on adult mouse forebrain, miR-7, miR-29a, miR-137, miR-200c, miR-322, and miR-339 were found highly present in synapses [198]. miR-151 and miR-23a were found to be involved in synaptic reorganization and transcription, respectively, in rat dentate gyrus [199]. The neurite-enriched miR-376b was found to regulate synaptic transmission and plasticity during postnatal period in the mouse hippocampus [200]. miR-16 regulated the neurotrophin receptor-mediated MAPK/ERK pathway in mouse hippocampal neurons and was demonstrated to play an important role in both synapse formation and plasticity [201]. miR-132 targeted MeCP2 in rat neurons [187, 202] and regulated BDNF–ERK–cAMP response element-binding protein (CREB) signaling pathway in rat cortex [203], implicated in dendritic development, synaptogenesis, and synaptic plasticity. In primary rat hippocampal neuronal cells, ectopic expression of miR-34a, miR-193a, or miR-326, modulated endogenous Arc protein expression in response to BDNF treatment [204]. In the nucleus accumbens (in rat basal forebrain), let-7d regulated the expression of BDNF, glutamate ionotropic receptor AMPA Type Subunit 2 (GRIA2), MeCP2, phosphorylation of CREB, supporting its involvement in regulating synapse maturation, growth, and dendritic arborization [205]. miR-19b targets PTEN, regulated dendritic development and synapse formation by modulating the AKT-mTOR signaling pathway in mouse hippocampal neurons [206]. miR-301a directly bound to PTEN in rat cortical neurons, conferring neuroprotection in oxygen-glucose deprivation-induced neuronal injury [207]. Glial-enriched miR-19a has been shown to target Mef2c and neurogenin 1 (Neurog1), known to enhance neuronal differentiation, in primary cultures from postnatal (day 1) rat cortex [208]. miR-134 [209] and miR-138 [210] were found to be involved in synaptic plasticity via targeting SIRT1 in murine neuronal cells. miR-7 targeted neuroligin 2 (NLGN2) and altered synaptic function [188]. miR-124 suppressed GluA2 expression via binding to its 3-UTR, resulting in calcium-permeable AMPARs (CP-AMPARs) formation in rat primary hippocampal neurons [211]. miR-132 and miR-212 affected synaptic transmission and plasticity possibly by regulating the number of postsynaptic AMPA receptors in mice [212]. In mouse hippocampal neurons, miR-146a was demonstrated to bind to microtubule associated protein 1B (MAP1B) mRNA and to inhibit mGluR-dependent AMPA receptor endocytosis [213], while miR-223 bound to the NMDA receptor subunit GluN2B and the AMPA receptor subunit GluA2 [214]. miR-125b (targeting NMDA receptor subunit NR2A) and miR-132 (targeting Rho GTPase-activating protein (p250GAP)) have been reported to exhibit opposing effects on dendritic spine morphology and synaptic function in mouse hippocampal neurons and both to target fragile X mental retardation protein (FMRP) [215, 216]. miR-137 regulated glutamate receptor and synaptic vesicle via binding to GRIA1 enhancing AMPA receptor-mediated synaptic transmission, or via targeting synaptotagmin-1 (Syt1), a synaptic vesicle membrane protein, in rat neurons [217]. miR-125a affected postsynaptic density protein 95 (PSD-95) expression and FMRP phosphorylation as described in mouse cortical neurons [218]. miR-501 mediated the activity-dependent regulation of the AMPA receptor subunit GluA1 expression in rat hippocampal neurons [179]. miR-204 directly targeted EphB2, a component of Eph/Ephrin synaptic signaling, and N-methyl-D-aspartate receptor subunit NR1 in aging mouse hippocampal neurons [219]. miR-188 targeted neuropilin 2 (Nrp-2) to potentiate spine development, miR-135, miR-191, and miR-29a/b targeted tropomodulin 2 (Tmod2), complexin-1/2 and ARP2/3 complex subunit 3 (Arpc3), respectively, and exerted an impact on actin cytoskeleton and spine morphology in rat hippocampal neurons [216]. miR-148a regulated synaptic transmission through targeting known synaptic genes, e.g., Snap25, syntaxin 6 (Stx6), vesicle associated membrane protein 1 and 2 (Vamp1 and Vamp2), calmodulin 1 (Calm1), heat shock protein family A (Hsp70) Member 1A (Hspa1), gap junction protein alpha 1 (Gja1), calcium/calmodulin-dependent protein kinase type II alpha chain (CaMKII), synaptotagmin (Syt), NMDA-R and Vamp in mouse hippocampi [220]. Overexpressed miR-210-5p associated with decreased synaptic number via targeting Snap25 in primary hippocampal neurons of rats [221]. miR-574 expression in hippocampi of mice associated with impaired synaptic and cognitive function in response to exposure to particulate matter [222].
In human or animal studies, miR-10b (human), miR-15a (mouse), miR-206 (mouse) [223–225], miR-30a (human) [226], miR-155 and miR-191 (human) [225] regulated neuronal maturation and promoted BDNF expression in neurons. Functional enrichment studies using human and mouse data demonstrated the involvement of miR-195 in synaptic function by rescuing in vivo ApoE4-associated cognitive deficits [227]. Knockdown of miR-1 in heart resulted in upregulated SNAP-25 expression in synapses of hippocampi from transgenic mice through transportation of exosomes [228].
Neuroinflammation (KE8)
Numerous miRs have been implicated in neuroinflammation leading to AD pathology, with microglia, specialized long-term resident macrophages, to play a key role in the inflammatory responses. Microglial activation is accompanied by expression of recognition, cytokine, and neuronal receptors. Microglia differentiate into to M1 (pro-inflammatory) or M2 (anti-inflammatory) phenotypes. M1 microglia release inflammatory mediators e.g., ROS, matrix metalloproteinase-9 and pro-inflammatory cytokines, e.g., TNFα, interleukin (IL)-6 and IL-1β, while M2 microglia release anti-inflammatory and protective cytokines, e.g., IL-10, transforming growth factor beta 1 (TGF-β), IL-4, and IL-13 [229].
In a review, several miRs are discussed in the context of their involvement in neuroinflammation observed in different models [229]. Let-7 family either inhibited or promoted inflammation. On one hand, let-7 induced polarization of macrophages into the anti-inflammatory M2 phenotype through targeting C/EBP-δ transcription factor and promoted M2 phenotype in microglia. On the other hand, let-7 stimulated inflammatory response via targeting the cytokines IL-6, IL-10, and toll-like receptor 4 (TLR4), and promoted activation of microglia and macrophages via targeting TLR7 [229, 230]. miR-155 and miR-124 were induced in macrophages and microglia in response to nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) dependent TLR signaling and the Th2 cytokines IL-4 and IL-10, and TLR-6 and myeloid differentiation primary response 88 (MyD88), respectively [229]. miR-107 regulated neuroinflammation via targeting granulin [229].
miR-27a inhibited inflammatory response via negatively regulating TLR4, TNFα, and IL-6 as shown in human serum [231]. miR-19b was implicated in inflammatory responses via regulating NF-κB signaling in human brain microvascular endothelial cells upon meningitic E. coli infection [232]. miR-30a was identified as an important inflammatory modulator in human microglia (of chronic active lesions of multiple sclerosis patients) targeting the proinflammatory cytokines, IL-1β, and inducible nitric oxide synthase [233]. miR-132 induced increase in proinflammatory lymphotoxin and TNFα via suppressing SIRT1 in human B cells of multiple sclerosis patients [234]. miR-142 has been suggested as key mediator of IL-1β via downregulation of the glial glutamate-aspartate transporter (GLAST) in human cerebrospinal fluid with active multiple sclerosis [235]. Overexpression of miR-29a/b cluster in immune cells has been linked to chronic inflammation [236] and aging via targeting specific central nervous system microglia modulators, IGF1 and C-X3-C motif chemokine ligand 1 (CX3CL1) in human cortical tissue [69].
miR-9 regulated the NF-κB pathway and induced microglial activation through targeting monocyte chemotactic protein-1-induced protein-1 (MCPIP1) in rat in vitro and in vivo models [237]. miR-125b regulated microglia activation via NF-κB-dependent inflammatory pathway in mouse microglial cells [238]. miR-27b directly targeted Bcl-2-associated athanogene 2 (Bag2) in macrophages regulating TLR-2/MyD88/NF-κB signaling pathway in murine macrophages-like cells [239]. miR-146a regulated TLR/MyD88/NF-κB and Janus kinase-signal transducer and activator of transcription protein (STAT) pathways in mouse microglial cells [240]. miR-125a regulated macrophage activation and inflammation through TLR2 and TLR4 in mouse bone marrow cells [241]. Inhibition of the expression of the miR-15a/16 cluster decreased the expression of inflammatory cytokines, IL-1β and TNFα, and increased the expression of G-protein-coupled receptor kinase 2 (GRK2), in the spinal cords of chronic constriction injury rats [242]. miR-30c targeted inflammatory mediators, including intercellular adhesion molecule 1 (ICAM-1), IL-1β, and TNFα, in spinal cord injury (rats) [243]. miR-101 induced inflammation in lipopolysaccharide-activated microglial cells in rats via targeting IL-1β, IL-6, and TNFα, and inhibiting mitogen-activated protein kinase phosphatase 1 [244]. Downregulation of miR-148a was linked to microglial activation in acute cerebral ischemic stroke via modulating the secretion of inflammatory mediators such as TNFα, IL-1β, and IL-1 in mouse microglial cells [245]. miR-181 was implicated in inflammatory responses of astrocytes, and its aberrant expression in mouse brain cells enhanced the production of pro-inflammatory (TNFα, IL-6, IL-1β, IL-8) and anti-inflammatory (IL-10) cytokines [246]. In mouse primary astrocytes, miR-7 targeted the endoplasmic reticulum (ER) stress protein-HERP2, which mediated ER stress-induced inflammation [247]. miR-137 was found to prevent inflammatory response and cognitive impairment via inhibiting Src-dependent MAPK signaling pathway in brain tissues of mice [248]. Lower levels of miR-143 in dorsal root ganglion neurons due to inflammation were reported in mice [249]. miR-34a along with miR-155 and miR-145 promoted inflammatory response by suppressing c-Maf in microglia from mice [250]. miR-23b modulated neuroinflammation by targeting inositol polyphosphate multikinase (IPMK) in rat BV2 microglial cells and HT22 neuronal cell lines [251]. Increased expression of miR-139 impaired inflammation via targeting eukaryotic translation initiation factor 3 subunit K (EIF3K) in serum of rats with hypoxic-ischemic brain damage [252]. In MPP+-induced Parkinson’s disease mouse model, downregulated miR-205-5p via targeting LncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) and lncRNA HOX transcript antisense intergenic RNA (HOTAIR) activated neuroinflammation in parkinsonism [253].
In vitro studies suggested that activated microglia induced miR-21 regulating IL6/STAT3 pathway in human models [254]. Lastly, miR-196a stimulated inflammation by regulating NF-κB signaling via annexin A1 (ANXA1) in endothelial cells or c-myc in human breast cancer cells [255].
Mechanistic cross-over between sAD and environmental neurotoxicity
Based on our hypothesis, sAD and environmental neurotoxicity may share common dysregulated processes, which can be controlled by common miRs. Using existing data on the potential function of the selected miRs implicated in AD pathogenesis and/or environmental neurotoxicity, we linked the retrieved miRs to the possible affected starting point or molecular/cellular KEs described in the tau-driven AOP [11]. Taking together, the AD-related miRs with their possible implication in neurotoxicity (human and/or animal), linked to the hypothetical starting point and/or KEs of the proposed AOP toward memory loss, are shown in Fig. 2.
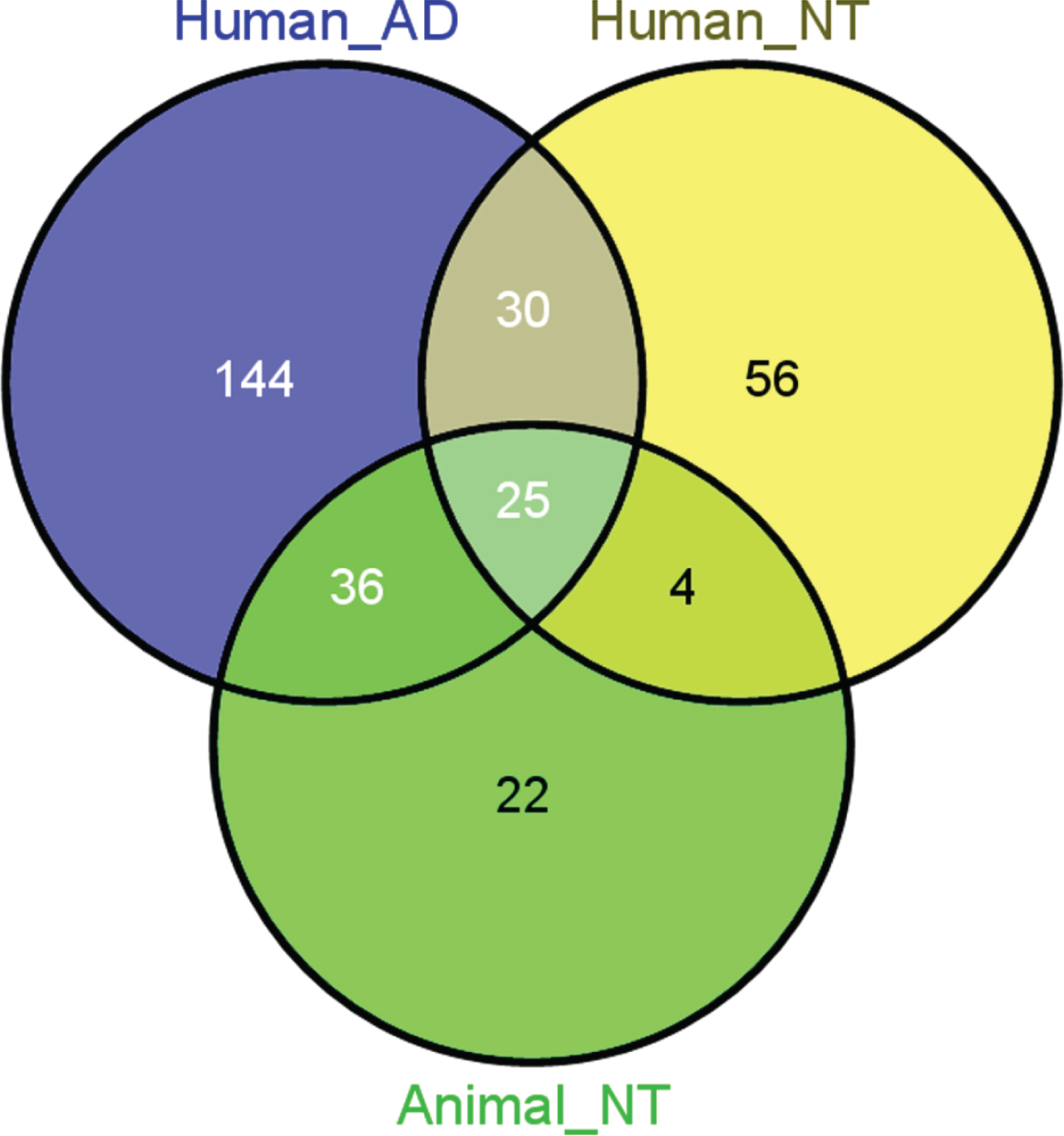
Overlap between miRs implicated in AD and in environmental neurotoxicity (NT), assigned as human and animal NT, respectively (Venn’s diagrams) [298].
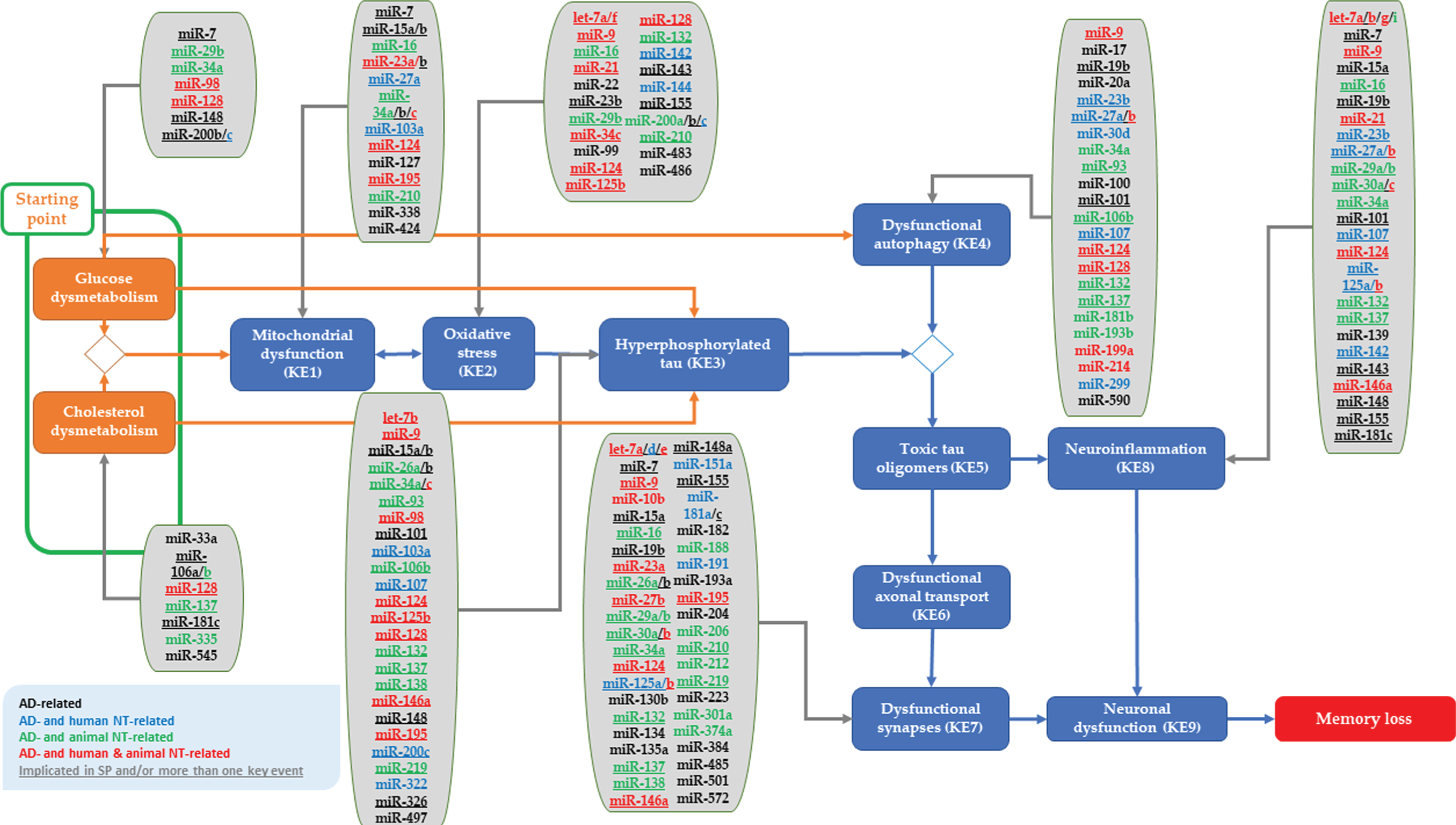
miRs implicated in AD (and/or neurotoxicity) plugged into the proposed tau-driven AOP toward memory loss. Underlined miRs are implicated in the regulation of more than one hypothetical starting point or key event of the plausible AOP. The relevance of these miRs to human neurotoxicity is indicated in blue color, to animal neurotoxicity in green color, and to both human and animal neurotoxicity in red. While miRs only linked to human AD are indicated in black color.
Functional miR analysis
The complete list of targets of the selected miRs is reported in Supplementary Table 2A and includes human miR–target interactions validated at experimental level retrieved by MiRTarBase. Functional analysis of the identified genes was performed considering the targets of the miRs involved in the proposed starting point and KEs as separate gene sets. PathDip outputs are available in Supplementary Table 2B. Biological relevant pathways were filtered, to finally select those genes that are proven to play a role in each specific starting point or KE. Figure 3 reports the list of curated sources containing the selected pathways in the case of glucose and cholesterol dysmetabolism, described as the hypothetical starting point of the AOP. REACTOME, KEGG, and WikiPathways are the most represented. Figure 4 reports the complexity of human miR–target interactions pathways for the hypothetical starting point. For representative purpose, only the hypothetical starting point is reported. Sankey diagram is a visualization technique that allows representing flows. It depicts how tens of miRs and hundreds of genes are potentially involved in few events. The number of interactions and their intersections enormously increases looking jointly at the starting point and the KEs. Table 2 summarizes only the number of unique biological relevant genes and miRs for each event or component of the proposed AOP.
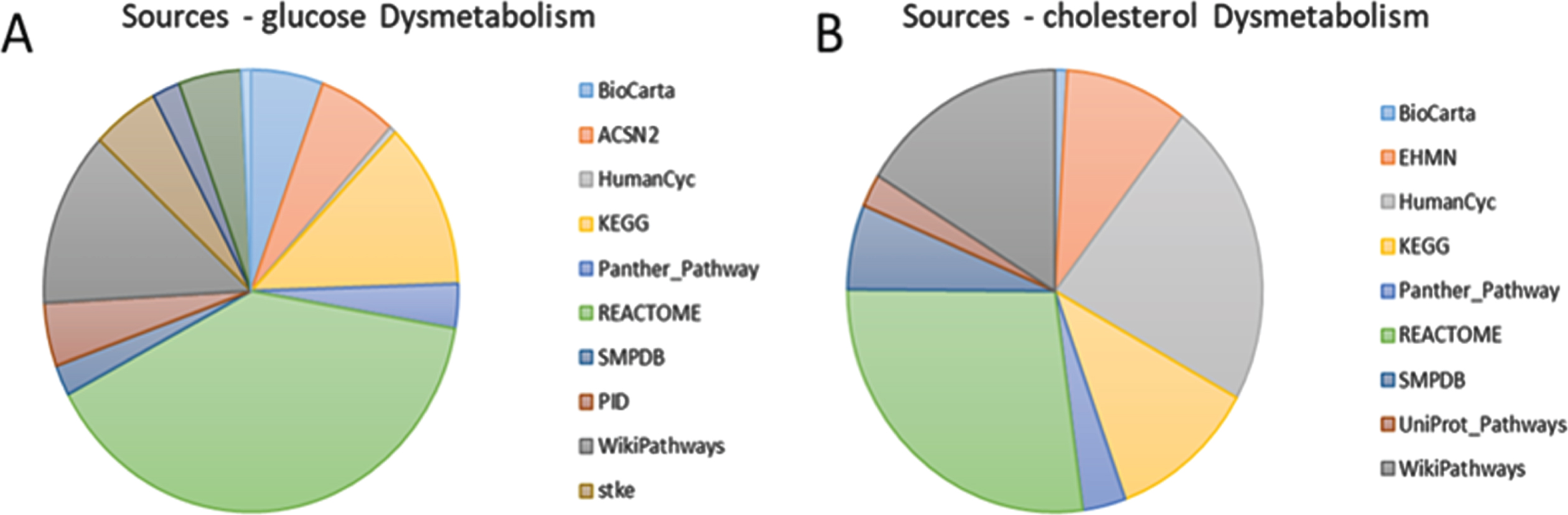
Sources reporting biologically relevant pathways for glucose dysmetabolism (A) and cholesterol dysmetabolism (B). The pie chart also describes the contribution given by each source.

The Sankey diagrams represent the miR–target interactions involving genes that are proven to be involved in the pathways of interest (cholesterol & glucose (dys)metabolism). SP-Cdys refers to starting point-cholesterol dysmetabolism and SP-Gdys refers to starting point-glucose dysmetabolism.

Networks describe respectively: the link between annotated genes and glucose metabolism related pathway (A), the interaction between miRs and selected gene targets (B), merged interactions (C).

Networks describe respectively: the link between annotated genes and cholesterol metabolism related pathway (A), the interaction between miRs and selected gene targets (B), merged interactions (C). aNT indicates “human neurotoxicity” and hNT “human neurotoxicity”.
Number of unique biological relevant genes, pathways of interest and miRs for each event or component of the proposed AOP. In the second column, only the number of pathways of interest obtained after filtering out those not related to the examined process are reported. Subsequent number of biological relevant genes and miRs are reported in the third and fourth column, respectively
Starting point (SP): glucose and cholesterol dysmetabolism
DipPath revealed 86 pathways related to “Glucose metabolism” regulated by 305 unique genes of which 12 unique miRs were potentially targeted based on their MTIs retrieved by MiRTarBase. Out of these 12 miRs, miR-29b(-3/5p), miR-98(-3/5p), and miR-128(-3/5p) were in overlap with literature search for their implication in AD and in brain glucose metabolism. miR-98-5p and miR-128-3p potentially target IRS2 and MTOR, respectively, while both miR-98-5p and miR-128-3p bind to IGF1 and MAPK6, among other genes. With regards to their link to neurotoxicity, miR-98 and miR-128 are involved in both human and animal neurotoxicity, while miR-29b only in animal neurotoxicity. Further, miR-302(a) has been shown in literature and by functional analysis to be involved in glucose metabolism; however, its relevance to human AD is not clear. While for the AD-related miR-29a(-3/5p) (animal neurotoxicity) and miR-126(-3/5p) (animal neurotoxicity), and for miR-3666 (unknown link to AD and neurotoxicity), which have been identified to be involved in glucose metabolism by this analysis, no data on their link to brain glucose metabolism has been earlier reported.
Similarly, 19 pathways were identified to be involved in “Cholesterol metabolism”, of which 94 unique genes were found to be potentially targeted by 22 unique miRs. Among them, miR-33a(-5p), miR-106a(-3/5p), miR-106b(-3/5p), miR-128(-2-5p), miR-181c(-3/5p), miR-335(-5p), and miR-545(-3p) were also confirmed by literature for their link to both AD and brain cholesterol metabolism. Among their potentially targeted genes, LDLR was controlled by miR-106a-3p, miR-106b-5p, and miR-335-5p, ABCA1 by miR-106a-3p and miR-106b-5p, and SREBF1/2 by miR-335-5p. In addition, miR-106b and miR-335 have been associated with animal neurotoxicity, while miR-128 with both human and animal neurotoxicity. Functional miR analysis revealed miR-1(-3/5p) (human neurotoxicity), miR-27a(-3/5p) (human neurotoxicity), miR-27b(-3p) (human and animal neurotoxicity), miR-30c(-5/3p) (human and animal neurotoxicity), miR-148a(-3/5p), miR-206 (animal neurotoxicity), miR-613 and miR-1229(-3p) to be involved in cholesterol metabolism. However, there is no evidence for the relevance of miR-1, miR-613 and miR-1229 to AD, while for the AD-related miR-27a, miR-27b, miR-30c, miR-148a and miR-206, there is no clear link to brain cholesterol metabolism.
Key events (KEs)
As reported in Fig. 7, some miRs could potentially target several genes with biological relevance to one of the KEs. The functional analysis revealed that most of the identified biological genes (n = 745) were relevant to processes regulating synaptic function.

Each key event occupies a portion of the diagram proportional to the number of biological relevant genes retrieved by the KE related gene set. The most external section of the diagram reports the miRs regulating these genes, according to miRTarBase.
KE1: Mitochondrial dysfunction
Fifty-six pathways were found related to “mitochondrial function”, and 25 unique miRs associated with these pathways are known to potentially interact with the 346 identified unique genes. Mitochondrial function-related miRs, including miR-7(-3/5p), miR-23b(-3p), miR-27a(-3/5p), miR-34a(-3/5p), miR-34c(-3/5p), miR-103a(-3p), miR-124(-3p), miR-195(-3/5p), miR-210(-3/5p), and miR-338(-5p) were commonly identified by literature and functional analysis. miR-7-3/5p, miR-23b-3p, miR-27a-5p, miR-34a-3p, and miR-210-3p possibly interact with ATP-related genes, such as ATP11 C, ATP7B, ATP7A, ATP1B3, ATP1A1, ATP8B2/3, and ATP8A2, among other miR targets. Besides their link to AD, miR-34a and miR-210 have been shown to be involved in animal neurotoxicity, while miR-23b, miR-27a, miR-34c, miR-103a, miR-124, and miR-195 in human (and/or animal) neurotoxicity. This analysis also revealed the implication of AD-related miR-143 and miR-183 and miR-330 (link to AD is currently unknown) in mitochondrial function, however, no evident data were found in literature for brain cells.
KE2: Oxidative stress
After filtering out the relevant pathways to “oxidative stress”, 51 pathways were obtained, regulated by 218 unique genes which are possibly targeted by 31 unique miRs. miR-9(-3/5p), miR-23b(-3p), miR-29b(-3/5p), miR-34c(-3/5p), miR-125b(-3/5p), miR-128(-3/5p), miR-132(-3/5p), miR-142(-3/5p), miR-200a(-3/5p), miR-200b(-3/5p), miR-200c(-3/5p), miR-210(-3/5p), miR-483(-5p), and miR-486(-5p) were indicated by literature, as well. Among their regulated genes, MAPK-related genes such as MAPK14, MAPK1, and MAPK3, are targeted by miR-9-3p, miR-125b-5p, miR-128-3p, miR-132-3p, miR-142-5p, miR-200a-3p, or miR-483-5p, and E2F1/2/3 genes by miR-9-3p, miR-23b-3p, miR-34c-5p, miR-125b-5p, miR-128-3p, miR-200b-3p, miR-200c-3p, or miR-210-3p. Among these miRs, miR-9, miR-34c, miR-125b, and miR-128 are implicated in human and animal neurotoxicity, whereas miR-29b, miR-132, miR-200a, and miR-210 in animal neurotoxicity, and miR-23b, miR-142, and miR-200c in human neurotoxicity. Notably, the identified miR-153(-3/5p), miR-196a(-5p), and miR-330(-3/5p) by this analysis were not confirmed by literature for their relevance to human AD.
KE3: Hyperphosphorylated tau
There were found 32 pathways linked to “hyperphosphorylated tau”, with 604 unique genes potentially targeted by 43 unique miRs. Commonly found miRs between functional analysis and literature, let-7b(3/5p), miR-9(-3/5p), miR-15a(-3/5p), miR-15b(-3/5p), miR-26a(-3/5p), miR-26b(-3/5p), miR-34a(-3/5p), miR-34c(-3/5p), miR-93(-3/5p), miR-98(-3/5p), miR-101(-3/5p), miR-103a(-3p), miR-106b(-3/5p), miR-124(-3p), miR-125b(-3/5p), miR-128(-3/5p), miR-132(-3/5p), miR-138(-3/5p), miR-146a(-3/5p), miR-200c(-3/5p), miR-219a(-3/5p), and miR-326. PTEN, CDK6, and/or PIK3R1 were targeted by let-7b-5p, miR-26a-3/5p, miR-34a-3/5p, miR-93-5p, miR-101-5p, miR-103a-3p, miR-106b-3/5p, miR-124-3p, and miR-128-3p. While GSK3B by let-7b-3p, miR-9-5p, miR-26a-3p, and miR-98-3p. Only miR-21(-3/5p), which is involved in AD and (human and animal) neurotoxicity, was not directly linked to KE3 in brain cells. Out of the overlapped KE3- and AD-related miRs, let-7b, miR-9, miR-34c, miR-98, miR-124, miR-125b, miR-128, and miR-146a are also involved in neurotoxicity (human and animal). While miR-26a, miR-34a, miR-106b, miR-132, miR-138, and miR-219 are implicated in animal neurotoxicity, and miR-103a and miR-200c in human neurotoxicity.
KE4: Dysfunctional autophagy
DipPath revealed 16 pathways related to “autophagy”, of which 441 unique genes were identified to be possibly targeted by 37 unique miRs. Among them, miR-9(-3/5p), miR-17(-3p), miR-20a(-5p), miR-23b(-3p), miR-27a(-3/5p), miR-27b(-3/5p), miR-34a(-3/5p), miR-93(-3/5p), miR-100(-3/5p), miR-103a(-3p), miR-107, miR-124(-3p), miR-128(-3/5p), miR-132(-3/5p), miR-181b(-3/5p), miR-193b(-3/5p), miR-199a(-5p), miR-214(-3/5p), miR-299(-5p), and miR-590(-3/5p) were shown to be autophagy-related by literature, as well. miR-93-5p, miR-181b-5p, and miR-214-5p potentially target ULK1 or ULK2, while miR-128-3p and miR-181b-5p bind to MTOR. Moreover, miR-183 and miR-496, both involved in animal neurotoxicity, were found to be related to KE4 but they were not confirmed by literature for their relevance either to autophagy in brain cells or to human AD.
KE7: Synaptic dysfunction
There were found 77 unique miRs possibly targeting the 476 unique genes that regulate the identified 42 relevant pathways to “synaptic function”. Most of them, including let-7d(-5p), miR-9(-3/5p), miR-15a(-3/5p), miR-16(-3/5p), miR-19b(-3/5p), miR-23a(-3/5p), miR-26a(-3/5p), miR-26b(-3/5p), miR-29a(-3/5p), miR-29b(-3/5p), miR-30a(-3/5p), miR-30b(-3/5p), miR-34a(-3/5p), miR-34b(-3/5p), miR-124(-3p), miR-125a(-3/5p), miR-125b(-3/5p), miR-130b(-3p), miR-132(-3/5p), miR-134(-3/5p), miR-135a(-3/5p), miR-138(-3/5p), miR-148a(-3/5p), miR-151a(-3/5p), miR-182(-5p), miR-188(-3/5p), miR-191(-5p), miR-193a(-3p), miR-195(-3/5p), miR-204(-3/5p), miR-206, miR-210(-3p), miR-212(-3/5p), miR-223(-3/5p), miR-301a(-3p), miR-326, miR-374a(-3/5p), miR-384, miR-485(-5p), and miR-501(-3p), were also indicated as KE7-related by literature. Among their targets, BDNF is possibly regulated by miR-30a-5p, miR-124-3p, miR-132-3p, miR-206, and miR-210-3p, PTEN by miR-23a-3p, miR-26a-3p, miR-29a-3p, miR-29b-3p, and miR-34a-3p, and CREB-related genes (CREB1/5, CREB3L2) by miR-9-5p, miR-124-3p, miR-125a-5p, miR-206, and miR-374a-5p. In addition, for the identified KE7-related let-7c, miR-1, miR-19a, miR-376b, and miR-574, there are no data on their link to human AD.
KE8: Neuroinflammation
Pathway analysis showed 62 pathways linked to “neuroinflammation”, which are regulated by 745 unique genes, which are potentially targeted by 47 unique miRs. Out of these identified miRs, let-7a(-5p), let-7b(-3/5p), let-7g(-5p), miR-7(-3/5p), miR-9(-3/5p), miR-21(-5p), miR-23b(-3p), miR-27b(-3/5p), miR-29a(-3/5p), miR-29b(-3/5p), miR-30c(-3/5p), miR-34a(-3/5p), miR-101(-3/5p), miR-107, miR-124(-3p), miR-125a(-5p), miR-125b(-3/5p), miR-132(-3/5p), miR-139(-3/5p), miR-142(-3/5p), miR-143(-3/5p), miR-146a(-3/5p), miR-148a(-3/5p), miR-181c(-3/5p), and miR-205(-3p) were also confirmed by literature for their relevance to KE8 in brain cells. Let-7b-3p, miR-9-3/5p, miR-34a-5p, miR-124-3p, and miR-146a-5p potentially bind to NF-κB-related genes. Moreover, for miR-196a no clear evidence for its link to human AD has been described earlier.
Considering all miRs with overlap between functional analysis and literature, we further checked for their potential targeted genes across the KEs of the proposed AOP. Forty miRs were identified, whose common targets were found in at least two different events of the proposed AOP (Fig. 8). In these 40 miRs, for some mature miRs, both 3p- and 5p-arm sequences are included. More details are provided in Supplementary Table 3.

The heatmap reports 40 miRs, whose common targets (in yellow) affect multiple events (hypothetical starting point and/or KEs) of the proposed AOP (in blue).
DISCUSSION
In late-onset sAD, neurodegeneration is postulated to be initiated 10-20 years before the first clinical signs become apparent [256]. Increasing evidence in the etiology of AD pathogenesis suggests other mechanisms, such as accumulation of damaged mitochondria, subsequent mediated-oxidative stress, or dysfunctional autophagy, rather than intracellular and extracellular plaque formation, to have fundamental roles in the early stages of the disease development [257–260]. Up to now, although numerous human or animal approaches have been developed for comprehending the etiopathology of sAD, the initiating events and early processes manifesting the disease remain unclear. Currently, both the inaccurate early diagnosis of sAD and the lack of effective treatments for AD [261] negatively affect not only the burden of the disease at individual level, but also the social and health care costs. Therefore, there is an urgent need for increasing the understanding of the early processes of sAD pathology and the impact of environmental and systemic risk factors on these processes, as well as for identifying human-relevant molecular biomarkers for sAD diagnosis before the appearance of the disease.
In this study, we considered the possible key events described in a recently published proposal for tau-driven AOP toward memory loss [11], and analyzed which human miRs were associated with the hypothetical starting point (i.e., glucose and cholesterol dysmetabolism) and KEs described in the proposed AOP. Such miRs are suggested as plug-ins for the plausible AOP for memory loss characterizing the early phase of sAD. In addition, we explored the mechanistic cross-over between sAD and environmental neurotoxicity, which we think that it may contribute to a better understanding of sAD initiation and early progression. Based on this, potential neurotoxicity-related molecular initiating events (MIEs) induced by environmental chemicals or drugs, were plugged into the tau-driven AOP, and it was hypothesized that neurotoxicants may accelerate disease progression by amplifying ongoing aging-related processes [11].
Several neurotoxic compounds have been involved in these molecular events or starting point, possibly leading to memory loss, the adverse outcome of the putative AOP. It is conceivable that modulation of the involved miRs in these early events at the onset of the disease may negatively affect cellular processes governing cellular homeostasis leading eventually to memory loss. Hence, miRs might be valuable early molecular biomarkers providing plausible mechanistic insight of the signaling pathways and KEs leading to memory loss, possibly governing the development of AD at early phase.
The reported AD-related miRs (Supplementary Table 1) have been retrieved from literature, with supporting evidence for their detection in human biofluids or in brain tissues. Regarding neurotoxicity, alteration in miR expression in human or animal neuronal cells, or in blood after exposure to chemical neurotoxicants, were considered. Since many of the publications did not provide the exact mature sequences of the investigated miRs, we indicated both -3p and -5p arms of the mature miR sequences to be linked to either AD or neurotoxicity. This point should be carefully taken into account if reproducibility of these suggested miRs needs to be evaluated.
Interestingly, the miR functional analysis revealed networks among pathways, miRs, and their targets per event of the proposed tau-driven AOP. Most of the identified miRs related to hypothetical starting point or to KEs confirmed the findings retrieved from literature, as well. As aforementioned, miR-128 was shown to be commonly altered in glucose and cholesterol metabolism, and in KE2, KE3, and KE4, of the proposed AOP. miR-128 is implicated in both AD and neurotoxicity (human and animal). Upregulation of miR-128 in plasma of MCI and AD patients [262], and in hippocampus of early AD (Braak III/V) patients [263], compared to controls, has been reported, and has been demonstrated also after exposure of human brain cells to aluminum sulfate [48] and of zebrafish to Cu [264].
With respect to KEs of the proposed AOP, miR-34a and miR-124, were found to act transversally across most events of interest. Both miR-34a and miR-124 were indicated to be involved in KE1, KE3, KE4, KE7, and KE8. Several studies have reported alteration of miR-34a expression in plasma [265], cerebrospinal fluid [76, 265], peripheral blood mononuclear cells [266], and brain [76, 267] of AD patients compared to controls. miR-34a has been also implicated in ketamine-induced hippocampal neurotoxicity (mouse) [268], in aluminum sulfate-induced microglial phagocytic responses (mouse) [269], and in lithium- and valproate-induced hippocampal neurotoxicity (rat) [64]. Likewise, changes in miR-124 expression have been observed in serum of MCI patients [121], in cerebrospinal fluid [270] and brain of AD patients [271]. miR-124 has been implicated in human neurotoxicity, after exposure of human stem-cell-derived neurons to propofol and after exposure of human neural progenitor cells to paraquat [47]. Besides human neurotoxicity, miR-124 has been also shown to mediate murine hippocampal ketamine-induced neurotoxicity [272].
In addition, miR-9 was identified across different KEs of the AOP and has been found to target genes that regulate processes related to KE2, KE3, KE4, KE7, and KE8. miR-9 plays a role in synaptic plasticity [193], neuronal differentiation, formation of cortex, neurogenesis, and brain development [273], as well as in apoptosis [274], autophagy, oxidative stress [117], phosphorylated tau [143], ROS accumulation [116], and neuroinflammation [275]. Regarding its implication in AD, alteration in miR-9 expression has been observed in cerebrospinal fluid [76, 276], serum [277], and brain of AD patients [48, 278]. miR-9 mediated iron- and aluminum sulfate- [116], and propofol-induced [279] neurotoxicity in human neuronal cells. Exposure to ethanol in mouse neural stem cells [58], fetal brain [280], and zebrafish [281], and exposure to mixture of fipronil and triazophos in zebrafish [282], have all been also shown to dysregulate miR-9 expression.
Furthermore, functional analysis suggested that some miRs were related to the KEs of the proposed AOP, but their relevance to human AD or brain cells was not confirmed by literature. In those cases, their direct links to neurotoxicity may indirectly provide additional information about their implication in early sAD processes, as suggested via the plausible plug-ins reported for the AOP toward memory loss [11]. For instance, altered expression of miR-30c in human embryonic stem cells derived neurons has been associated with exposure to propofol (sedative drug) [279], which is known for its neurotoxic effects. miR-30c expression has been found altered in blood [283], cerebrospinal fluid [76], and serum [270] of AD patients; however, no link of miR-30c has earlier been reported in literature to brain cholesterol dysmetabolism. The functional analysis revealed that miR-30c(-1-3p) targets a member of the Cytochrome P450 Family 51, CYP51A1, which has been previously shown to be involved in cholesterol biosynthesis pathway [27]. Notably, in our proposed AOP, propofol was suggested as possible molecular initiating event, MIE8, for cholesterol dysmetabolism (hypothetical starting point) by mediating the effects of impaired cholesterol biosynthesis via targeting the β-hydroxy β-methylglutaryl-CoA (HMG-CoA) reductase, a rate-controlling enzyme of metabolic pathway of cholesterol.
Another example is provided by the observed changes in miR-574 expression, which have been associated with exposure of human neuronal progenitor cells to paraquat [47] and with exposure of mouse brain to particulate air pollution [222]. miR-574 was found to be related to synaptic function by functional analysis; however, no association with human AD has been previously reported in literature. Interestingly, functional analysis revealed that miR-574 targets microtubule-associated proteins, such as MAP2, of which dysregulation has been linked to AD in animal models [284]. Zheng et al. [285] have shown that paraquat exposure of human astrocytes induced detrimental effects on their synaptic function.
Moreover, changes of miR-1(-3p) expression in human blood has been associated with particulate exposure [46], known to confer neurotoxicity; however, no link has earlier been reported to brain cholesterol dysmetabolism (as revealed by functional analysis) and human AD. Previously, among other neurotoxic environmental contaminants, particulate air pollution has been suggested as plug-ins for KE1, KE2, and KE3, of the proposed AOP, possibly mediating the effects by targeting SH containing proteins (MIE9), GSK-3β (MIE13), PSD-95 (MIE22), or NF-κB (MIE26) [11].
Taking into account only the AD- and neurotoxicity-related miRs, 40 miRs, whose potential target genes were commonly regulated in more than one KE of the proposed AOP, were identified. These miRs have been confirmed for their relevance to the specific KEs by literature and functional analysis, while their link to neurotoxicity (human and/or animal) may provide additional mechanistic information for these commonly dysregulated processes in both AD and neurotoxicity. Their shared genes may also indicate their importance in the dysregulation of these interesting processes described by the plausible AOP for memory loss.
As aforementioned, around 70% of the known human mature miRs are expressed in brain [72, 286], regulating gene expression involved in different processes required for nervous system functions. These brain-enriched miRs may govern specific cellular phenotypes across the different neural cells, including neurons, astrocytes, oligodendrocytes, and microglia [208]. Thereby, the understanding of the cell-type specific miR-target interactions may strengthen their implication in pathological processes manifesting AD and neurotoxicity. Among the 40 identified miRs, commonly found in AD and neurotoxicity, derived from our functional and literature analyses, miR-128 is the most abundant brain-enriched and neuron-specific miR, regulating the gene expression of several targets involved in brain functioning [72]. miR-34a and miR-124 are highly expressed in brain cells, specifically in neurons promoting neuronal transcriptome and neurogenesis, among other functions of their targets [72, 208]. miR-9 and miR-132 are also brain-enriched miRs, of which their expression is restricted to hippocampus and medal frontal gyrus [72], while miR-9 together with miR-124 have been reported to regulate neuronal lineage differentiation [286]. miR-142, miR-200c and miR-146a have been reported as specifically glia-enriched miRs, with the latter to inhibit neuronal differentiation and neuroligin 1-dependent synaptogenesis [208]. Additionally, miR-143 is highly expressed in astrocytes [208]. Nevertheless, more research is needed to support cell-type-specific miRNA functions.
In many epidemiological studies, several human circulating miRs, detected in blood, plasma, serum, or cerebrospinal fluid, have been suggested as candidate biomarkers for the diagnosis of AD [76, 287–292]. Circulating blood-based miRs are remarkably stable in biofluids, easily accessible by non- or minimally invasive procedures, and straightforwardly measured by quantitative methods (e.g., RT-qPRC) [293]. However, conflicting findings on miRs’ regulation are also reported, which may limit their candidacy for further use as early biomarkers of sAD. For example, up- and downregulation of miR-9 expression in several human (in vivo or in vitro) studies related to AD have been reported [48, 295]. In most cases, inconsistencies are mainly attributed to the inadequate size and heterogenicity of the study populations, absence of validation of findings, different analytical and normalization methods.
Another crucial limitation of studies describing potential miR involvement in AD development, is the AD-stage misclassification of study participants (i.e., early or late stage of AD). Most of the studies often group the participants with subjective or MCI to possible early AD stage. Nevertheless, there is no consensus on the applied method/test for the characterization of the cognitive state of the study participants. In the current clinical practice, among different neuropsychological tests, the MMSE is a widely used prescreening tool for the diagnosis of dementia in combination with clinical examination [296]. Based on MMSE scores, ranging from 0-30, a quick assessment of cognitive function can be performed revealing cognitive deficits, e.g., problems in attention, orientation, memory, and/or language. Although this test was not developed for the detection or prognosis of early AD status, it is often used by general health practitioners and scientists to define the cognitive status of the individuals [297]. Remarkably, no consistent cut-offs in the MMSE scores among the different studies are applied, which makes comparison of studies’ findings challenging.
In conclusion, existing data on AD-related and neurotoxicity-related miRs may provide an effective approach to deepen our understanding of the mechanisms underlying sAD initiation and progression, even at the preclinical AD-stage. Functional analysis and literature search confirmed processes, commonly (de)regulated by AD- and neurotoxicity-related miRs, which are described in the tau-driven AOP for memory loss. These miRs might be valuable molecular biomarkers providing plausible mechanistic insight into key events leading to memory loss, possibly governing the early onset of AD.
Footnotes
ACKNOWLEDGMENTS
This work was supported by EU Interreg, Vlaanderen-Nederland (https://grensregio.eu/projecten) (Project Herinneringen), ToxGenSolutions BV (https://toxgensolutions.eu), and 3Rs Management and Consulting ApS (![]() ).
).