
Abstract
Background:
Despite decades of research, our understanding of Alzheimer’s disease (AD) etiology remains incomplete. In recent years, appreciation has grown for potential roles for the microbiota in shaping neurological health.
Objective:
This study aimed to examine associations between the microbiota and AD in a human cross-sectional cohort.
Methods:
Forty-five AD patients and 54 matched controls were recruited in Vancouver, Canada. Fecal and oral samples underwent 16S microbiota sequencing. A wide array of demographic and clinical data were collected. Differences between participant groups were assessed, and associations between microbes and clinical variables were examined within the AD population.
Results:
The gut microbiota of AD patients displayed lower diversity relative to controls, although taxonomic differences were sparse. In contrast, the AD oral microbiota displayed higher diversity, with several taxonomic differences relative to controls, including a lower abundance of the families Streptococcaceae and Actinomycetaceae, and a higher abundance of Weeksellaceae, among others. The periodontitis-associated oral microbe Porphyromonas gingivalis was 5 times more prevalent among patients. No significant associations between gut or oral microbes and cognition were detected, but several correlations existed between microbes and mood disorders and BMI among patients, including a strong positive correlation between Alphaproteobacteria and depression score.
Conclusion:
The gut microbiota of AD patients was not overtly different from controls, although it displayed lower diversity, an overall marker of microbiota health. The oral microbiota did display marked differences. Cognition was not associated with a microbial signature, but other relevant AD factors including mood and BMI did demonstrate an association.
INTRODUCTION
Alzheimer’s disease (AD) is an incurable progressive neurodegenerative disease and is the leading cause of dementia. In recent years, the microbiota-gut-brain axis has increasingly been implicated in a variety of neurological conditions [1, 2], including AD. Human and animal studies have shown links between microbes and AD, although definitive mechanisms remain elusive. Microbially-induced inflammation, increased gut permeability, a reduction in anti-inflammatory short chain fatty acid production, altered enteric and vagal nerve signaling, inhibition of autophagy-mediated amyloid clearance, and modulation of neurotransmitter levels are among the leading putative mechanisms by which commensal microbes might influence disease etiology and pathophysiology [3, 4]. Microbial metabolites are also critical to microglia maturation and function [5]. It has even been shown that amyloid-β may have an anti-microbial role [6], potentially suggesting that amyloid plaques are the result of an innate immune response aimed at eliminating pathogens from the CNS, although the relevance of this in human AD remains largely speculative.
In addition to intestinal microbes, a role for the oral microbiota has been suggested in AD [7]. Chronic oral cavity inflammation may contribute to systemic and CNS inflammation, promoting AD-related pathology [4]. Indeed, periodontitis is a recognized risk factor for development of dementia and AD [8, 9]. Specifically, the periodontitis-implicated oral microbe Porphyromonas gingivalis has been associated with elevated AD risk and with amyloid-β accumulation [10–13].
Current studies of the microbiome in human AD have been relatively small in size and very limited in their geographical distribution. In this study, we aimed to add important knowledge to this field by characterizing the fecal and oral microbiota in a well-matched clinical cohort from Vancouver, Canada. Microbes were compared between patients and controls, and associations with clinical variables were closely examined.
MATERIALS AND METHODS
Cohort recruitment
45 patients with AD were recruited from the University of British Columbia Hospital’s Centre for Alzheimer Disease and Related Disorders at the Vancouver Coastal Health Authority, Centre for Brain Health, University of British Columbia (UBC) between February 2018 to December 2019. Patient inclusion criteria were: 1) age 50–85 years, 2) a diagnosis of probable AD dementia, or dementia of mixed AD/vascular etiology, based on the McKhann criteria [14], 3) age of onset between 50–80 years. Thirty-nine of the patients had AD dementia as their primary diagnosis, and 6 had mixed AD/vascular dementia. Exclusion criteria included: 1) a diagnosis of a non-AD dementia, 2) a concurrent diagnosis of a gastroinflammatory disease, 3) antibiotic use within the previous 3 months, 4) probiotic use within the previous month, 5) a current illness, including a cold, flu, or bacterial infection. This study was approved by UBC’s Clinical Research Ethics Board and all participants provided written informed consent to be involved in the study.
Control selection
To maximize the availability of potential controls for optimal matching, controls were pooled from two studies which ran concurrently at the UBC Centre for Brain Health, both initiated by our research team. Namely, 42 potential controls were recruited as part of this current AD study running between February 2018 to September 2019, and another 97 controls were available from a parallel study of the microbiome in Parkinson’s disease [15] that ran between January 2017 to September 2019. A total of 139 potential controls were therefore available for selection. Both studies used identical sample collection, processing, storage, extraction, and sequencing methodology. Inclusion criterion was age 50–85. Exclusion criteria were the same as for AD patients, with the additions of 1) a current diagnosis of AD or a related neurological disorder, 2) a known family history of early-onset AD or related disorder, 3) a score lower than 26 on the Montreal Cognitive Assessment. From the 139 potential controls, a final matched control study group was then selected using coarsened exact matching [16]. Briefly, male and female AD patients were separately segregated into age tertiles (i.e., 6 total groups), and 1.2 controls per patient were selected from the pool of 139 controls. In subgroups with surplus controls, the tiebreaker used was older age, as this resulted in optimal age-matching. This strategy resulted in a final analyzed control group of 54 individuals, perfectly balanced by sex with the AD group, and slightly but not significantly younger in age.
Data collection
During a 3 h study visit at the Clinic, information was collected on demographics, AD-related medication use, and various lifestyle factors. Cognition was tested via the Montreal Cognitive Assessment (MoCA) [17]. Depression, anxiety, and fatigue were respectively assessed via the Beck’s Depression Inventory-II [18], the State-Trait Anxiety Inventory [19], and the Fatigue Severity Scale [20]. The presence of functional constipation and irritable bowel syndrome (IBS) were assessed using the Rome-III Constipation Module [21]. Data were stored on RedCap using secure university-wide encryption.
Statistical analysis
Differences between participant groups were assessed using Mann-Whitney U tests for continuous variables, and Fisher’s exact tests for categorical variables. Microbiome-specific analyses and statistical methods are discussed below. In cases of multiple hypothesis testing, Benjamini-Hochberg false discovery rate (FDR) correction was employed. FDR-corrected p-values < 0.05 were considered statistically significant.
Biological sample collection
Oral swabs and blood samples were collected during the study visit. The mucosa of the oral cavity was sampled by swabbing the salivary glands on both sides of the mouth and below the tongue for 5 to 10 s using Purflock Ultra Standard Tips (VWR 18052-164). The cotton tip of the swab was cut with sterile scissors into a cryovial, and frozen at –80°C until DNA extraction. 12 mL of blood was collected into 6 mL serum and 6 mL EDTA BD Vacutainer tubes, spun at 2,000 x g at 5°C, and serum was aliquoted and frozen at –80°C until analysis. At the conclusion of the study visit, participants were supplied an OMNIgene-GUT fecal collection kit (DNA Genotek) and a pre-paid envelope and requested to provide a fecal sample at the next available opportunity. The collection tubes in this kit contain a stabilization buffer which halts microbial growth and stabilize DNA, providing a snapshot of the microbiome at the time of collection. Samples were mailed back to the center, aliquoted into cryovials, and frozen at –80°C.
C-reactive protein measurements
Serum C-reactive protein (CRP) was measured by high-sensitivity ELISA (ThermoFisher KHA0031) according to the manufacturer’s recommended protocol.
DNA extraction
DNA from fecal samples was manually extracted using QIAamp PowerFecal DNA Kits (QIAGEN 12830-50) using the manufacturer’s recommended protocol. DNA from oral swabs was extracted using MOBIO Powersoil DNA Isolation Kits (QIAGEN 12888-50), automated on a KingFisher robot (ThermoFisher).
Microbiota sequencing
The bacterial microbiota from fecal and oral DNA extractions was sequenced using indexed barcoded 515F/806R primers targeting the V4 hypervariable region of the 16S rDNA gene (F: GTGCCAGCMGCCGCGGTAA, R: GGACTACHVHHHTWTCTAAT). Library preparation was performed with Phusion High Fidelity PCR kits (New England Biolabs M0530L) and PCR reactions were cleaned and normalized using SequalPrep Normalization Plate Kits (ThermoFisher A1051001). Pooled libraries were run on an Illumina MiSeq instrument using V3 technology with 300 bp paired-end reads. Fecal DNA extractions and library preparation were performed in the Finlay lab. Oral swab DNA extractions and library preparation were commercially performed by Microbiome Insights. Fecal and oral samples were sequenced on separate MiSeq runs.
Microbiota analysis
Fecal and oral sequencing runs were analyzed separately, using the same parameters. Demultiplexed sequences were trimmed to 240 bp and quality-filtered using DADA2 in QIIME2 [23]. Any features mapping outside the kingdom Bacteria, or which represented < 0.05% of total reads, were filtered from the analyses. Filtered feature tables were exported into phyloseq [24] in R for downstream analysis. For diversity analyses, a rarefaction depth of 8,227 was used for fecal samples, and 5,132 for oral samples. Taxonomy was assigned using the 99% Greengenes database. Differential abundance analysis was performed using DESeq2 [25] in R. Associations between bacterial taxa and clinical variables were examined using Spearman correlations with Benjamini-Hochberg FDR correction. Nine extraction-blank and no-template controls were included across the sequencing runs, returning an average of 29 raw unprocessed reads (range 3–62), and an average of 4 DADA2-processed reads (range 1–10), ruling out contamination during DNA extraction/ sequencing as possible confounders. Raw sequencing data, including controls, has been deposited with NCBI SRA under accession PRJNA770746.
Analysis of shared fecal and oral taxa
To examine the extent to which fecal microbes were observed in the oral cavity and vice-versa, any amplicon sequence variant (ASV) seen in both the fecal and the oral rarefied datasets was computationally extracted. Six such ASVs were identified, described in the Results. Differences between each taxon and the sum of all 6 was compared between patients and controls with Mann-Whitney U-tests.
RESULTS
Demographic and clinical data
Participant demographics and characteristics are summarized in Table 1. AD patients had a median MoCA score 9 points lower than controls (AD 18 versus controls 27). Patients had significantly lower body mass index (BMI) and fewer years of education, both of which are associated with AD [26–28] and likely represent true disease features rather than mismatching. Mood disorder scores (depression, anxiety, fatigue) were consistently but not significantly worse among patients. Patients and controls were well-matched for smoking status, rural living, and various gastrointestinal measures. All but eight enrolled patients were on an AD specific medication at the time of the visit.
Participant demographics and characteristics. Continuous variables are presented as median (interquartile range). Dichotomous variables are represented as a percentage. Statistical significance was determined by Mann-Whitney U test (continuous variables) and Fisher’s exact test (categorical variables)
Fecal microbiota
Family-level summary results for patients and controls are available in Supplementary Table 1. The overall community composition of the fecal microbiota was not significantly different between AD patients and controls (beta-diversity metrics Jaccard, Bray-Curtis, Unweighted Unifrac all p > 0.05; Unweighted Unifrac PCoA plot shown in Fig. 1A). However, there was significantly lower alpha diversity in the microbiota of AD patients (Chao1 and Faith’s PD, Fig. 1B, C). Alpha diversity measures were slightly, but not significantly, negatively correlated with age (Chao1 Spearman’s ρ= –0.049, p = 0.634; Faith’s PD ρ= –0.085, p = 0.411). Alpha diversity was not correlated with MoCA score. Differential abundance analysis in DESeq2 revealed only a single taxon, the family Erysipelotrichaceae, that differed between patients and controls after FDR correction at any taxonomic level, with its relative abundance being higher in AD patients (Fig. 1D).
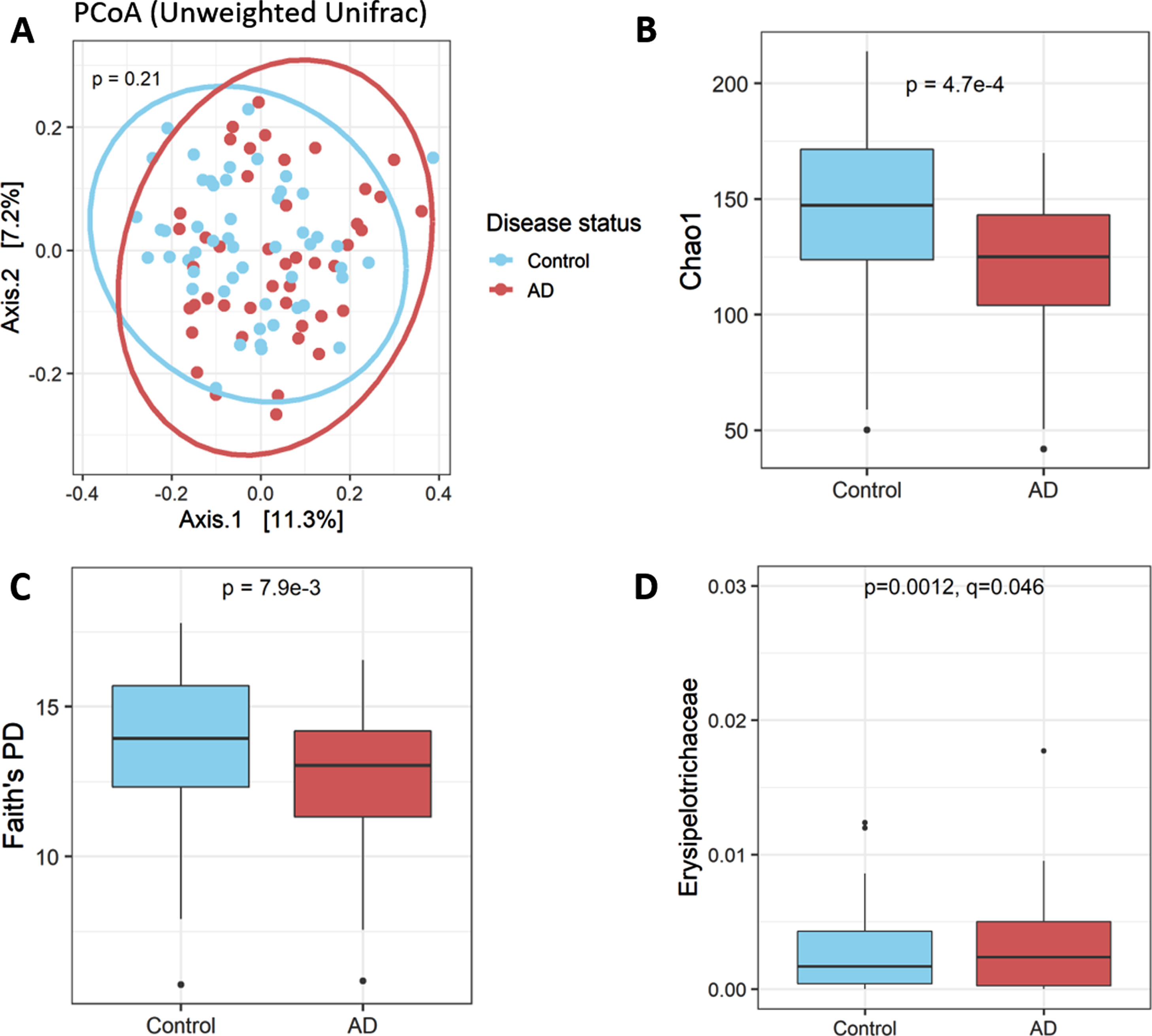
The gut microbiota in AD patients and matched controls. A) The gut microbiota of AD patients and controls did not differ on beta diversity metrics (Unweighted Unifrac PCoA plot shown). B) The gut microbiota of AD patients had lower alpha diversity, as demonstrated by Chao1 index C) as well as Faith’s Phylogenetic Diversity index. D) Only a single taxon, the family Erysipelotrichaceae, differed significantly between patients and controls, observed at a higher relative abundance among patients.
Correlations between bacterial taxonomic abundances and clinical variables were examined within the AD patient subgroup. No bacteria were correlated with MoCA score, or with age, age of AD onset, disease duration, years of education, or serum CRP levels. Use of donepezil was not associated with differences in any microbiota taxa. No other AD-modifying medication was used at a rate high enough to provide meaningful analysis. No taxonomic differences were observed based on sex, neither within patient groups nor in the entire cohort irrespective of disease status. Erysipelotrichaceae was positively correlated with BMI score (Fig. 2A). Interestingly, several positive correlations were observed between bacteria and mood disorder scores in AD after multiple testing correction. The phylum Alphaproteobacteria, as well as the family Odoribacteraceae and the specific genus Butyricimonas within this family, were all correlated with higher BDI-II depression score (Fig. 2B-D). Odoribacteraceae was also correlated with higher STAI anxiety score (Fig. 2E).

Correlations between gut microbes and clinical features in AD patients. A) Erysipelotrichaceae was positively correlated with body mass index among patients. B-D) Depression score, as measured by Beck’s Depression Inventory-II, was positively correlated with Alphaproteobacteria, Odoribacteraceae, and Butyricimonas in patients. E) Odoribacteraceae was also positively correlated with anxiety score, as measured by the State-Trait Anxiety Inventory. Slight transparency and positional jitter have been added to B-D in order to show overlapping data points.
Oral microbiota
Unlike the fecal microbiota, the overall community composition of the oral microbiota did differ significantly between patients and controls on most beta diversity metrics (Jaccard p = 0.001, Bray-Curtis p = 0.13, Unweighted Unifrac p = 0.001; Unweighted Unifrac PCoA plot shown in Fig. 3A). Furthermore, in direct contrast to the fecal microbiota, the oral microbiota of AD patients demonstrated higher alpha diversity relative to controls (Fig. 3B). Several taxa differed between the two population groups in differential abundance analysis across several taxonomic levels. AD patients had a lower abundance of the phylum Firmicutes, driven largely by the family Streptococcaceae (Fig. 3C, D), which is the most dominant family in the oral cavity. Patients also had a lower abundance of the family Actinomycetaceae and the species Rothia mucilaginosa (family Micrococcaceae) (Fig. 3E, F). In contrast, AD patients had increased relative abundance of the family Weeksellaceae (Fig. 3G). However, no significant correlations were observed between any oral taxa and clinical features within the AD subgroup. A family-level taxa table for all families observed at a relative abundance > 0.1% is available in Supplementary Table 2.
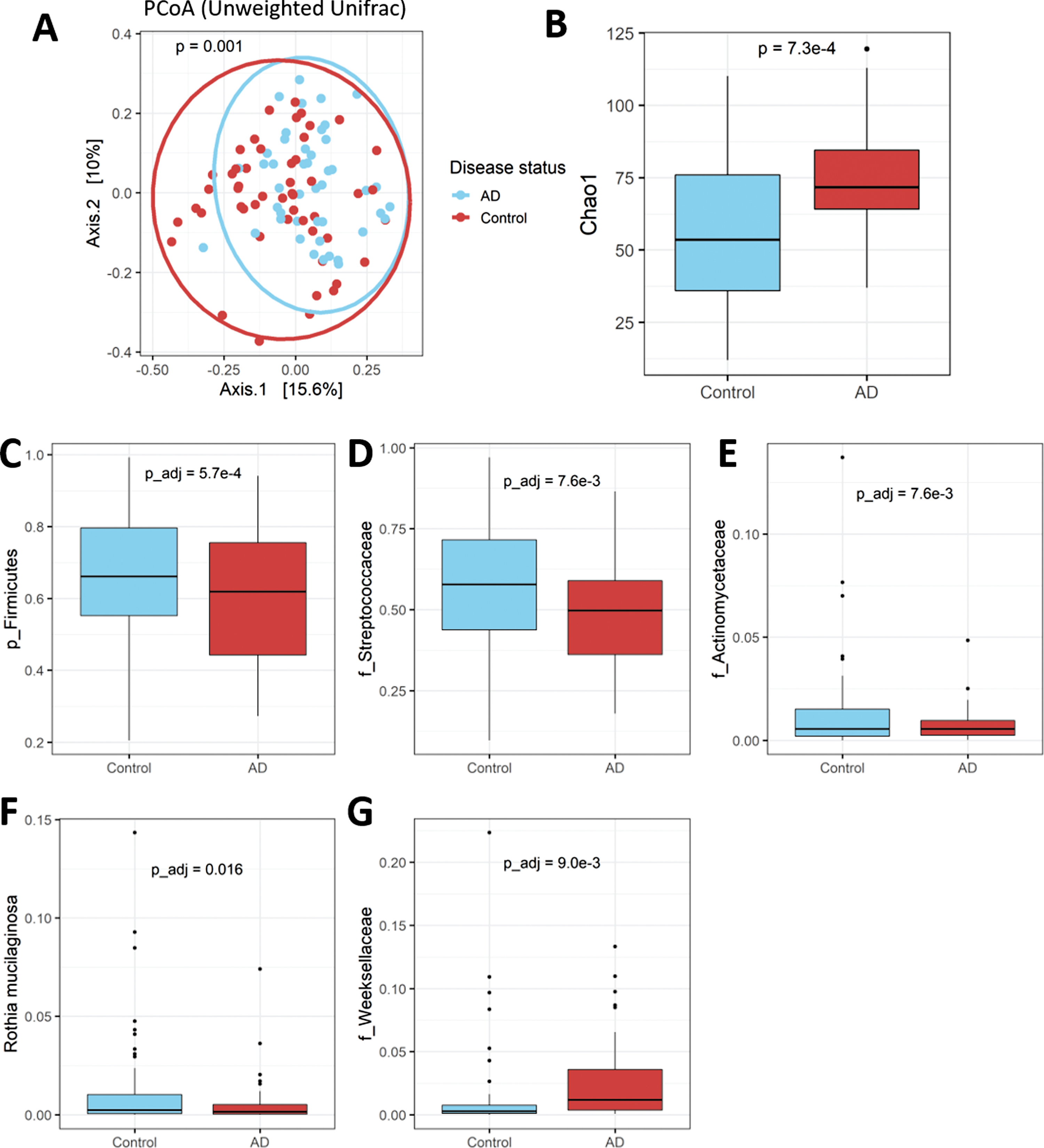
The oral microbiota in AD patients and matched controls. A) The oral microbiota of AD patients and controls differed on most beta diversity metrics (Unweighted Unifrac PCoA plot shown). B) In contrast to the gut microbiota, the oral microbiota of patients had higher alpha diversity (Chao1 index shown). C-G) Several taxonomic differences were observed between patients and controls, including, in AD patients: C) lower abundance of the phylum Firmicutes, D) the family Streptococcaceae, E) the family Actinomycetaceae, and F) the species Rothia mucilaginosa; and G) higher abundance of the family Weeksellaceae.
The periodontitis-associated oral microbe P. gingivalis has been frequently associated with AD [10–13]. The Greengenes 16S database did not provide species-level resolution of the Porphyromonas genus; therefore, we manually extracted all ASVs from the unfiltered dataset that mapped to this genus (n = 11 total) and manually aligned them against the full-length P. gingivalis 16S rDNA gene using BLAST. Two ASVs matched with > 99% identity to P. gingivalis. Their abundance was summed to provide a total relative abundance of P. gingivalis for each participant. Overall, the relative abundance of P. gingivalis was extremely low in the cohort (mean 0.0078%, range 0–0.34%). However, interestingly, it was far more prevalent in AD, found in 13/45 patients (28.9%) versus 3/54 controls (5.6%) (Fisher’s exact test p = 0.0022). There was no significant difference in MoCA score among AD patients with or without P. gingivalis, with indeed a trend towards higher MoCA score among patients with this microbe (18.4 versus 16.2, p = 0.24). Serum C-reactive protein level, as a marker of systemic inflammation, did not differ among patients with or without P. gingivalis (p = 0.57). There was a strong trend towards longer disease duration among patients with P. gingivalis (4.4 years without, 6.5 years with, p = 0.085), despite no difference in absolute age (p = 0.48). Therefore, it may be that the lifestyle changes leading to poorer oral health among patients contributes to stable establishment and proliferation of this oral microbe.
Finally, we examined the extent to which fecal microbes were detected in the oral cavity and vice versa, as studies have shown that decompartmentalization of the gastrointestinal tract is associated with poorer health [29, 30]. Six unique ASVs overlapped between the oral and fecal datasets, mapping taxonomically to: two Streptococcus species with unknown classification, Haemophilus parainfluenzae, Veillonella parvula, Veillonella dispar, and an unclassified species in the genus Dialister. All of these are oral microbes that were highly abundant in the oral cavity of participants; no fecal ASVs were detected in the oral cavity. To examine oral-fecal decompartmentalization, abundances of these shared ASVs were compared in the gut microbiota samples between AD patients and controls. None of them differed between participant groups, nor did their total sum differ (Fig. 4). Therefore, oral-gut decompartmentalization does not appear to be a significant factor in AD.

No evidence of oral-fecal decompartmentalization of the microbiota in AD. Six amplicon sequencing variants (all oral-cavity in origin) were shared between fecal and oral samples in the study. A) In sum, these taxa did not differ in relative abundance in the gut microbiota between patients and controls; B) nor did any of them differ individually.
DISCUSSION
In this study, we describe the fecal and oral microbiome in a well-matched clinical AD cohort. The fecal microbiota of AD patients was not dramatically altered from controls, with the notable exception of lower alpha diversity. Erysipelotrichaceae was found at higher relative abundance among AD patients. This bacterium has been associated with pro-inflammatory cytokines, obesity, and metabolic disorders in humans [31]. We did indeed observe a positive correlation between Erysipelotrichaceae and BMI score. It has been speculated that a Western diet can lead to increased abundance of this family, which may in turn lead to gut and systemic inflammation [31]. However, as this taxon has not previously been reported as elevated in AD, the generalizability of our observation is uncertain.
Several fecal microbes were associated with worse depression and anxiety scores among the AD population, most notably Alphaproteobacteria and Odoribacteriaceae. Better understanding the complex nature of these symptoms is critical to the care and well-being of AD patients, as depression and anxiety affect an estimated 42% and 39% of patients respectively [32]. There have been several proposed mechanisms linking the microbiota and mood disorders, including modulation of the hypothalamic–pituitary–adrenal axis, interfering with GABAergic or serotonergic signaling, and increasing systemic inflammation [33–35]. The bacteria we observed to correlate with mood symptoms in AD were not associated with depression in large meta-analyses in non-AD populations [36, 37]. Importantly, however, depression and anxiety in AD may vary in subtype and etiology, and be markedly different from mood disorders in individuals without AD [38]. Likewise, the relationship between the microbiota and mood disorders in AD may also be unique to this population and requires further investigation.
In contrast to the fecal microbiota, the oral microbiota differed significantly between study groups. Overall, the oral microbiota of AD patients demonstrated higher alpha diversity and was less dominated by the family Streptococcaceae. Moreover, a significantly higher prevalence of the periodontitis-associated P. gingivalis was observed among patients. However, no oral microbes were associated with any relevant disease features. It has been speculated that a dysbiotic oral microbiota can contribute to systemic inflammation, and therefore we specifically tested for correlations between oral bacterial taxa and serum C-reactive protein; however, no significant relationships were found.
Several studies have investigated the fecal and oral microbiome in AD in recent years, without any definitive patterns or conclusions emerging. At least eight studies have explored the fecal microbiome in human AD cohorts [39–46] (summarized in Supplementary Table 3). Repeated observations include an increase in Bifidobacterium or its phylum Actinobacteria in AD patients in three studies [39, 44]; however, another study reported decreased Bifidobacterium in patients [45]. In our study, we saw a slight but non-significant increase in the Bifidobacteriaceae family among patients (see Supplementary Table 1). Pro-inflammatory Proteobacteria and genera in this phylum such as Escherichia-Shigella have been observed to be elevated in AD patients by at least three studies [40, 43]. Additional observations of altered taxonomic abundance appear sporadic and typically only seen by a single study; this includes our current observation of elevated Erysipelotrichaceae among individuals with AD. Two studies [41, 45] observed, as we did, a decrease in alpha diversity among the microbiota of AD patients.
At least four studies [47–50] have investigated the oral microbiome in similar populations. As with the fecal microbiome, results are variable with no consistent trends. We observed increased alpha diversity in the oral microbiota of AD patients, which was also seen by one other study [49]; however, two studies observed the opposite result [47, 50]. Holmer et al. [49] observed no taxonomic differences in the ten most common oral genera between cognitively healthy individuals and participants with AD, mild cognitive impairment, or subjective cognitive decline; however, they did observe significand differences in several less-prevalent genera. Wu et al. [47] noted higher levels of Streoptococcaceae among AD patients, in contrast to our current observation. All other taxonomic differences observed by these studies and ours appear to be unique and likely sporadic.
AD results in many alterations to behavior, lifestyle, exercise, and diet, all of which can greatly impact both the gut and oral microbiota. Given the wide disparity in existing studies, it is plausible that these lifestyle changes—combined with geographical differences between studies [51]—may be driving the subtle and inconsistent changes seen in the microbiota of patients across cohorts, rather than there being any consistent AD-relevant microbiota signature. Notably, 6/8 of the published human gut microbiome studies to date have come from the same geographic location (mainland China), and all have been fairly small, with a maximum of 100 AD patients and median of 31.5 (Supplementary Table 3). Additionally, all but one study [46] employed 16S amplicon sequencing, which has limited taxonomic resolution and fails to adequately capture functional-level data; this is indeed a limitation of our own study. As a further complexity, different subtypes of AD might have variable involvement of inflammation and the gut or oral microbiome and only large cohort studies will be able to identify such subgroups [52]. AD is typically associated with poorer oral hygiene, which we posit explains the increased microbial diversity and taxonomic alterations we observe in the oral cavity of our patients. A potential limitation of our study is the fairly broad sampling of the oral cavity (swabbing the side of the mouth and below the tongue); more targeted sampling of the gingivae may be more informative in this specific population. Our study did confirm the overrepresentation of the oral microbe P. gingivalis among AD patients; however, the strongest clinical correlate with this microbe was disease duration, possibly implying that deteriorating oral health caused by AD-related behavioral changes may create conditions hospitable for this microbe, rather it being causally implicated in the etiology of human AD.
In conclusion, this study adds to the body of literature from the past 5 years that shows a subtle alteration in the gut microbiota profile of AD patients compared to matched controls. However, the lack of consistent results from these studies, even on fundamental factors such as diversity, indicates that the gut microbiota may not be highly implicated in the pathophysiology of AD, at least not at the level that 16S amplicon sequencing can reliably identify. We did observe several novel correlations between mood disorder symptoms and various microbes within the AD population, suggesting that further research into the relationship between gut microbes and more subtle neurological features of AD and possible subgroups may be warranted. The oral microbiota showed larger differences between patients and controls than the gut microbiota, however again results are highly inconsistent between existing studies. The differences we observe, including higher microbial alpha diversity in the AD oral cavity, may be explained by poorer oral health practices among patients. Whether the gut or oral microbiota is involved in the etiology of AD will require large prospective longitudinal studies before the development of cognitive decline.
Footnotes
ACKNOWLEDGMENTS
We thank all participants for their generous contributions to the study without which the research could not have been carried out. This work was supported by grants from the Canadian Consortium on Neurodegeneration in Aging (CCNA), the Canadian Institutes of Health Research (CIHR), the Pacific Parkinson’s Research Institute, and Parkinson Canada/Parkinson Society British Columbia. M.S.C. is supported by a CIHR Vanier Scholarship. S.A.C. is supported by the Marg Meikle Professorship for Research in Parkinson’s Disease.