
Abstract
Background:
It can be challenging to discriminate between progressive supranuclear palsy (PSP) and frontotemporal dementia (FTD). However, a correct diagnosis is a precondition for targeted treatment strategies and proper patient counseling. There has been a growing interest to identify cerebrospinal fluid (CSF) biomarkers, including neurofilament light chain (NfL).
Objective:
This systematic review evaluates the existing literature on neurofilament light in CSF aiming to validate its utility for differentiating FTD from PSP.
Methods:
A systematic literature search was conducted. A broad range of synonyms for PSP, NfL, and FTD as well as associated MeSH terms, were combined and used as keywords when searching. Relevant data were extracted and assessed for risk of bias.
Results:
Nine studies including a total of 671 patients with FTD, 254 patients with PSP, 523 healthy controls, and 1,771 patients with other disorders were included in the review. Four studies found a significantly higher level of CSF NfL in FTD (n = 445) compared to PSP (n = 124); however, in three of these studies the difference was only significant in certain FTD variants. Four studies found no significant difference in CSF NfL between PSP (n = 98) and FTD (n = 248). One study found a significantly higher level of NfL in PSP (n = 33) compared to FTD (n = 16).
Conclusion:
In the majority of patients in the studies included in this review, a higher level of NfL in CSF was found in patients with FTD compared to patients with PSP; however, results were inconsistent and prospective studies including large study cohorts are needed.
INTRODUCTION
There is an urgent need for biomarkers in neurodegenerative disorders to discriminate among clinical overlapping diseases, for evaluating disease progression and prognosis, and in obtaining more homo-genous groups in therapeutic trials contributing to the development of efficacious disease-modifying treatment strategies.
Pathologically, progressive supranuclear palsy (PSP) and frontotemporal dementia (FTD) are considered frontotemporal lobe (FTLD) spectrum disorders, sharing both histopathological but also clinical characteristics [1]. The behavioral variant of frontotemporal dementia (bvFTD) is a clinical syndrome characterized by a progressive deterioration of personality, social comportment and cognition, whereas nonfluent primary progressive aphasia (nfvPPA) and semantic variant primary progressive aphasia (svPPA) are language disorders [2]. The most common clinical features in patients with PSP include vertical supranuclear gaze palsy, postural instability with frequent falls, parkinsonism, and language and cognitive impairment with frontal dysfunction [3]. The clinical diagnostic criteria for PSP (Hoglinger et al., The Movement Disorder Society Criteria) include nonfluent/agrammatic aphasia and frontal cognitive/behavioral presentation as possible clinical features [4], making the FTD subtypes behavioral variant FTD (bvFTD) and nfvPPA relevant differential diagnoses for PSP. PSP with predominant frontotemporal dysfunction (PSP-FTD) has been reported in 5–20% of all cases of PSP [5]. PSP is a tauopathy, characterized by tau-immunopositive tufted astrocytes, neurofibrillary tangles, gliosis, and neuronal loss particularly affecting the basal ganglia, diencephalon, and brainstem. In common with other FTLD disorders is the accumulation of tau in the frontal and temporal lobes [3]. Pathological differences between FTD and PSP are seen in the predominant tau isoform accumulated in neurons and glial cells, with mainly 4R-tau and 3R-tau in PSP and FTD, respectively. In addition, accumulation of other proteins than tau can cause FTD, including TAR DNA binding protein of 43 kDa (TDP-43) and fused in sarcoma (FUS), giving rise to three pathologic subtypes of FTLD designated FTLD-tau, FTLD-TDP, and FTLD-FUS [1].
The diagnostic workup in PSP can be challenging, due to a wide phenotypic heterogeneity and an extensive clinical overlap with other neurodegenerative disorders including Parkinson’s disease, FTD, and corticobasal degeneration [5]. Therefore, biomarkers are needed to support the early clinical diagnosis and improve the diagnostic accuracy.
Based on the neuropathology, disease-associated proteins such as tau, TDP-43, and FUS are obvious biomarker candidates for the pathological processes in FTLD. However, previous studies of cerebrospinal fluid (CSF) tau and CSF TDP-43 in FTLD have shown conflicting results and poor diagnostic accuracy and so far, no studies have demonstrated the presence of the FUS-related FET proteins in CSF [6]. Indicators of other pathophysiological processes including neuroinflammation, glial cell activation, oxidative stress, and neurodegeneration have also previously been suggested as potential CSF biomarker candidates including YKL-40 and uric acid [7, 8]. The concentration of neurofilament light chain (NfL) in CSF and blood, an unspecific but sensitive marker of axonal damage, is one of the most thoroughly investigated and is elevated in several neurodegenerative disorders including PSP, multiple system atrophy, amyotrophic lateral sclerosis (ALS), multiple sclerosis, and Huntington’s disease [9–12].
Neurofilament proteins are major cytoskeletal components of the neuronal axons and maintain axonal caliber and integrity. They are classified according to their molecular weight into three different forms, neurofilament light, medium, and heavy chain [13]. The most important technique to measure NfL in CSF is a commercially available ELISA assay (NF-light®, UmanDiagnostics AB®, Umeå, Sweden). According to the manufacturer, the antibodies in the kit are highly specific for NfL with no cross-reactivities to other CSF components. The detection range is from 100 ng/L to 10,000 ng/L. The intra-assay coefficient of variation is estimated to a maximum of 6%. For measurement of NfL in blood the ultra-sensitive single-molecule array (Simoa) digital immunoassay is recommended [14].
This systematic review evaluates the existing literature on neurofilament light in CSF and blood aiming to validate its utility for differentiating FTD from PSP.
METHODS
Protocol
Prior to the data search, a protocol of the re-view was prospectively registered at the International prospective register of systematic reviews (PROSPERO). The systematic review was conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement [15].
Eligibility criteria
Studies were eligible if they met the following criteria: 1) original studies (not reviews or meta-analysis), 2) published in English, 3) investigated NfL in CSF or blood in patients diagnosed with PSP and FTD, and 4) FTD and PSP patients were classified according to internationally agreed diagnostic consensus criteria. Studies were excluded if 1) they were case reports or abstracts, 2) it was not possible to separate PSP from other types of atypical parkinsonism, or 3) no comparison of NfL between FTD and PSP was made.
Search
We performed a PubMed and Embase search. Both databases were searched from the date of inception of the database until April 12, 2021, and we limited searches to studies published in English. A broad range of synonyms for PSP, NfL, and FTD as well as associated MeSH terms, were combined and used as keywords when searching. For detailed search strategy, please see Supplementary Table 1.
Study selection
Two authors (NB, LS), independently screened the search results, excluded irrelevant publications based on the title and abstract, then read the full texts of potentially relevant studies, assessed eligibility, and extracted data. In addition, relevant studies from reference lists were included. The studies were discussed with a third author in case of disagreements.
Data items
The following data were extracted: the first auth-or’s name, year of publication, number of cases and controls, age, sex, diagnostic criteria used, sample type, analytical methods for NfL measurements, pre-analytical sample handling, confounders adjusted for, and main results.
Synthesis of results
Data were synthesized in a descriptive manner as well as illustrated in figures and tables. The synthesis was structured around the level of NfL in CSF and blood and its ability to discriminate PSP and FTD. Conducting a metanalysis have been taken into consideration but because of the limitations and characteristics of the data in the included studies, we concluded that presenting the results in a descriptive manner in a systematic review, would be reasonable.
Risk of bias assessment
When assessing the risk of bias, in the included studies, one author completed the risk of bias assessment, and a second author subsequently reviewed this. We evaluated the following items for sources of bias: study design, sample size, demographics of the study population in each diagnostic group, adjustments for potential confounders, proper matching of study participants regarding disease duration, pre-analytical and analytical sample handling, and blinding of laboratory technicians performing the NfL analyses. In addition, in order to evaluate the quality and get an overview of each study two authors (NB, LS) independently assessed the risk of bias using a modified version of the Newcastle-Ottawa scale, please see Table 1 and Supplementary Note 3.
Modified Newcastle-Ottawa quality assessment scale for case control studies
*for one star. / for no star. A study can be awarded a maximum of one star for each numbered item within the Selection and Exposure categories. A maximum of two stars can be given for Comparability. NOS consists of 8 items with 3 subscales, and the total maximum score of these 3 subsets is 9. We considered a study which scored 7–9 as high quality, 4–6 high risk of bias and 0–3 very high risk of bias, since a standard criterion for what constitutes a high-quality study has not yet been universally established.
RESULTS
Study selection
The initial search in the PubMed and Embase database resulted in 467 publications. 92 systematic reviews were subsequently excluded. 375 publications were screened by title and abstracts, and 351 were excluded, since they did not meet one or more of the inclusion criteria or met one or more exclusion criteria. Full text assessment was done in 24 publications and 15 of these were excluded, since they did not compare the NfL level between PSP and FTD. Nine publications from 2014 to 2020 were selected for analysis all measuring CSF NfL. We found no articles published comparing blood NfL in FTD and PSP. A schematic flow chart of the literature search is illustrated in Fig. 1.
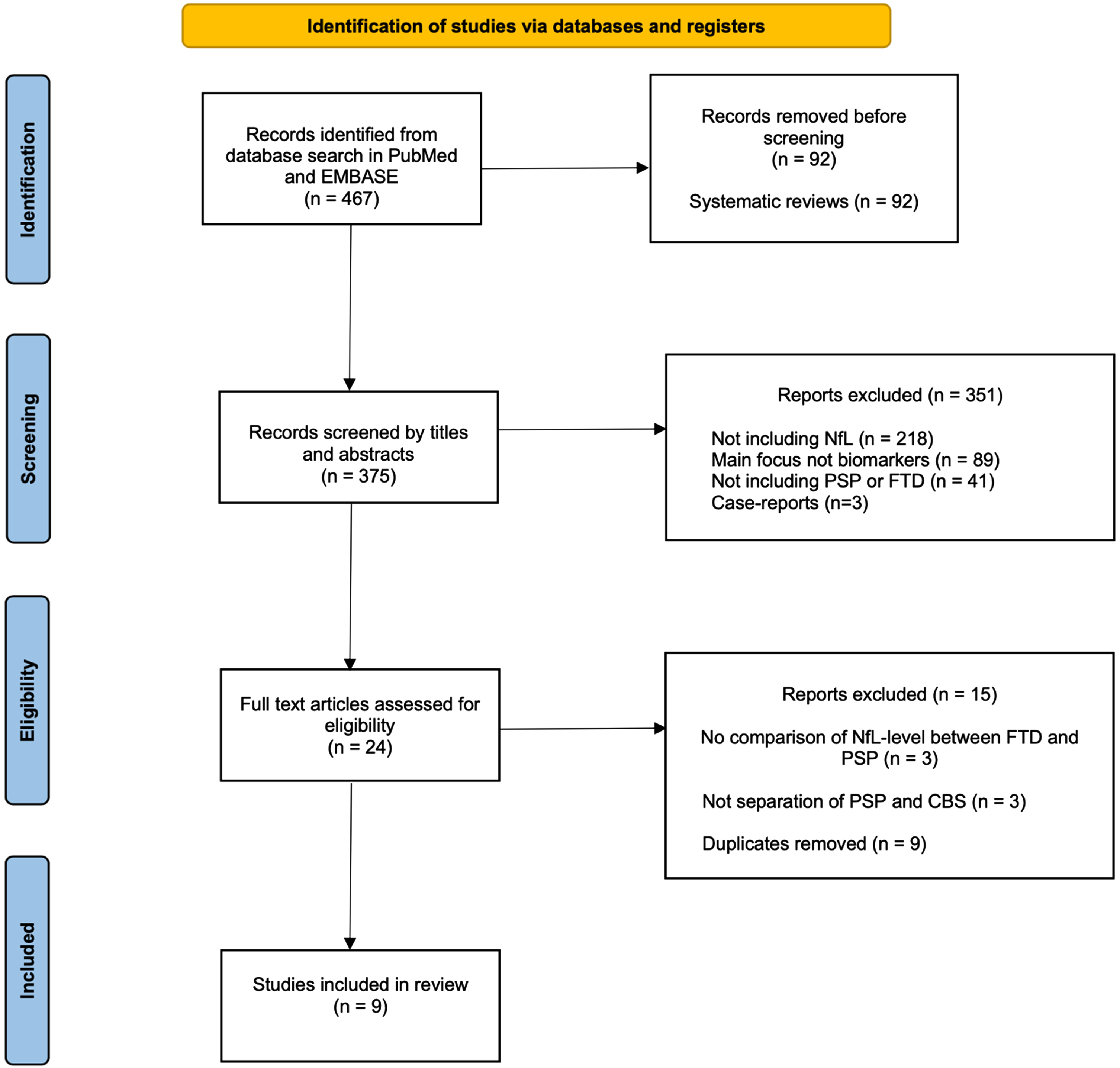
Flow diagram of the search strategy. Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram of the search strategy. The database search initially resulted in 467 records. 92 systematic reviews were excluded. 375 records were screening of title and abstract and 351 were subsequently excluded. Full text assessment was done in 24 publications and 15 of these were excluded. 9 publications were selected for analysis. For detailed search strategy, please see the Supplementary Material.
Study characteristics
The nine studies included a total of 683 patients with FTD, 254 patients with PSP, and 523 healthy controls. Six of the included studies divided the patients into FTD subtypes, resulting in following distribution: 392 bvFTD, 92 svPPA, and 96 nfvPPA. Additionally, 1,771 patients with other disorders were described: Alzheimer’s disease (AD), Down syndrome, Down syndrome with AD, ALS, dementia with Lewy bodies, corticobasal syndrome, prion disease, mild cognitive impairment, posterior cortical atrophy, vascular dementia, Parkinson’s disease with mild cognitive impairment, and multiple system atrophy.
NfL level in FTD compared to PSP
All the included studies [7, 16–23] found an in-creased level of NfL in CSF in patients diagnosed with PSP or FTD, compared to healthy controls.
Four studies [17, 21–23] found a significantly higher level of NfL in FTD (n = 419) than PSP (n = 124). However, in two of the studies the difference was only significant in certain FTD subtypes. Abu-Rumeileh et al. (2020) [17] showed only significantly higher NfL levels in PPA (svPPA and nfvPPA) (n = 8) versus PSP (n = 23), and Meeter et al. [22] found only significantly higher NfL levels in bvFTD (n = 164) versus PSP (n = 58), in the last study patients were not age matched, with older age in the PSP group.
Four out of nine studies found no significant difference in the level of NfL in CSF in PSP (n = 98) compared to FTD (n = 248) [16, 18–20]. Delaby et al. [19] described a higher level of NfL in PSP versus FTD, but without a significance level desig-nated.
Magdalinou et al. [7] found a significantly higher level of NfL in PSP (n = 33) than in FTD (n =16). However, the FTD patients were significantly younger and had shorter disease duration compared to PSP patients. Of the nine studies included in our review, this study had the lowest number of included FTD patients.
In those five studies [7, 21–23] that showed a significant difference in the concentration of NfL in CSF between PSP and FTD, there was an overlap in the mean CSF NfL level±one standard deviation or the median, interquartile range (IQ) between the two diagnostic groups in four of the studies [7, 22]. Only Abu-Rumeileh et al. (2020) [17] did not show an overlap between the NfL concentration in PSP (n = 23) and PPA (n = 8). Scherling et al. [23] only reported values of NfL concentrations in a scatterplot. Demographics are shown in Table 2. NfL values in PSP, FTD subtypes, and healthy controls are illustrated in Fig. 2.
NfL values in patients with PSP, FTD, and healthy controls in the included studies
(N), number of patients; IQR, interquartile range; PSP, progressive supranuclear palsy; FTD; frontotemporal dementia; CSF, cerebrospinal fluid; NfL, neurofilament light. The highest NfL value reported in each study was chosen for this table, independently of disease subtype. NfL levels indicated in pg/mL. #NfL-values are stated in mean (standard deviation). $Only p-values for statistically significant results reported. €No p-value for the comparison of the NfL level in FTD versus PSP reported in the study.
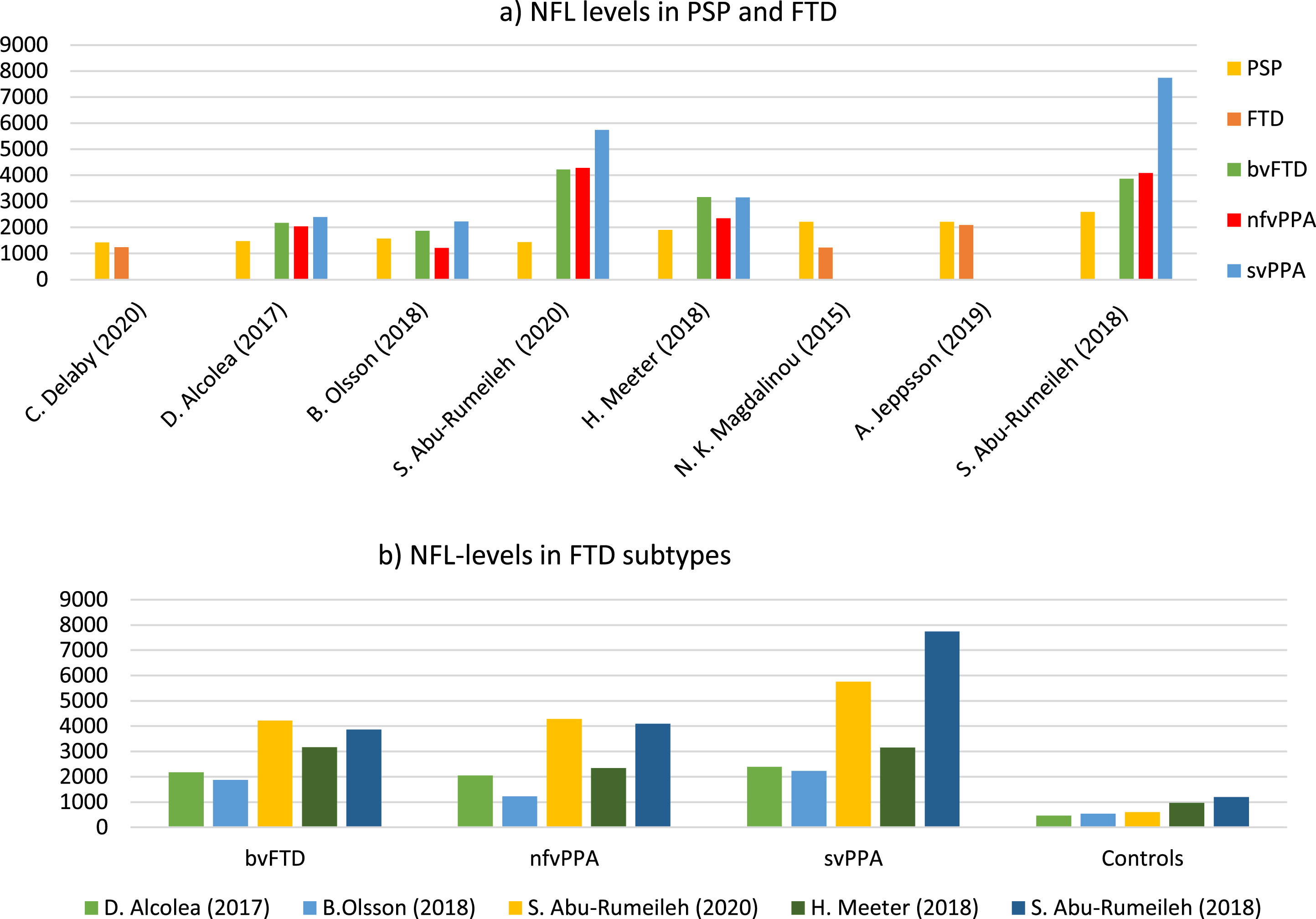
NfL levels in PSP, FTD, and FTD subtypes in the included studies. a) NfL levels in PSP, FTD, and if reported FTD subtypes, for each study included in the review. One study (C. Scherling et al., 2014) is not included, since no NfL concentration was available. NfL levels indicated in pg/mL. NfL values and study characteristics for each study illustrated in Fig. 2, are listed in Table 2. b) NfL levels in the different FTD subtypes and control groups. NfL levels of FTD subtypes were reported in 6 out of 9 of the included studies (each study indicated by a given color). NfL, neurofilament light; PSP, progressive supranuclear palsy; FTD, frontotemporal dementia; bvFTD, behavioral FTD; PPA, primary progressive aphasia; nfvPPA, nonfluent variant PPA; svPPA, semantic variant PPA.
Risk of bias
Study design
All the nine studies were case-control studies, and data were collected retrospectively in all of the studies except one [7].
Sample size
Six [16–19, 23] of the included studies had a small sample size (n < 30) of PSP patients and three [7, 20] had a small sample size of FTD patients. The sample size ranged from 12 to 58 with PSP and 16 to 219 with FTD.
Demographics
In six studies [7, 23], the PSP and FTD groups were not age matched. Two studies [20, 21] did not report whether age differed significantly between groups and only in Abu-Rumeileh et al. (2020) [17] were the PSP and FTD groups age matched. There was an equal distribution of sex between the diagnostic groups in the included studies; however, two studies [20, 21] did not report if sex differed significantly.
Inclusion and exclusion criteria
All the studies had either no inclusion criteria described, or a given certainty level for FTD or PSP as the only inclusion criteria. Exclusion criteria for patients with FTD and PSP were limited. Six studies [17–21, 23] did not report any exclusion criteria. Magdalinou et al. [7] excluded patients younger than 40 years and older than 80 years as well as patients with severe cerebrovascular disease or systemic diseases affecting the central nervous system. Abu-Rumeileh et al. (2020) [17] excluded patients if they had evidence of significant co-morbidity, e.g., AD, dementia with Lewy bodies, prion disease, or vascular dementia. Meeter et al. [22] excluded patients with CSF results suggesting AD.
Regarding the selection of healthy controls, six of the studies [7, 23] reported that the included healthy controls had a normal neurological and/or neuropsychological examination. In the remaining three studies, Meeter et al. [22] included healthy controls with subjective memory complaints but negative AD biomarkers, Jeppsson et al. [20] included healthy controls who had a Mini-Mental State Examination score of 26 or higher and underwent an orthopedic intervention, and Alcolea et al. [18] included cognitively normal controls but not further specified.
Disease duration
In four of the included studies [17, 19–21], no information on the disease duration was reported. Two studies [18, 23] reported no significant difference in disease duration between PSP and FTD and in two studies the disease duration was significantly longer for patients with PSP compared to FTD [7, 16].
Diagnostic criteria
The studies used different clinical criteria for diagnosing PSP and FTD. Fives studies [7, 23] used the NINDS-SPSP clinical criteria [24], two studies [16, 17] used the MDS criteria [4], and two studies [19, 21] did not report the clinical criteria used for the diagnosis of PSP. Six of the studies [16–19, 22] used the criteria by Rascovsky [25] and Gorno-Tempini [26] for diagnosing bvFTD and PPA, respectively. Two studies [20, 23] used the criteria by Neary [2] in diagnosing FTD and one study [7] used the clinical criteria by the Lund and Manchester Groups [27]. The studies required different certainty levels when diagnosing PSP and FTD, respectively. Five studies [7, 22] included probable or definite, two [18, 23] included in addition possible PSP, and two studies [19, 21] did not report a certainty level.
Pre-analytical and analytical variables
All the nine studies used the enzyme-linked im-munosorbent assay (ELISA), for the measurement of NfL in CSF. However, the ELISA kit were from different manufacturers. Five studies [7, 23] used the ELISA kit produced by UMAN DIAGNOSTICS, Umeå, Sweden, two studies [16, 17] used the one by IBL, Hamburg, Germany, and two Swedish studies [20, 21] used an in-house ELISA.
Standard operating procedure (SOP) was followed in five of the included studies [7, 22] and in the remaining studies [16, 23], no information regarding SOP was reported. In four studies [7, 22], the laboratory technicians analyzing the CSF were blinded.
DISCUSSION
To our knowledge, this is the first systematic review summarizing the existing literature on the concentration of NfL in CSF and blood from patients with PSP and FTD.
The studies reporting the highest NfL level in FTD had the largest sample sizes and accounted for 61% of all FTD patients and 63% of all PSP patients included in the nine studies, making a significantly higher CSF NfL in FTD compared to PSP the most consistent finding. In the only study reporting a higher CSF NfL level in PSP, the sample size of FTD patients was small (n = 16), and in addition, the FTD patients were significantly younger and had shorter disease duration compared to the included PSP patients. Thus, this review suggests a higher level of CSF NfL in FTD compared to PSP, which is in line with previous studies having demonstrated more elevated CSF NfL in FTD compared to other forms of dementia [28].
CSF NfL is a validated diagnostic and prognostic marker of axonal damage in ALS [29] and in a previous study, CSF NfL has been shown to correlate with the burden of TDP-43 pathology [14]. The suggestion of a higher CSF NfL in FTD compared to PSP in this review could reflect that one of the pathological subtypes of FTLD is associated with TDP-43, whereas PSP is a tauopathy. The different topographical distribution of pathology in FTD and PSP with a more cortical pathological burden in FTD or a possible occurrence of subclinical motor neuron degeneration in a subset of FTD patients offer other possible explanations [28, 30]. Several [7, 23] of the included studies had low sample sizes in either one or both diagnostic groups. The low sample size in some of the studies may be due to the low prevalence of both PSP [31] and FTD [32] or because the main focus of the studies was not on PSP or FTD. A large variation in CSF NfL concentrations was seen in both PSP, FTD, and healthy individuals emphasizing the need for large study cohorts. In several of the studies [7, 23], the diagnostic groups were not age matched with a tendency for PSP patients to be older. Studies have shown a positive correlation between age and NfL in both CSF and blood [33, 34], thus the age difference between the groups could influence on the NfL levels. The diversity in clinical criteria and certainty level used for the diagnosis of PSP is also a limitation. The NINDS-SPSP clinical criteria [24] focus on the classical presentation of PSP, PSP-Richardson’s syndrome, while the MDS criteria [4] aim to improve sensitivity for early and variant PSP presentations. Regarding certainty level, most studies [7, 22] included probable or definite PSP, two studies [18, 23] in addition, included possible PSP (NINDS-SPSP criteria). Including possible PSP reduces the specificity of the diagnosis, thus increasing the risk of misdiagnosed patients in the study cohort of PSP.
The stability of CSF NfL in PSP and FTD during the disease course is yet unknown, thus it is unknown if the level of NfL increases or decreases as the diseases progress, and if the change is linear. In four of the included studies [17, 19–21] no information on the disease duration was reported. Two studies [18, 23] reported no significant difference in disease duration between PSP and FTD and in two studies the disease duration was significantly longer for patients with PSP compared to FTD [7, 16].
In none of the included studies disease severity was assessed with for instance rating scales. Thus, potential difference in disease severity and progression between the groups could lead to a skew in the reported NfL values.
Limitations regarding pre-analytical and analytical sample handling mainly include the use of the SOP. Five of the [7, 22] included studies followed SOP, and in the remaining studies [16, 23], no information regarding SOP was reported. All nine studies used ELISA for the measurement of NfL in CSF. However, the ELISA kits were from different manufacturers. Interesting is that the two studies [16, 17] using the ELISA kit by IBL, Hamburg, Germany, found higher NfL levels in all the diagnostic groups compared to the other studies included, which is also illustrated in Fig. 2a and 2b.
A limitation of this review is that a metanalysis was not conducted. Performing a meta-analysis could potentially have contributed to a more precise estimate on the difference in the NfL concentration between PSP and FTD. Conducting a meta-analysis have been taken into considerations but because of the limitations and characteristic of the data in the included studies, we concluded that presenting the results in a descriptive manner in a systematic review would be reasonable. Another limitation of this review is that it did not include FTD motor neuron disease (FTD-MND) or logopenic variant PPA (lvPPA). The reason for the exclusion of FTD-MND is that it encompasses distinct clinical and pathological features diverse from the other FTD subtypes and PSP. Clinical discrimination between FTD-MND and PSP does not normally represent the same challenges as differentiating the other FTD subtypes from PSP. The lvPPA was excluded because it is most frequently associated with AD pathology [35].
It is relevant to discriminate PSP from FTD because a correct diagnosis might result in better-targeted treatment strategies and improve the possibility of evaluating disease progression and progno-sis as well as providing proper patient counseling. In this systematic review we found that there was a trend on group level, towards higher levels of NfL in the FTD patients compared to the PSP patients. We found a broad variance in the NfL levels and therefore an extensive overlap in the concentrations of NfL in FTD and PSP, making the clinical use of CSF NfL in the diagnostic work unquestionable. However, results were inconsistent, and an inadequate number of studies were available with low sample sizes in most of the studies. Only two of the studies included patients with neuropathologically confirmed diagnoses. Also, the results were inconsistent, and we found an inadequate number of studies available with low sample sizes in most of the studies. Only two of the studies included patients with neuropathologically confirmed diagnoses.
NfL is a sensitive but unspecific marker of axonal injury, its potential diagnostic value lies in the ability to discriminate between neurological diseases with a different degree of axonal damage or with a different progression rate or disease severity and more disease-specific biomarkers are wanted. NfL has previously been shown to be an independent prognostic marker for ALS and is considered a promising marker of therapeutic effect of drugs reducing axonal damage. Further studies are needed to establish the value of CSF and blood NfL in PSP and FTD [29, 36]. Blood NfL is a potential future direction offering the possibility of repeated measurements potentially increasing the diagnostic and prognostic value of NfL. Recent studies have shown that NfL in the blood can be reliably measured and that it correlates well with NfL in CSF [14]. Based on this review the diagnostic value of NfL in PSP and FTD remains to be established and prospective studies including large study cohorts of preferably all the FTLD spectrum disorders are thus needed.