
Abstract
Background:
Although cigarette smoking is an important modifiable factor of cognitive impairment, the roles of the Alzheimer’s disease (AD) core pathologies in modulating this process have not been fully delineated.
Objective:
This study aimed to explore associations of cigarette smoking with cognition and cerebrospinal fluid (CSF) AD biomarkers.
Methods:
A total of 1,079 non-demented participants were included from the Chinese Alzheimer’s Biomarker and LifestylE (CABLE) study. Associations of cigarette smoking with cognition and CSF AD biomarkers were explored by multiple linear regression models. The mediation analyses with 10,000 bootstrapped iterations were conducted to explore the mediation effects.
Results:
Heavy cigarette smokers (pack-years > 20) had poorer global cognition as well as higher levels of CSF p-tau and t-tau compared with the non-smokers (p < 0.01). Time-dose effect analysis among smokers also suggested that both cognitive impairment and tau pathologies markedly deteriorated with greater cumulative cigarette exposure, independently of the Aβ pathology (p < 0.01). In addition, smokers with older age or APOE ɛ4 showed more obvious influences on CSF tau pathologies but not on cognition. Overall, the influence of smoking on cognition was partially mediated by tau pathologies (estimated proportion: 12%), which still remained in late-life (10% ∼11%) and increased in APOE ɛ4 carriers (18% ∼24%). Encouragingly, long-term smoking cessation mitigated both cognitive impairment and tau pathologies (p < 0.05).
Conclusion:
Cigarette smoking was associated with both cognitive impairment and tau pathologies, which were accompanied by time-dose effects. Tau pathology might be a key mediator for influences of cigarette smoking on cognitive impairments.
INTRODUCTION
Cigarette smoking is the leading modifiable risk factor for numerous negative health outcomes [1]. Associations of cigarette smoking were found with poorer global cognitive functioning and several cognitive domains [2, 3]. Meanwhile, longitudinal studies also indicated that cigarette smoking might increase the risk of cognitive decline and dementia [4–7]. Globally, nearly 4.7 million Alzheimer’s disease (AD) cases worldwide could be attributable to smoking [8]. Furthermore, our recent study also regarded avoidance of smoking exposure as one of the preliminary clinical suggestions for AD with Class I recommendation and level B evidence [9]. However, the biological mechanisms by which cigarette smoking increases the risk of cognitive impairments and AD have not been fully delineated.
As the main type of dementia, AD is pathologically characterized by extensive neuronal loss, as well as the accumulation of extracellular amyloid plaques and intracellular neurofibrillary tangles in brain [10]. Although a limited number of animal studies suggested that smoking might influence these pathological changes in AD [11–13], there is still a lack of large clinical studies to prove that and to systematically explore the relationships among AD pathology, cognition, and smoking. Ample evidence has demonstrated that the levels of cerebrospinal fluid (CSF) amyloid-β42 (Aβ42), phosphorylated tau protein (p-tau), and total-tau protein (t-tau) could well reflect AD pathology and serve as core biomarkers for a preclinical diagnosis [10]. Therefore, exploring the associations between cigarette smoking and these CSF AD core biomarkers will help reveal how smoking affects these pathological changes and subsequently causes cognitive impairment. In the present study, after controlling various confounding factors, we aimed 1) to explore the relationships of cigarette smoking with cognition and CSF AD biomarkers; 2) to test whether the influences of cigarette smoking on cognition were mediated by AD core pathology; and 3) to examine whether smoking cessation was helpful in alleviating cognitive impairment and brain pathology in a large sample of 1,079 Chinese adults without dementia.
MATERIALS AND METHODS
The CABLE database
All non-demented participants in our study were included from the Chinese Alzheimer’s Biomarker and LifestylE (CABLE) database. The CABLE database is an ongoing large and independent cohort initiated in 2017 [14]. CABLE aimed to determinethe genetic and environmental modifiers of AD biomarkers and their utility in early diagnosis in the northern Chinese Han population. All participants were recruited at Qingdao Municipal Hospital, Shandong Province, China. The exclusion criteria include: a) central nervous system infection, head trauma, neurodegenerative diseases other than AD (e.g., epilepsy, Parkinson’s disease), or other major neurological disorders; b) major psychological disorders; c) severe systemic diseases (e.g., malignant tumors); d) family history of genetic diseases. All participants underwent clinical and neuropsychological assessments, biochemical testing, as well as blood and CSF sample collection. Comprehensive questionnaire, electronic medical record system, and laboratory inspection management system were used to collect demographic information, AD risk factor profile, and medical history. All participants in the CABLE were aged between 40 to 90 years old and each individual underwent comprehensive clinical, neuropsychological, psychosocial, and psychiatric evaluations to determine their cognitive diagnoses in compliance with the National Institute on Aging–Alzheimer’s Association (NIA-AA) workgroup diagnostic criteria [15]. Global cognitive levels were measured by China Modified Mini-Mental State Examination (CM-MMSE) and Montreal Cognitive Assessment (MoCA). Behavioral or psychological symptoms were assessed by Hamilton Rating Scale for Depression (HAMD) and Hamilton Rating Scale for Anxiety (HAMA). The CABLE database was conducted in accordance with the Helsinki declaration, and the research program was approved by the Institutional Ethics Committee of Qingdao Municipal Hospital. All subjects or their proxies provided written consents.
Study participants
A total of 1,307 adults who failed to meet the criteria of AD had available basic information. Ninety-six participants without complete smoking data were excluded. Fifty-one participants with significant symptoms of anxiety or depression were removed. Eighty-one participants were excluded because of missing CSF data. Finally, a total of 1079adults who failed to meet the criteria of AD and had no significant symptoms of anxiety (HAMA scores≤7) and depression (HAMD scores≤7) were included from CABLE database. Basic information of participants included age (continuous), sex (female versus male), years of education (continuous), CM-MMSE score (continuous), MoCA score (continuous), Apolipoprotein E ɛ4 (APOE ɛ4: carriers versus non-carriers) status, and body mass index (BMI: weight [kg]/height2 [m]), alcohol use habit (yes versus no) and medical factors (yes versus no: histories of hypertension, diabetes mellitus, hyperlipemia, atrial fibrillation, and stroke). The information on CSF AD core biomarkers (continuous) included levels of Aβ42 (n = 1,036), p-tau (n = 1,013), and t-tau (n = 1,029).
Smoking information
All smoking data were collected in the form of a self-reported questionnaire. Smokers were defined as individuals with current or former smoking on most or all days. Three smoking exposure variables were applied in our study. a) Smoking frequency was quantified by cigarettes per day. b) Smoking duration was defined as the age at the time of data collection minus the year they started smoking (for current smoking), or the age they stopped smoking minus the year they started smoking (for former smoking). c) A more composite indicator, pack-years, was calculated as the cigarettes per day multiplied by duration, divided by 20 (cigarettes per pack). In this study, the pack-years was analyzed as the primary smoking exposure indicator, and the other two smoking exposure indicators (cigarettes per day and duration) were used in post hoc sensitivity analyses. Smokers were further stratified into light and heavy smoking subgroups in some following analyses according to the three smoking exposure variables respectively (cigarettes per day > 20; duration > 20 years; pack-years > 20).Duration of smoking cessation was defined as the age at the time of data collection minus the year they quitted smoking.
CSF AD biomarkers
Fasting CSF samples were collected through the standard operational process of lumbar puncture and processed within two hours. Each specimen was centrifuged at 2000×g for 10 min, and stored in an enzyme-free EP (Eppendorf) tube at –80°C until subsequent assays were performed. The thaw/freezing cycle was limited not to surpass two times. CSF Aβ42, p-tau, and t-tau were determined with the ELISA kit (Innotest β-AMYLOID (1–42) [catalog number: 81583], PHOSPHO-TAU (181p) [catalog number: 81581] and hTAU-Ag [catalog number: 81579]; Fujirebio, Ghent, Belgium). All analyses were performed by professional experimenters who were blind to clinical and smoking information. The within-batch coefficient of variation (CV) was < 5% (mean CV: 4.4% for Aβ42, and 2.7% for p-tau, 4.2% for t-tau). The inter-batch CV was < 20% (mean CV: 5.1% for Aβ42, 2.6% for p-tau, and 4.1% for t-tau). There was no difference in CV between non-smoking and smoking groups (within -batch CV: Aβ42, p = 0.7810; p-tau, p = 0.5871; t-tau, p = 0.6552; inter-batch CV: Aβ42, p = 0.7541; p-tau, p = 0.3745; t-tau, p = 0.6152).
APOE ɛ4 genotyping
The DNA was extracted from blood samples with QIAamp® DNA Blood Mini Kit (250). And the extracted DNA was separated and stored in an enzyme free EP tube at –80°C until the APOE genotyping was completed in this study. Two specific loci related to APOE status (rs7412 and rs429358) were selected for genotyping with restriction fragment length polymorphism technology.
Statistical analysis
The data were shown in the form of mean±SD (standard deviation) or the proportions of them. The outlier values situated outside three SD were excluded prior to subsequent analyses. In terms of skewed data (Shapiro-Wilk test < 0.05), the Box-Cox transformations were utilized to construct these data with approximately normal distributions via “car” package of R software. Chi-square test (for categorical variables) and Mann-Whitney U test (for continuous variables) were used to compare the demographic characteristics. Given that MoCA score is more sensitive than MMSE score, especially in the early stages of the disease [16], it was utilized to reflect global cognitive function in the following analyses. The relationships of cigarette smoking with cognition and CSF AD biomarkers were explored in five steps.
Step 1. As for the smoking status, the two-group comparative analyses of cognition and CSF AD biomarkers were conducted using Mann-Whitney U test. Given that light smoking exposure might have a great confounding effect on the results due to irregular smoking behavior, we further performed a three-group comparative analysis (non-smoking, light smoking and heavy smoking) by Kruskal-Wallis test followed by multiple comparisons with the Holm’s adjustment. Then, with non-smoking as a reference, the associations of light and heavy smoking with cognition or CSF AD biomarkers were further verified in multiple linear regression models, after adjusting for covariables.
Step 2. In the smoking group, the time-dose effects of smoking exposure (continuous variables: pack-years) on cognition or CSF AD biomarkers were explored in multiple linear regression models, after adjusting for covariables.
Step 3. The influences of age or APOE ɛ4 status on the associations of smoking exposure with cognition or CSF AD biomarkers were tested in the two analyses. Firstly, interaction terms for APOE ɛ4 status or age were additionally added into multiple linear regression models. Secondly, subgroup analyses for different ages (Mid-life < 65 years and Late-life≥65 years) or APOE ɛ4 status (APOE ɛ4 carriers and APOE ɛ4 non-carriers) were further performed if the interaction effects were statistically significant (p < 0.1) in above interaction analyses.
Step 4. Next, to explore whether the association between cigarette smoking (the continuous variable: pack-years) and cognition was mediated by AD pathologies, linear regression models were fitted based on the method proposed by Baron and Kenny [17]. Mediation effects were established if the following criteria were simultaneously met: 1) pack-years was significantly related to CSF AD biomarkers; 2) pack-years was significantly related to cognitive measures; 3) CSF AD biomarkers were significantly related to cognitive measures; 4) the association between pack-years and cognition was attenuated when CSF AD biomarkers (the mediator) were added in the regression model. Furthermore, the attenuation or indirect effect was estimated, with the significance determined using 10,000 bootstrapped iterations (“mediate” package in R 3.5.1 software). Each path of the model was controlled for age, gender, years of education, and APOE ɛ4 status. A p value < 0.1 for indirect effect (IE) was considered significant.
Step 5. Finally, to test the associations of smoking cessation with cognitive impairment and AD pathology, we next separated the individuals with smoking cessation (n = 140) from current smokers and stratified them by duration of cessation (cessation groups: 1–5 years, 6–10 years > 10 years). With current smoking as a reference, the associations of different cessation groups with cognition or CSF AD biomarkers were tested in multiple linear regression models after adjusting for covariables.
Above multiple linear regressions (in Step 1, 2, 3, 5) were calculated in two models adjusting for different covariates (Model 1: adjusting for age, sex, years of education and APOE ɛ4 status; Model 2: additionally adjusting for BMI, alcohol use, medical factors [history of hypertension, diabetes mellitus, hyperlipemia, atrial fibrillation and stroke], CV [for CSF biomarkers’ analysis], MMSE scales [for CSF biomarkers’ analysis] and pack-years). The multicollinearity was assessed using tolerance, Variance Inflation Factor (VIF) and Pearson’s correlation coefficients. No multicollinearity existed in each model of the current study. Bonferroni method was used for multiple correction. The “lm”, “mediate”, “car” and “ggplot2” packages in R 3.5.1 software were used to perform the above analyses. A two-tailed p < 0.05 was considered significant except where specifically noted.
RESULTS
Participants’ characteristics
As shown in Table 1, a total of 1079 participants were included in our study consisting of 722 non-smoking participants and 357 smokers. The average age of participants was 61 years (SD = 11.44); 431 (39.9%) participants were males; and 184 (17.1%) participants were APOE ɛ4 carriers. Smokers tended to be male and to have the alcohol drinking habit (p < 0.05). Moreover, except for BMI and history of hypertension, we found no significant differences in age, years of education, APOE ɛ4 status and medical history (diabetes mellitus, hyperlipemia, atrialfibrillation and stroke) between the two groups (p > 0.05).
Characteristics of participants from CABLE database
APOE, apolipoprotein E gene; BMI, body mass index; CM-MMSE, China-Modified Mini-Mental State Examination; MoCA, Montreal Cognitive Assessment; CSF, cerebrospinal fluid; Aβ, amyloid-β; p-tau, phosphorylated tau protein; t-tau, total tau protein; SD, standard deviation Bold indicated that the results were statistically significant. * Intergroup comparisons were tested by Mann-Whitney U test.
The relationship of cigarette smoking with cognition
As for the smoking status, though two-group comparative analyses did not find significant cognitive decline in smokers (p = 0.095) (Table 1), three-group comparative analyses showed that heavy smokers (pack-years > 20) had much more significant cognitive impairment than non-smoking participants (p < 0.001) (Fig. 1A). Furthermore, these associations were attenuated but still remained significant after adjusting for various covariates, (Model 1, p = 0.034; Model 2, p = 0.015) (see Supplementary Table 1).
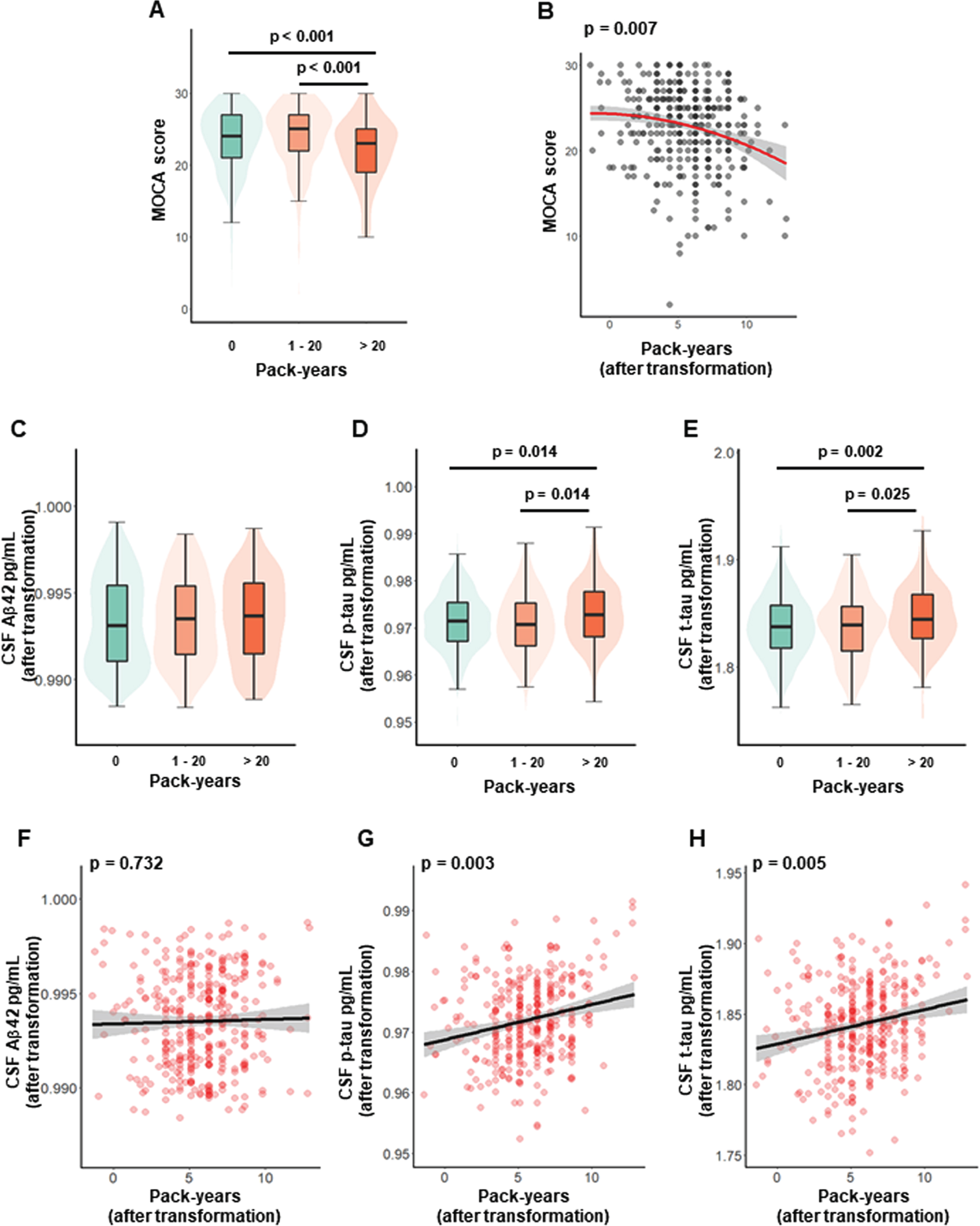
Relationships of cigarette smoking with cognition and CSF AD biomarkers. The three-group comparative analysis for cognitive levels (A). Time-dose effects of smoking exposure on cognition (B). The three-group comparative analyses for levels of CSF AD core biomarkers (C-E). Time-dose effects of smoking exposure on levels of CSF AD core biomarkers (F-H). The three-group comparative analyses were tested by Kruskal-Wallis test followed by multiple comparisons with the Holm’s adjustment. The time-dose effects were tested by multiple linear regression models after adjusting for age, sex, years of education and APOE ɛ4 status. MoCA, Montreal Cognitive Assessment; CSF, cerebrospinal fluid; Aβ, amyloid-β; p-tau, phosphorylated tau protein; t-tau, total tau protein.
Then time-dose effect analysis showed that cognitive levels decreased significantly with the increase of smoking pack-years in Model 1 (β= –12.28, p = 0.007) (Fig. 1B). These associations were attenuated but still remained significant after additionally adjusting for all covariables (β= –0.24, p = 0.010) (see Supplementary Table 2).
Influences of age or APOE gene on the relationship between cigarette smoking and cognition
After adding the interactions of smoking exposure with APOE ɛ4 status or age into multiple linear regression models, the associations between smoking exposure and cognition still remained significant, but the interactions did not show influences on cognition (pack-years×age, p = 0.940; pack-years×APOE, p = 0.170) (see Supplementary Table 3).
The relationships of cigarette smoking with CSF AD biomarkers
As for the smoking status, the two-group comparative analyses showed that smokers had higher t-tau levels than non-smokers (p = 0.019). This tau-related trend was more obvious in the three-group analyses which showed that heavy smokers (pack-years > 20) had higher levels of both p-tau (p = 0.014) and t-tau (p = 0.002) than non-smokers (Fig. 1D, E). However, no significant difference in Aβ42 was found in either two-group (p = 0.152) or three-group (p = 0.247) comparative analyses (Table 1, Fig. 1C). Then all these results did not change significantly after adjusting for covariables (Model 1, Aβ42, p = 0.286; p-tau, p = 0.010; t-tau, p = 0.007; Model 2, Aβ42, p = 0.243, p-tau, p = 0.009; t-tau, p = 0.001) (see Supplementary Table 1).
In time-dose effect analyses, higher pack-years was associated with increased levels of p-tau (β= 4.25*10–4, p = 0.003) and t-tau (β= 18.04*10–4, p = 0.005), but not with Aβ42 (β= 0.18*10–4, p = 0.732) (Model 1: Fig. 1F-H). All these results did not change significantly in Model 2 after adjusting for allcovariables (see Supplementary Table 2).
To further confirm whether above associations between cigarette smoking and tau-related biomarkers could be affected by Aβ42 levels, we repeated the above analyses after additionally controlling for Aβ42 levels (Model 2). These associations with tau-related biomarkers still existed (see Supplementary Tables 4 and 5).
Influences of age or APOE gene on the relationships between cigarette smoking and CSF AD biomarkers
In the interactive analyses, we found that the interactions between smoking exposure and APOE ɛ4 status or age had influences on tau-related biomarkers (p < 0.1), but not on Aβ42 (Supplementary Table 3). Further subgroup analyses also confirmed these influences (Fig. 2). Specifically, the pack-years was significantly associated with p-tau and t-tau in late life (p-tau, p = 0.035, t-tau, p = 0.007) and APOE ɛ4 carriers (p-tau, p = 0.032, t-tau, p = 0.003). However, attenuated and less significant associations were found in midlife and APOE ɛ4 non-carriers.
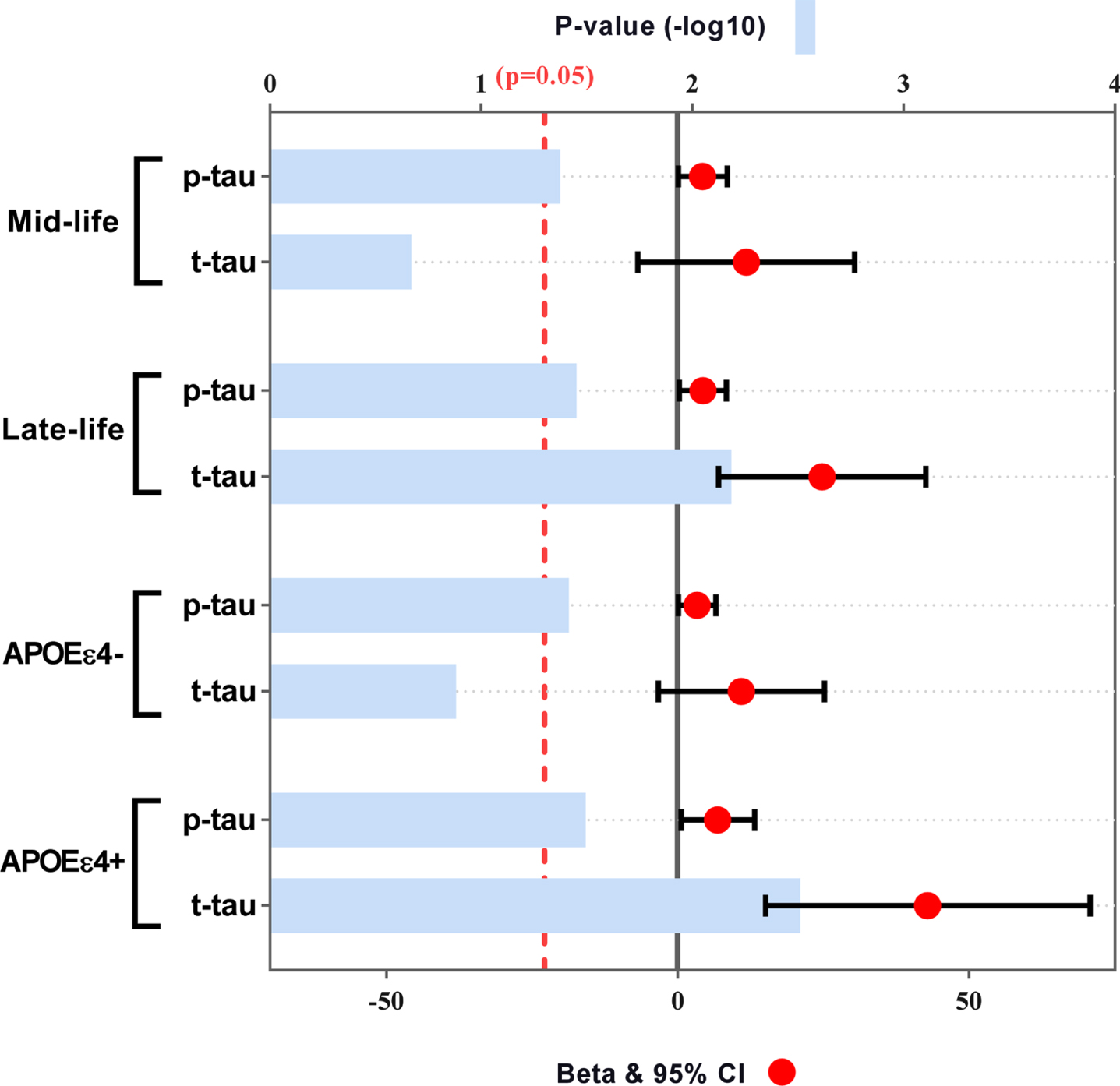
Subgroup analyses of age and APOE ɛ4 status for the associations of smoking exposure with tau-related biomarkers. Subgroup analyses were tested by multiple linear regression models after adjusting for age, sex, years of education and APOE ɛ4 status (in age subgroups). APOE, apolipoprotein E gene; CSF, cerebrospinal fluid; p-tau, phosphorylated tau protein; t-tau, total tau protein.
Causal mediation analyses
The above findings indicated that cigarette smoking was not only a significant risk factor for cognitive impairment, but also a potential modulator of tau pathologies. Therefore, we further explored whether tau pathologies could mediate the influences of cigarette smoking on cognition. The results demonstrated that the relationship between smoking exposure and global cognitive impairment was mediated by tau pathologies, including p-tau (Fig. 3A) and t-tau (Fig. 3B). The effect was considered partial mediation and the approximate proportion of mediation was 12%. Furthermore, this partial mediation effect still remained significant in late-life with the approximate proportion of mediation varying from 10% to 11% (Fig. 3C, D), and it increased in APOE ɛ4 carriers with the approximate proportion of mediation varying from 18% to 24% (Fig. 3E, F).
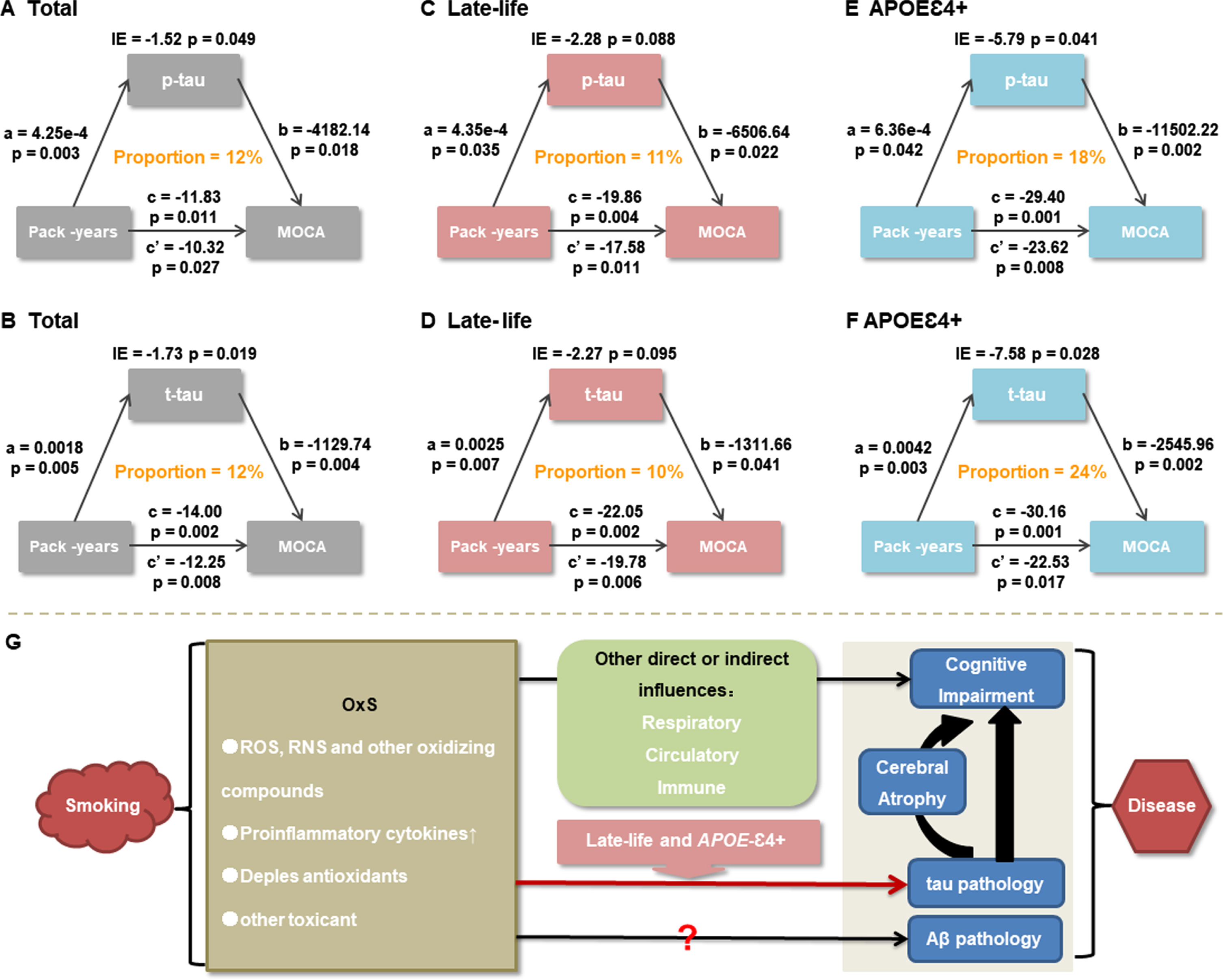
Causal mediation analyses. The relationship between cigarette smoking and MoCA scores was mediated by tau pathologies, including p-tau and t-tau (A, B). These mediation effects still remained significant in late-life (C, D), and increased in APOE ɛ4 carriers (E, F). A schematic graph depicting associations among cigarette smoking, AD core pathologies, and cognition was shown. Combining our study with previous studies, it could be reasonably inferred that tau pathology acted as a key mediator for influences of cigarette smoking on cognitive impairment independently of Aβ pathology. Older age and APOE ɛ4 might enhance this pathway. However, this mediation effect was deemed partial and there might be other direct or indirect pathways via which cigarette smoking contributed to cognitive impairments (G). CI, confidence interval; CSF, cerebrospinal fluid; p-tau, phosphorylated tau protein; t-tau, total tau protein; APOE, apolipoprotein E gene; IE, indirect effect; MoCA, Montreal Cognitive Assessment; OxS, oxidative stress; ROS, reactive oxygen species; RNS, reactive nitrogen species.
Associations of smoking cessation with cognition and CSF tau-related biomarkers
The above robust relationships of cigarette smoking with cognitive impairment and tau pathologies naturally raised the hypothesis of whether smoking cessation can mitigate these changes. We thus separated the individuals with smoking cessation from current smokers (Table 2). Compared with the current smokers, individuals with smoking cessation (especially with a longer duration of cessation) had better cognitive performance (cessation > 10 years, p < 0.001) and decreased CSF t-tau levels (cessation > 10 years, p = 0.032) after adjusting for age, sex, years of education, and APOE ɛ4 status (Model 1).These results still remained significant after further adjusting for other confounding factors including pack-years (Model 2).
Associations of smoking cessation with cognition and tau-related biomarkers
MoCA, Montreal Cognitive Assessment; CSF, cerebrospinal fluid; p-tau, phosphorylated tau protein; t-tau, total tau protein. aMultiple linear regression models after adjusting for age, sex, years of education, and APOE ɛ4 status. bMultiple linear regression models after adjusting for age, sex, years of education, APOE ɛ4 status, BMI, alcohol use, medical factors (history of hypertension, diabetes mellitus, hyperlipemia, atrial fibrillation and stoke), CV (for CSF biomarkers) and MMSE score (for CSF biomarkers) and pack-years. Bold indicated that the results were statistically significant.
Sensitivity analysis
The post hoc sensitivity analyses using the other two smoking exposure indicators (cigarettes per day and duration) also got generally similar results (Supplementary Figures 1–3). Since the percentage of female smokers in our study was small. To exclude the possible confounding effects of gender, we repeated the above calculation after excluding femaleparticipants. The results did not change significantly (results were not shown). To avoid the influence of missing data on the results, we repeated our analyses in a subset with complete CSF data (n = 955). The results were barely changed (results were not shown).
DISCUSSION
The present study systematically explored the interrelationships of cigarette smoking, cognitive impairment, and CSF AD core biomarkers. In non-demented adults, we found that a) cigarette smoking was a risk factor for cognitive impairment with atime-dose effect; b) cigarette smokers had greater CSF tau pathologies with a time-dose effect; c) the influence of cigarette smoking on cognition was partially mediated by tau pathologies; and d) smoking cessation might mitigate cognitive impairment and tau pathologies. The study detailed the possible mechanism by which cigarette smoking was involved in cognitive impairment, and further demonstrating that cigarette smoking was a significant modifiable risk factor for cognitive impairment diseases (such as dementia due to AD) or tau-related neurodegenerative diseases.
Our results are consistent with the findings from previous cross-sectional and longitudinal studies in which smoking or cigarette smoking showed harmful effects on global cognitive functioning and multiple neurocognitive domains including attention, learning, memory, processing speed, and impulse control in adolescents as well as in mid-life or late-life adults [2–5]. As for the associations between cigarette smoking and tau pathologies, though few clinical studies focused on the AD core pathologies, in vitro and animal studies still supported our results. Hellstrom-Lindahl et al. reported that nicotine treatment of the human neuroblastoma cells (SH-SY5Y) caused increased levels of p-tau and t-tau [12]. Chronic nicotine administration has been shown to exacerbate tau pathology in a transgenic model of AD (3xTg-AD) [11]. Decreased levels of acetylated-tubulin and increased levels of p-tau were also observed in the hippocampus of the smoking rats [13]. Besides, a well-documented association between smoking and regional brain atrophy in humans also indirectly supported our results [18], as brain atrophy was found to be closely related to tau pathologies and cognitive levels [19]. In that sense, in addition to cognition, our results also provide a possible mechanism for the association between smoking and brain atrophy.
Generally speaking, the primary pathophysiological mechanism that may contribute to the neurobiological and neurocognitive abnormalities observed in smokers is oxidative stress [20] (Fig. 4H). It was worth noting that although tau pathologies might modulate the relationship of smoking toxicity (such as oxidative stress) with cognitive impairment, this effect was only partial mediation (10%–24%). This suggested that this relationship might be also modulated by many other direct or indirect mechanisms involving the circulatory system [21], the respiratory system [22], the immune system [23], and so on. In support of this hypothesis, we found that the influences of cigarette smoking on cognitive levels were attenuated when we further corrected for the medical covariates in the present study. In addition, as for Aβ pathology, we found no association of cigarette smoking with it. However, previous animal and autopsy studies also indicated some possible influences of smoking on Aβ pathology [4]. Therefore, this pathway still needs to be further explored in future studies. Anyway, given that the results about tau pathology persisted after controlling for CSF Aβ42 levels, our study at least confirmed that the influences of cigarette smoking on tau pathologies were not completely dependent on Aβ pathology.
Notably, interaction analysis, subgroup analysis, and mediation analysis all suggested that older age and APOE ɛ4 seemed to enhance the mediation effects of tau pathologies, compared with younger adults and APOE ɛ4 non-carriers. We hypothesized that this might be because aging or APOE ɛ4 increased the vulnerability of brain tissue to smoking toxicity. Consistent with our hypothesis, similar influences of interaction between smoking and APOE ɛ4 status was found on glucose metabolism, and smokers with APOE ɛ4 showed lower glucose metabolism than smokers without APOE ɛ4 and non-smoking individuals [2].
Overall, the above discussion suggested that smoking aggravated not only cognitive impairment but also pathological changes in the brain. Encouragingly, in our study, smoking cessation had positive effects on both cognition and tau pathology. However, these effects seemed to occur so slowly that it might take at least 10 years before these effects became apparent. Consistently, partial recovery of cerebral atrophy was also found after smoking cessation, whereas this was also a long process and could take an average of 25 years or more [18]. Therefore, all this evidence might serve as strong motivational arguments to encourage smoking cessation, but what should also be emphasized was that smoking cessation should be started as early as possible.
Some strengths enhance the reliability of our study, including the largest sample to date examining smoking-related CSF AD core biomarker associations, a blind quality control of CSF data and the ability to statistically control for a variety of potential confounds. There are also some limitations in this study. Firstly, this is a cross-sectional study and thus whether there is reverse causation needs to be considered. In other words, it is possible that the changes in cognitive function or brain pathologies may in part be a precipitating factor for initiating smoking.However, in the present study, the average age at which smokers initiated smoking is 27.02 (SD = 11.08) and ninety percent of smokers initiated smoking before the age of 40. Therefore, it is unlikely that cognitive impairment or AD pathologies precipitate smoking given they typically show up in older age after most have already begun smoking. Secondly, the recalled nature of the smoking history data is another limitation. However, recall errors would generally tend to add noise to the data and attenuated the degree of associations between smoking and cognitive function or AD pathologies. On such a basis, the degree of associations in our studies is likely to be even stronger than the one observed here. Furthermore, in order to minimize the influences of recall errors on results’ reliability, we used multiple exposure indicators rather than a single one for analyses and got consistent results. Thirdly, the lack of imaging or postmortem data was also a limitation of our study, which needs to be verified in the future.
In conclusion, cigarette smoking was associated with both cognitive impairment and tau pathologies, which were accompanied by time-dose effects. Tau pathology might be a key mediator for influences of cigarette smoking on cognitive impairments. This further emphasizes the importance of smoking cessation as a modifiable factor in the early prevention of AD or tau-related neurodegenerative diseases.
Footnotes
ACKNOWLEDGMENTS
The authors thank all participants of the present study as well as all members of staff of the CABLE study for their role in data collection. This study was supported by grants from the National Key R&D Program of China (2018YFC1314700), Shanghai Municipal Science and Technology Major Project (No.2018SHZDZX01) and ZHANGJIANG LAB, Tianqiao and Chrissy Chen Institute, and the State Key Laboratory of Neurobiology and Frontiers Center for Brain Science of Ministry of Education, Fudan University.