
Abstract
Background:
Down syndrome (DS) is frequently associated with Alzheimer’s disease (AD)-related neuropathological changes. There are few observations on the spectrum of mixed proteinopathies in DS patients.
Objective:
This study aimed to evaluate multiple disease-associated proteinopathies in a series of DS cases.
Methods:
We analyzed the distribution of neurodegenerative disease associated proteins in postmortem brain samples from 11 DS cases (6 females, median age 57, range 38–66 years). Sections were stained for phosphorylated tau, 3-repeat and 4-repeat tau, amyloid-β, alpha synuclein, phosphorylated TDP-43, and p62. A comprehensive anatomical mapping and staging were applied for all proteins.
Results:
Tau and amyloid-β pathology was prevalent in all cases and compatible with that typically seen in AD with some subtle deviations. Four of 11 cases presented with Lewy-related pathology (LRP). Two cases followed the Braak staging (stage 4 and 5) whereas 2 cases presented with an atypical distribution. Two cases showed limbic predominant age-related TDP-43 encephalopathy (LATE) (stage 1 and stage 2) neuropathologic change. Two cases exhibited aging-related tau astrogliopathy (ARTAG).
Conclusion:
In addition to subtle deviations from AD regarding the morphology of amyloid-β deposition and distribution of neuronal tau pathology, we find that the spectrum of mixed-pathologies in DS show distinctive features such as deviations from the Braak staging of LRP and that LATE neuropathologic change and ARTAG pathology can be seen in individuals younger than in sporadic AD cases. Our observations support the notion that DS has distinctive pathogenic pathways from sporadic AD.
Keywords
INTRODUCTION
Down syndrome (DS) is caused by triplication of chromosome 21 which harbors the gene for amyloid precursor protein, the major component of amyloid-β (Aβ) plaques [1]. Individuals with DS are at high risk for developing dementia of the Alzheimer’s disease (AD)-type [2, 3], thus most neuropathologic studies to date have focused on AD-like pathology including Aβ plaques and phosphorylated tau containing neurofibrillary tangles (NFTs) [3–9]. However, there is an increasing understanding that neurodegenerative proteinopathies rarely involve deposition of a single protein [10–14]. Indeed, abnormal deposits of Aβ, tau, alpha-synuclein (aSyn), and transactive response DNA binding protein 43 kDa (TDP-43) are often present in a single diseased brain in various combinations [10]. In elderly individuals, the number of pathologies correlate with the degree of AD-type neuropathologic change [10, 11]; hence, due to severe AD type pathology, mixed proteinopathies are expected in DS patients. Nevertheless, the spectrum of proteinopathies in DS patients has only been addressed in a single study that focused on the evolution of Braak NFT stage and Thal amyloid pathology [2].
Braak and colleagues proposed an aSyn staging system for Parkinson’s disease (PD), which hypothesized that aSyn pathology first appears in the enteric nervous system, dorsal motor nucleus of the vagal nerve, and the olfactory bulb [15, 16]. However, this initial staging system has not been able to satisfactorily classify a significant number of cases [17–19], and other hypothetical propagation models have been recently proposed [20–22]. Regarding DS patients, the frequency of Lewy-related pathology (LRP) including Lewy bodies and Lewy neurites differs considerably from report to report [2, 23–26] and none have fully evaluated all strategic anatomical regions in the brain of DS patients.
Individuals with DS are prone to develop AD-like pathology early in their life, and thus represent an interesting model of premature aging [2, 27]. Recently, characteristic aging-related pathologies such as age-related tau astrogliopathy (ARTAG) and limbic-predominant age-related TDP-43 encephalopathy (LATE) neuropathologic change (NC) have been proposed [28–30]. In addition, argyrophilic grain disease is also frequently detected as a concomitant pathology with other age-related neurodegenerative conditions [31, 32]. If accelerated aging is part of the neurodegenerative process in DS patients, these age-related pathologies would be expected to be observed frequently and in an advanced stage in DS patients.
To address these hypotheses, we performed a comprehensive, systematic immunohistochemical study of multiple disease-associated proteinopathies in a series of 11 DS cases. We focused specifically on the morphology and distribution patterns of the deposits in each proteinopathy and the presence or absence of age-related changes and compared our findings with a previous study [2].
MATERIALS AND METHODS
Case selection
We examined cerebral and cerebellar specimens from 11 clinically diagnosed DS cases (five males, six females) ranging from 38 to 66 years (mean age 54.7±8.6) from the University Health Network Neurodegenerative Brain Collection (UHN-NBC). Selected case details are provided in Table 1. All brains had been obtained at autopsy through appropriate consenting procedures with Local Ethical Committee approval. This study was approved by the UHN Research Ethics Board (Nr. 20-5258) and University of Toronto (Nr. 39459) and was performed per the ethical standards established in the 1964 Declaration of Helsinki, updated in 2008.
Summary of the clinical, autopsy and immunohistochemical findings of the cases
–, negative;±, indefinite; +, positive. AD, Alzheimer’s disease; AGD, argyrophilic grain dementia; Am, amygdala; ARTAG, age-related tau astrogliopathy; BS, brainstem; CAA, cerebral amyloid angiopathy; CBL, cerebellar; DN, dentate nucleus; F, female; Fr, frontal lobe; Int, intermediate; LR, Lewy-related; M, male; N, nucleus; NA, not available; NFD, neurofibrillary degeneration; Pa, parietal lobe; PC, Purkinje cell; PN, pontine nucleus; TDP-43, transactive response DNA binding protein 43 kDa; Te, temporal lobe; WM, white matter. LR pathology indicates brainstem, limbic, or neocortical type; TDP-43 pathology indicates the stage of LATE (limbic predominant age-related TDP-43 encephalopathy).
Tissue sampling and pathological assessment
Formalin-fixed paraffin-embedded tissue sections from the following brain regions from all cases except for the middle temporal cortex, anterior cingulate cortex, and were investigated: middle frontal cortex, middle temporal cortex (n = 10), inferior parietal cortex, anterior cingulate cortex (n = 7), hippocampus, amygdala, basal ganglia including caudate nucleus, putamen and globus pallidus, thalamus, cerebellar cortex, brainstem including midbrain, pons, and medulla oblongata (n = 10).
Immunohistochemistry was performed using the following primary antibodies: phosphorylated tau (clone AT8, 1:200, Invitrogen/ThermoFischer, Carlsbad, USA); Aβ (Clone 6F/3D, 1:50, Dako, Glostrup, Denmark); aSyn (clone 5G4, 1:1000, Analytikjena, Jena, Germany) [33]; phosphorylated TDP-43 (clone 11-9, 1:2000, CosmoBio, Tokyo, Japan); and p62 (clone 3/P62 LCK LIGAND, 1:800, BD Transduction Laboratories, Franklin Lakes, USA). Immunostaining for 3-repeat-tau (3R-tau, clone 8B6/C11, 1:5000, MilliporeSigma, Bedford, USA) and 4-repeat-tau (4R-tau, clone 1E1/A6, 1:200, MilliporeSigma) was additionally performed in cases that exhibited neuronal immunoreactivity for AT8 in the midbrain and/or pons and/or dentate nucleus. Antigen retrieval was performed using Dako PT Link with low pH solution (aSyn, TDP-43, p62, 3R- and 4R-tau antibodies) and/or 80% formic acid (1 h for anti-Aβ, 5 min for anti-aSyn, and 1 min for anti-TDP-43, 3R- and 4R-tau antibodies). Immunostaining was performed using the Dako Autostainer Link 48 and EnVision FLEX+ Visualization System, according to manufacturer’s instructions. Subsequently, all sections were counterstained with hematoxylin.
Following Aβ and tau immunostaining, all cases were staged according to Braak’s stage of NFT burden [34], Thal phase of Aβ deposition [35], and the density of neuritic plaques was evaluated in accordance with the Consortium to Establish a Registry for Alzheimer’s disease (CERAD) criteria [36]. Based on these results, the level of AD neuropathological change was divided into four categories (Not, Low, Intermediate, High) following the National Institute on Aging-Alzheimer’s Association (NIA-AA) guidelines [37]. Additionally, the type and stage of cerebral amyloid angiopathy (CAA) [38, 39] and the Aβ deposition patterns in the cerebellum were evaluated using Aβ-stained specimens, and the distribution of tau-pathology such as oligodendroglial coiled bodies, pretangles, argyrophilic grains suggestive of argyrophilic grain dementia (AGD) and thorn-shaped astrocytes (TSAs) suggestive of ARTAG were evaluated using tau-immunostained specimens [40]. The severity of LRP was assessed according to the Consensus Guidelines for Dementia with Lewy body, the Braak stages in the development of PD-related pathology, and the Lewy pathology consensus criteria (LPC) using aSyn immunohistochemistry [15, 41]. The pathological type of TDP-43 proteinopathy was assessed following the stages of LATE-NC [30].
Semiquantitative grading system of tau and Aβ pathology
In addition to the staging systems described above, we semi-quantitatively graded the severity of immunohistochemical findings of tau and Aβ. The severity of overall tau (AT8) pathology including pretangles, NFTs, neuropil threads was evaluated using a five-point scoring system, similar to that employed by Davidson et al. [2], as follows: Grade 0, no tau pathology present; Grade 1, very few and scattered neuronal cytoplasmic tau immunoreactivity and neuropil threads in each low power (×10 microscope objective) field; Grade 2, a mild number of neurofibrillary tangles and neuropil threads in each low power field; Grade 3, a moderate number of neurofibrillary tangles and neuropil threads in each low power field; Grade 4, many densely packed neurofibrillary tangles and neuropil threads in each low power field. We also evaluated the oligodendroglial pathology in the cerebral white matter (WM) using a similar scoring system.
The severity of Aβ pathology within brain parenchyma (as amyloid plaques) and cerebral vessels (as CAA) was evaluated using a five-point scoring system [2, 43] as follows: Plaque Grade 0, no Aβ plaques in parenchyma/layer; Grade 1, a few Aβ plaques in parenchyma/layer occupying each low power (×10 microscope objective) field; Grade 2, a moderate number of Aβ plaques in parenchyma/layer occupying each low power (×10 microscope objective) field; Grade 3, many dispersed Aβ plaques in parenchyma/layer occupying each low power (×10 microscope objective) field; Grade 4, very many densely packed Aβ plaques in parenchyma/layer occupying each low power (×10 microscope objective) field. CAA Grade 0, no CAA in blood vessel walls in leptomeninges or brain parenchyma; Grade 1, occasional blood vessels with CAA in leptomeninges and/or within brain parenchyma, usually not occupying the full thickness of the wall; Grade 2, a moderate number of blood vessels with CAA in leptomeninges or brain parenchyma in leptomeninges or within brain parenchyma, some occupying the full thickness of the wall; Grade 3, many blood vessels with CAA in leptomeninges or brain parenchyma, most occupying the full thickness of the wall; Grade 4, most or all blood vessels with severe CAA in leptomeninges or within brain parenchyma, occupying the full thickness of the wall.
Statistical analysis
Fisher’s exact test was used for categorical variables (sex and pathological findings) and Mann-Whitney U test was used for continuous variable (age at death). Levels of significance were two-tailed at p < 0.05.
RESULTS
Tau pathology
All cases showed phosphorylated tau (AT8) immunoreactivity characterized by the presence of diffuse cytoplasmic staining of nerve cells and neuropil threads. 10 out of 11 cases showed NFTs, dystrophic neurites, and oligodendroglial coiled-bodies in the WM, and 2 cases showed TSAs (Table 1). The representative pathological findings are shown in Fig. 1 and the severity of total burden of tau pathologies in each anatomical region is shown in Table 2. The distribution of neurofibrillary degeneration was compatible with the Braak stage V to VI in 9 cases and stage III in 1 case (Table 1). The youngest case (Case 1) showed only very mild pathology in the transentorhinal region considered to be the Braak stage 1 (Fig. 1A). However, this case showed few pretangles in frontal, temporal, and parietal cortices (Fig. 1B) and dentate nucleus (Fig. 1C). Interestingly, neuronal cytoplasmic immunoreactivity and threads were seen in the pontine and dentate nucleus in six and five cases, respectively. No case exhibited cerebellar cortex and WM involvement. Neuronal tau consisted of both 3 R and 4R-tau isoform immunoreactivity including in the midbrain, pons, and dentate nucleus (Fig. 1D–F). The amount of WM oligodendroglial coiled-bodies and threads were mild in all 10 cases where these were observed (Fig. 1G).

Representative photomicrographs of tau pathology. A–D, G–I) Immunohistochemistry for phosphorylated tau (AT8). Immunohistochemistry for 3-repeat (E) and 4-repeat tau (F). A–C) Neuronal cytoplasmic immunoreactivity and neuropil threads in the entorhinal cortex (A), parietal cortex (B), and dentate nucleus (C) in Case 1. D–F) The findings in substantia nigra in Case 3. A moderate number of tangles and neuropil threads is observed in AT8-immunostained specimen (D). Both 3-repeat (E) and 4-repeat (F) tau immunoreactivity are observed in the same region. G) Threads and coiled-bodies in the temporal white matter in Case 2. Subpial thorn-shaped astrocytes in the frontal cortex in Case 7 (H) and in the amygdala in Case 8 (I). Scale bar = 100μm (D–F, H, I), 50μm (A–C), 20μm (G).
Phosphorylated tau (AT8) deposition grading in each anatomical region

NA, not available; NFT, neurofibrillary tangle.
Two cases exhibited subpial TSAs; one in the depth of the frontal cortical sulci (Case 7, aged 58, Fig. 1H), and another in the amygdala (Case 8, aged 61, Fig. 1I). In Case 7, unlike observed in chronic traumatic encephalopathy, we did not observe patchy accumulation of perivascular neuronal and astrocytic tau pathology in the grey matter of the depth of sulci where subpial TSAs were observed. In both cases, TSAs involved only one anatomical region (frontal cortex and amygdala, respectively) and were not identified in the subependymal area, perivascular area, grey matter, or WM. Also, other astrocytic tau pathologies, including tufted astrocytes, astrocytic plaques, globular astroglial inclusions, and ramified astrocytes, were not observed. Additionally, we did not observe argyrophilic grains in the limbic area. In summary, tau pathology was compatible with that seen in sporadic AD; however, we noted deviations such as 1) the consistent presence of oligodendroglial coiled-bodies; and 2) the distinctive anatomical distributions of neuronal tau pathologies such as involvement of the dentate and pontine nucleus and presence of pretangles in the cortical regions in low Braak stage.
Aβ pathology
Parenchymal Aβ deposition was identified in all cases in the form of focal deposits with or without neuritic corona and diffuse plaques. In 10 cases this distribution was compatible with Thal phase 5 and in 1 case with phase 3 (Table 1). The representative pathological findings are shown in Fig. 2 and severity in each anatomical region is shown in Table 3. All cases except for the youngest (Case 1) exhibited Grade 3 to 4 plaque pathology considered to be the CERAD grade C in the cerebral neocortex (Fig. 2A, B). In the cerebellum, 5 out of 10 cases showed a stripe-like deposition pattern with focal fibrillar structure perpendicular to the surface (Fig. 2C, D). In Case 3 (Table 1), in the Purkinje cell (PC) layer, occasional PCs accumulated dense Aβ deposition completely filling the cytoplasm and expanding to the dendritic arborization in a fine-granular pattern (Fig. 2E). In addition, in Case 2, 3, 4, and 11 (Table 1), occasional Bergman glial cells showed dense Aβ deposition (Fig. 2F). Both neuronal and astrocytic deposition were negative for AT8 and p62 immunostaining. Granular cell layer involvement was identified in seven cases (Table 1) and no case showed cerebellar WM involvement.
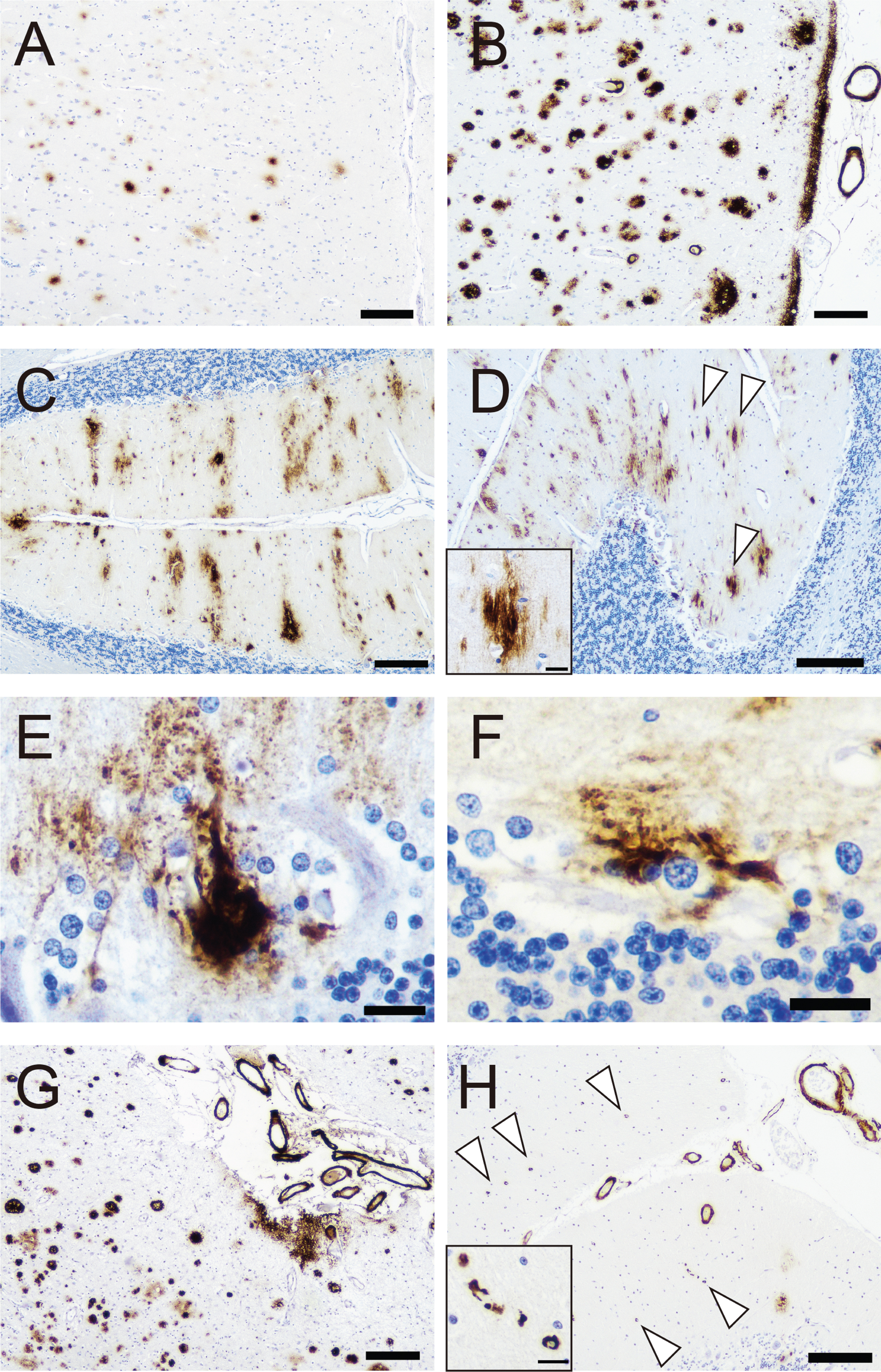
Representative photomicrographs of amyloid-beta (Aβ) pathology. A–H) Immunohistochemistry for Aβ (6F/3D). Neocortical Aβ deposition in Case 1 (A) and Case 3 (B). Cerebellar Aβ deposition in Case 3 (C–F). C) Stripe-like Aβ deposition in the molecular layer. D) Fibrillar Aβ deposition in the molecular layer (Arrowheads, Inset is higher magnification view of a deposit). Purkinje cell-like (E) and astrocytic (F) Aβ deposition in the Purkinje cell layer. G, H) Cerebral amyloid angiopathy (CAA) in Case 7. G) Leptomeningeal CAA is predominant, and no capillary involvement is identified in the cerebral cortex. H) In contrast, many capillary CAA lesions are identified in the cerebellum (arrowheads, inset is the higher magnification view of the capillary lesion). Scale bar = 200μm (A–D, G, H), 20μm (E, F, inset of D and H)
Amyloid-β distribution grading in the cerebral and cerebellum

CAA, cerebral amyloid angiopathy; CBM, cerebellum; GCL, granular cell layer; ML, molecular layer; PCL, Purkinje cell layer; WM, white matter.
CAA pathology was observed in 10 out of 11 cases (Fig. 2G, H). Although the severity did not significantly differ between the cerebrum and cerebellum, the capillary involvement (Fig. 2H) was more frequently identified in the cerebellum (two and four cases, respectively). In summary, Aβ pathology strongly resembled that seen in sporadic AD.
α-synuclein pathology
Four cases (two females) exhibited LRP (Case 3, 4, 7 and 10). The representative pathological findings are shown in Fig. 3 and severity in each anatomical region is shown in Table 4. The mean age of the patients was 55.8±6.3 years (range 49–65). All cases showed LRP in the amygdala and substantia nigra. In contrast, only three cases showed LRP in the locus coeruleus (Case 4, 7, and 10) and two cases in the dorsal vagal nucleus (Case 4 and 7). Additionally, compared to the amygdala (Fig. 3A) and substantia nigra (Fig. 3B), the LRP severity was low in the dorsal vagal nucleus (Fig. 3C). Therefore, two cases did not clearly follow Braak staging in the brainstem nuclei and were interpreted as being atypical (Case 3 and 10). However, according to the LPC [19], we classified the atypical cases as Brainstem predominant (Case 3 and 10), and the two further ones as Limbic (Case 7) and Neocortical (Case 4). In summary, aSN pathology was not unequivocally classifiable following the current criteria.
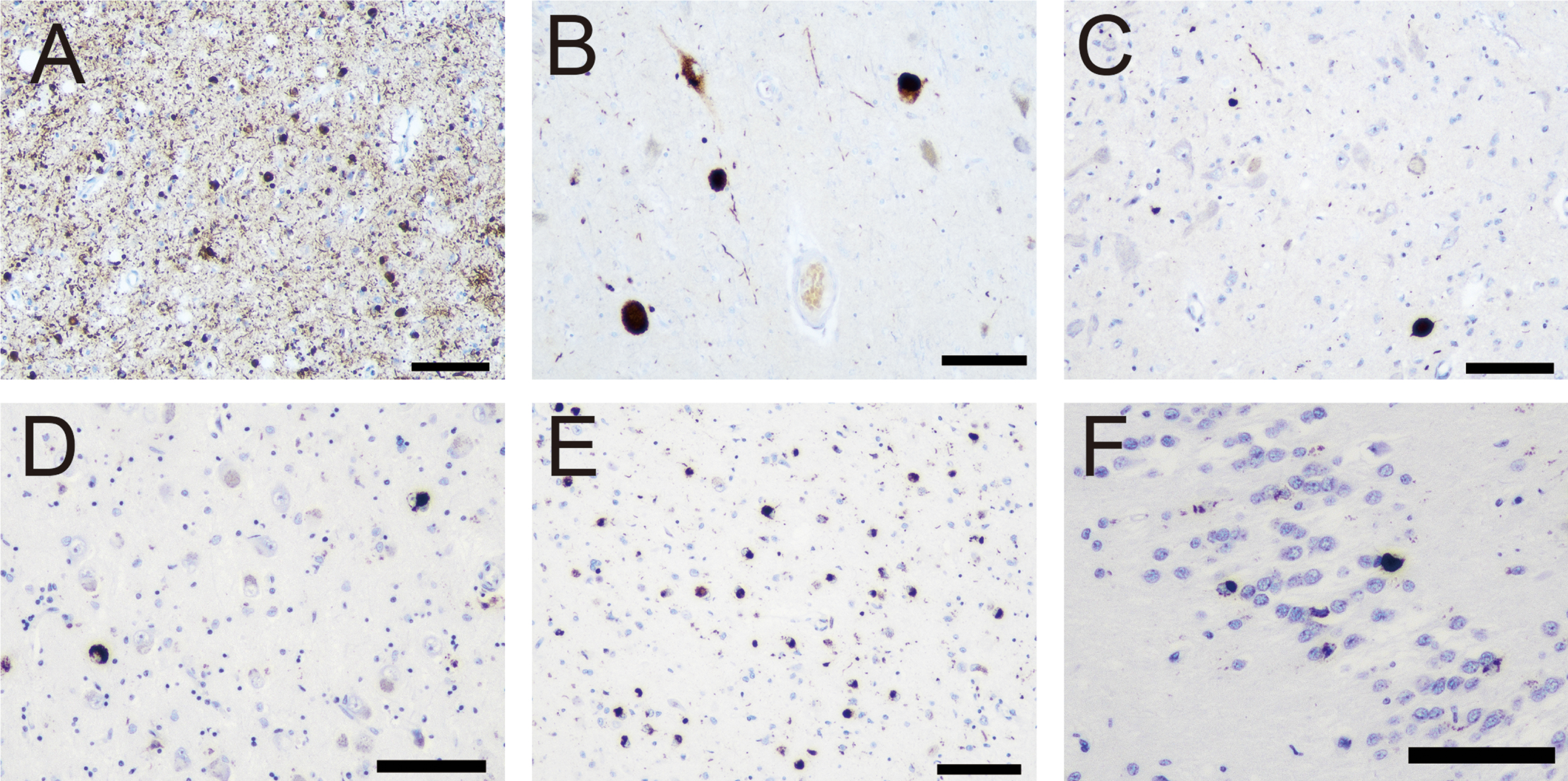
Representative photomicrographs of α-synuclein and phosphorylated transactive response DNA binding protein 43 kDa (p-TDP-43) pathologies. A–C) Immunohistochemistry for α-synuclein (Case 7). D–F) Immunohistochemistry for p-TDP-43. Numerous Lewy bodies (LBs) and neurites (Grade 4) in the amygdala (A) and severe LBs and cytoplasmic immunoreactivity with scattered neurites (Grade 3) are observed in the substantia nigra. C) In contrast, only scattered neurites (Grade 1) are identified in the dorsal vagal nerve nucleus. D) Sparse neuronal cytoplasmic inclusion bodies in the amygdala in Case 10. Numerous neuronal cytoplasmic inclusion bodies and sparse neurites in the amygdala (E) and sparse neuronal cytoplasmic inclusion bodies in the dentate gyrus (F) in Case 11. Scale bar = 100μm (A–D).
α-synuclein (5G4) deposition grading in each anatomical region
LPC, Lewy pathology consensus criteria; NA, not available.
TDP-43 pathology
Two cases (Case 10 and 11), aged 65 and 66, exhibited TDP-43 pathology. The representative pathological findings are shown in Fig. 3. In case 10, sparse neuronal cytoplasmic inclusion bodies were observed only in the amygdala (Fig. 3D). In contrast, in Case 11, both neuronal cytoplasmic inclusions and neurites were identified in the amygdala (Fig. 3E) and sparse cytoplasmic inclusions were identified in the hippocampus (Fig. 3F). In summary, these findings were consistent with the LATE-NC stage 1 and 2, respectively [30]. We did not observe hippocampal sclerosis.
In summary, four cases exhibited AD + one co-proteinopathy, three of them with different levels of LRP (Case 3, 7, and 4, ranging from brainstem, limbic, and neocortical type, respectively), and moderate level of TDP-43 pathology in Case 11 (LATE-NC stage 2), and one case exhibited AD + two co-proteinopathies (Case 10), including brainstem (atypical) LRP and low level of TDP-43 pathology (LATE-NC stage 1).
DISCUSSION
The neuropathology of DS is generally thought to resemble that seen in sporadic AD [2–9]. However, our work highlights neuropathological features suggestive of distinct aspects of pathological protein accumulation. In particular, we showed that 1) AD pathology, including morphology of Aβ deposition and distribution of neuronal tau pathology, shows subtle deviations from AD and 2) the spectrum of mixed-pathologies shows distinctive features such as that concomitant LRP does not follow the Braak staging and that typical age-related LATE-NC and ARTAG pathology can be seen in younger individuals [28–30].
To our best knowledge there has been only one study that systematically addressed the topic of different proteinopathies in DS cases [2]. A comparison of our work to this previous study is shown in Table 5. Patients in our study showed a higher mean age at death, proportion of cases over 40 years of age, and females (not statistically significant). In both studies, pathological findings including severe AD, CAA, LRP, and TDP-43 were found only in the patients aged over 40 years. The frequency of NIA-AA AD level-High, CAA pathology in our study was slightly higher than that of the previous study, but none of these reached statistical significance. In contrast, although not statistically significant, the frequency of LRP in our study was more than twice as high as that of the previous report in both all cases and those aged over 40 years (p = 0.05 and 0.08, respectively). In addition, we also highlighted important differences of distribution of LRP. Finally, although not statistically significant, the frequency of co-proteinopathy pathology in our study was also higher than in the previous report in both all cases and those aged over 40 years (p = 0.05 and 0.19 in AD + one co-proteinopathy, p = 0.42 and 0.48 in AD + two co-proteinopathy, respectively). In contrast, ARTAG pathology was found only in our study and this difference was statistically significant (p = 0.03 in both all cases and in those aged over 40 years). No AGD-like pathologies were observed in either study. One reason for the differences in the reported co-pathologies could be that we evaluated the amygdala also, which is thought to be an epicenter of mixed protein pathology [44].
Comparison of the results between our study and the previous study [2]
*p < 0.05 versus Previous study (Fischer’s exact test). AD, Alzheimer’s disease; AGD, argyrophilic grain dementia; ARTAG, age-related tau astrogliopathy; CAA, cerebral amyloid angiopathy; F, female; LR, Lewy-related; M, male; N, number; NA, not available; TDP-43, transactive response DNA binding protein 43 kDa; y.o., year-old.
Interestingly, half of the DS patients (Case 3 and 10) with LRP did not follow Braak staging. Although we were able to classify these as Brainstem predominant following the LPC [19], we emphasize that not all levels of brainstem were affected and thus these would be more compatible with substantia nigra predominant LRP. Moreover, one of these (Case 10) would be more compatible with amygdala predominant LRP. However, following the recent criteria [19], due to the presence of comparably less amount of LRP in the substantia nigra, we had to classify as Brainstem predominant. Recently, Borghammer et al. proposed a hypothetical body-first versus brain-first model of LRP [21, 22]. In the body-first model, the initial pathogenic aSyn originates in the enteric nervous system with secondary spreading to the brain, which is equivalent to Braak’s hypothesis [15, 16]. In contrast, in the brain-first model, the first pathology is proposed to appear in structures inside the central nervous system without prior involvement of the autonomic nervous system. The most probable “brain-first” locations are the amygdala, followed by the substantia nigra, and locus coeruleus [21, 22]. The distribution of aSyn pathology in our study was consistent with the latter pattern. Lippa et al. reported that aSyN pathology was identified in 50% of amygdala samples from DS patients and all LRP-positive cases exhibited the amygdala involvement, which is consistent with our results [23]. Furthermore, this is compatible with the result that the frequency of aSyn pathology in our patients was more than twice that reported by Davidson et al. who did not evaluate the amygdala [2]. Thus, most likely aSyn pathology in the DS patients follows the brain-first hypothesis and originates in the amygdala with secondary spreading to the lower brainstem [21, 22]. Toledo et al. reported that coincidence of AD pathology was associated with increased LRP in PD cases. Thus, AD pathology may play an important role in the deposition of aSyn in the amygdala in DS patients [17]. Unfortunately, we were not able to evaluate peripheral organs to evaluate the hypothesis that aSyn pathology propagates from the body to brain in DS.
Davidson et al. report that the pattern of progression of AD pathology in patients with DS is consistent with AD [2]. However, we observed deviations including observing neuronal tau pathology in relatively unusual locations including the pontine and dentate nucleus in several cases. Additionally, despite being Braak NFT stage I, the youngest case showed neuronal tau pathology in the neocortex and dentate nucleus. In spite the lack of tufted astrocytes, these distribution patterns of neuronal tau pathology are reminiscent of that seen in progressive supranuclear palsy [45, 46]; however, the expression pattern of 3R- and 4R-tau was consistent with that of in AD in our DS cohort. In summary, the development of tau pathology in DS patients may differ from typical AD, especially in the early stages, leading us to speculate that early prominent alterations in Aβ processing might induce the additional cortical neuronal pathology. On the other hand, this difference may be associated with altered physiological tau phosphorylation seen during brain development in DS individuals [27].
We found age-related pathologies consistent with ARTAG and LATE-NC in DS patients. All the cases exhibiting these lesions were younger than is typically seen [28–30], suggesting a possible accelerated rate of brain aging in DS. However, the frequency of ARTAG with AD pathology in this study was significantly lower than that in AD cases (25% versus 63%, p = 0.02) [29]. In particular, we did not observe lobar WM ARTAG, which is a frequent observation in typical AD [29]. Additionally, the lesions observed in this study were small, localized to only one anatomical region and no lesions other than the subpial TSAs were identified. We did not observe tau pathological changes reminiscent of chronic traumatic encephalopathy. Furthermore, as in the previous report [2], no AGD-like pathologies were identified. The reasons for these differences are not clear. We can speculate that ARTAG and AGD lesions are less detected because only a small amount is formed, and they show a less progressive course of accumulation as compared to AD lesions. Thus, the inconspicuousness of these aging-related lesions may also support the pathophysiology of accelerated aging in DS patients. We detected limbic TDP-43 pathology in the two oldest patients. There was a lack of TDP-43 pathology in the frontal and temporal cortex, suggesting that our observation is compatible with LATE-NC, not with frontotemporal lobar degeneration-TDP [30]. Therefore, it is tempting to speculate that DS initiates accelerated TDP-43 pathology in addition to Aβ accumulation.
Consistent with several previous studies [2–9], severe Aβ plaque and CAA pathology were observed in all DS patients aged over 40. In the cerebellum, we observed stripe-like deposition and focal fibrillar structures in the molecular layer, as reported in DS and various forms of AD [37, 47–49]. Furthermore, Purkinje cell-like and astrocytic Aβ deposition in the PC layer were also identified, similarly to the studies of DS [50]. Surprisingly, a similar stripe-like deposition pattern is reported in genetic Creutzfeldt-Jakob disease (E200K mutation in the PRNP gene), suggesting the association between abnormal protein deposition and the structure of the modular compartment of the cerebellar cortex [51]. Li et al. reported that about 65% of cerebellar Aβ deposits in the molecular layer were in physical contact with PC dendrites, and most of those deposits enveloped PC dendrites [6]. Thus, it is speculated that this characteristic stripe is formed by abnormal protein deposition along the PC dendrites in the molecular layer. Hence, on the immunostained specimen, it is considered that this striped pattern becomes more clearly visible as the amount of abnormal protein deposition increases, which may be useful for estimating severity in the cerebellum.
CONCLUSION
We find that DS is a complex multi-proteinopathy exhibiting subtle deviations from AD. It is hypothesized that the pathologic aggregation of one protein can work synergistically to initiate or otherwise promote the aggregation of different protein species [44]. Therefore, since Aβ deposition is the leading proteinopathy in DS patients, treatment using anti-Aβ antibodies may more effective in DS patients including preventing the development of co-proteinopathies [52]. The etiology of Aβ pathology in DS cases is clearer than in sporadic AD cases, and therefore DS is thought to be a better model system for examining the effects of inhibition of Aβ deposition. Our observations in DS showing distinctive features support the notion that pathogenic pathways in DS are more purely Aβ driven as compared to their complex pathogenic scenarios of sporadic AD.