
Abstract
Background:
The cellular and molecular alterations associated with synapse and neuron loss in Alzheimer’s disease (AD) remain unclear. In transgenic mouse models that express mutations responsible for familial AD, neuronal and synaptic losses occur in populations that accumulate fibrillar amyloid-β 42 (Aβ42) intracellularly.
Objective:
We aimed to study the subcellular localization of these fibrillar accumulations and whether such intraneuronal assemblies could be observed in the human pathology.
Methods:
We used immunolabeling and various electron microscopy techniques on APP x presenilin1 - knock-in mice and on human cortical biopsies and postmortem samples.
Results:
We found an accumulation of Aβ fibrils in lipofuscin granule-like organelles in APP x presenilin1 - knock-in mice. Electron microscopy of human cortical biopsies also showed an accumulation of undigested material in enlarged lipofuscin granules in neurons from AD compared to age-matched non-AD patients. However, in those biopsies or in postmortem samples we could not detect intraneuronal accumulations of Aβ fibrils, neither in the lipofuscin granules nor in other intraneuronal compartments.
Conclusion:
The intralysosomal accumulation of Aβ fibrils in specific neuronal populations in APPxPS1-KI mice likely results from a high concentration of Aβ42 in the endosome-lysosome system due to the high expression of the transgene in these neurons.
INTRODUCTION
The cellular and molecular alterations associated with synapse and neuron loss in Alzheimer’s disease (AD) have not yet been fully elucidated. Mutations in the amyloid-β precursor protein (AβPP) gene or its duplication or mutations in the genes encoding its cleaving enzymes presenilins (PS) 1 or 2 induce early-onset familial AD (FAD) [1]. AβPP is successively cleaved by a β- and a γ-secretase, with PS as catalytic subunit, leading to the generation of amyloid-β (Aβ) peptides. FAD mutations lead to an increase in the relative production of the aggregation-prone Aβ42 peptide [2]. Intraneuronal accumulation of Aβ42 was described in the endosome-lysosome system (ELS) of AD patients and of transgenic mouse models of AD [3–8]. In transgenic mouse models of AD, thioflavin S-positive accumulations of Aβ42 were observed in neuronal cell bodies specifically in areas with neuronal loss [9, 10]. We aimed to study the subcellular localization of these fibrillar accumulations and whether such intraneuronal assemblies could be observed in the human pathology.
Neuronal loss occurs only in few mouse models that were genetically modified to express FAD mutations [9, 10]. For instance, neuronal loss can be observed in APPxPS1-KI mice, but not in single APP transgenic or PS1-KI mice [9]. Interestingly, the loss of neurons is observed specifically in neuronal populations accumulating fibrillar Aβ42 intracellularly such as the CA1 pyramidal cell layer of the hippocampus, but not in neurons of the adjacent CA3 that displays only extracellular deposits [9]. In order to gain insights on the subcellular localization of Aβ fibrils, we analyzed CA1 pyramidal neurons in APPxPS1-KI mice using immuno-labeling and various electron microscopy (EM) techniques. To verify the specificity of the organelle alterations with respect to Aβ accumulations, we also analyzed CA3 pyramidal neurons, or CA1 pyramidal neurons in single APP transgenic or PS1-KI mice.
Having observed an accumulation of Aβ fibrils in enlarged lipofuscin granule-like lysosomes in APPxPS1-KI mice, we also analyzed if similar enlargements of lysosomes and accumulations of Aβ fibrils could be observed in neurons of AD patients. An accumulation of large, lysosome-derived lipofuscin granules was previously noticed in EM analyses of neuronal cell bodies from AD patients [11–13]. However, to our knowledge there was no quantitative comparison with neurons from non-AD patients. We quantified the numerical density and size of lipofuscin granules at the EM resolution in frontal cortical biopsies obtained from a neurosurgical collection of age-matched AD and non-AD patients. Their well-preserved ultrastructure allows the identification and morphometry of neuronal components [14–16]. In AD patients, Aβ42 accumulates in multivesicular bodies rather than in lipofuscin granules [7]. However, this study was performed using pre-embedding immunohistochemistry (IHC), and antibodies may have failed to reach the core of the granules. To overcome this potential limitation, we performed post-embedding IHC on postmortem samples from the frontal cortex or subiculum of AD patients. This technique gives to the antibodies access to cross sections of lipofuscin granules to analyze the undigested material. We indeed observed an enlargement of lipofuscin granules in neurons from AD patients with an accumulation of undigested material. However, we could not detect Aβ fibrils in these lipofuscin granules.
MATERIALS AND METHODS
Cases
Brain cortical biopsies were collected for diagnostic purpose between 1974 and 1986 [15, 16]. They were taken from the middle frontal gyrus of the right hemisphere of AD patients (n = 3; gender: male; age (mean ± SD): 60 ± 5 years), and non-AD patients (n = 4 cases; normal pressure hydrocephalus; gender: 3 males/1 female; age (mean ± SD): 60 ± 4 years). One of the AD patients had a familial form of AD (M146L mutation of the PS1 gene) [17]. The biopsies were immediately fixed in Karnovsky’s solution, postfixed in osmium and embedded in Araldite [15, 16].
Three brains were investigated that had been collected in a brain donation project and stored in the national brain bank NeuroCEB (bioresource research impact factor number BRIF BB-0033-00011). Case 1 was from a 67-year-old man (Braak neurofibrillary stage VI, Thal amyloid phase 5); case 2 was from a 95-year-old woman (Braak stage V, Thal phase 5); and case 3 was from a 81-year-old woman (Braak stage VI, Thal phase 5) [18, 19]. The prefrontal cortex (cases 1 and 2) and subiculum (case 3) were re-sampled from the hemisphere, which had been stored in buffered formalin 10% (formaldehyde 4%; cases 1 and 3) or paraformaldehyde 4% (case 2). Two hundred-μm-thick sections were cut in PBS with a vibratome and used for post-embedding IHC.
Preparation of the mouse samples
The APPxPS1-KI mice contain both an APP transgene bearing the Swedish K670N/M671L and London V717I mutations under the control of a Thy1 promoter, and M233T and L235P mutations knocked-in into the endogenous PS1 gene [9]. They develop an early-onset neuropathological phenotype, including the formation of extracellular Aβ42 deposits and intracellular accumulations of Aβ42 [9]. Furthermore, they develop, in the CA1 area, a progressive loss of synapses starting at 4 months, associated with a loss of neurons starting at 6 months [9, 21]. PS1-KI littermates, which do not develop behavioral-cognitive deficits nor extra- and intracellular accumulations of Aβ42, and in which no loss of hippocampal synapses or neurons has been detected, were used as controls as in previous studies [9, 20–22]. Additionally, we analyzed single APP transgenic mice that do not present with any intracellular accumulation of AβPP cleavage products nor any loss of CA1 pyramidal neurons, but develop extracellular Aβ deposits [9]. Light microscopy IHC was performed in three-month-old APPxPS1-KI (4 mice per immunostaining procedure) and PS1-KI (n = 3) mice. Post-embedding IHC was performed in four-month-old APPxPS1-KI and PS1-KI mice (2 mice per genotype). Pre-embedding IHC was performed in three-month-old APPxPS1-KI mice (n = 2). Morphological studies using EM were carried out in three-, four-, and six-month-old APPxPS1-KI mice (4 mice per age), six-month-old PS1-KI mice (n = 4), and six-month-old APP transgenic mice (n = 3).
All mice received an overdose of sodium pentobarbital (120 mg/kg) and trans-cardiac perfusion was performed with phosphate buffer (PB) containing 4% paraformaldehyde for IHC and pre-embedding IHC, 4% paraformaldehyde and 0.1% glutaraldehyde for post-embedding IHC, and 2% paraformaldehyde and 2% glutaraldehyde for EM morphological analysis. After 1 h at 4°C, the brain was extracted from the skull and post-fixed before being stored in PBS.
For paraffin embedding, hemibrains were dehydrated in graded ethanol solutions followed by xylene. They were then incubated in Tissue-Tek Paraffin wax (4509, Sakura) overnight. Frontal sections were cut at a thickness of 5μm in the middle part of the dorsal hippocampus.
Frontal sections of 50μm thickness for light microscopy IHC and of 200μm thickness for EM were cut in PBS with a vibratome in the middle part of the dorsal hippocampus.
Experiments on animals were conducted in accordance with European ethical standards (European Communities Council Directive 2010/63/EU on the protection of animals used for scientific purposes). The project was approved by the local ethics Committee (Charles Darwin Committee; project # 8761).
Electron microscopy
For post-embedding IHC, the sections were processed using a Reichert AFS vibratome (Leica, Vienna, Austria). After washes in PBS, they were progressively transferred in 30% and 50% methanol (the latter with 0.5% uranyl acetate) while the temperature was lowered to –20°C (24°C / h). Sections were rinsed in 50% methanol (15 min) and then dehydrated through graded methanol while lowering the temperature to –45°C at the rate of 15°C / h. The sections were then infiltrated with Lowicryl HM20 (Polysciences). The resin was polymerized by exposure to UV light for 48 h. Seventy-nm-thick ultrathin sections were collected on formvar-coated nickel grids.
For morphological analysis, the sections were transferred into a 1% osmium solution for one hour. After several washes, they were stained “en bloc” with a 5% uranyl acetate solution for one hour. After several washes, the samples were dehydrated in graded ethanol solutions followed by acetone. They were incubated in progressive concentrated epoxy resin before being embedded in pure resin. Ultrathin sections were collected on copper grids.
The sections were contrasted by incubation with lead citrate and analyzed with a Hitachi HT 7700 electron microscope operating at 70kV (Elexience, Verrieres-le-Buisson, France).
Immunolabeling
For light microscopy IHC, paraffin sections were first dewaxed in xylene and rehydrated. The biotinylated 4G8 antibody (1:10000; Ozyme) was used to recognize the intra- and extracellular accumulations of Aβ-containing peptides [9]. It is directed at the mid region of the Aβ peptide, and therefore can detect AβPP, but displays a strong preference for Aβ aggregates [23]. The sections were left 10 min in 0.01 M citrate buffer at 95° and for 10 min in 50% formic acid for retrieval of Aβ epitopes for immunostaining using the 4G8. Slides were then incubated in a PB blocking solution containing 0.9% NaCl (PBS), 0.2% triton, and 5% normal goat serum for 1 h and left overnight in the same solution to which was added the antibodies. After washes, sections were directly incubated with Elite complex (Vector labs) for 90 min. Revelation was performed using di-amino benzidine as chromogen. After mounting in Eukitt, the sections were analyzed using an Olympus BX56 microscope.
Post-embedding IHC was performed as described previously [24] using the 4G8 antibody (1:500) followed by secondary anti-mouse antibodies coupled to 10 nm gold particles (1:50; EM.GMLH10; BBI Solutions). The sections were contrasted and analyzed as described above.
Vibratome sections for light microscopy IHC were incubated in a PB blocking solution as above. Sections were then incubated overnight with anti-LAMP1 antibodies (1:1000; sc-19992; Santa Cruz). After washes, sections were incubated with secondary biotinylated antibodies and Elite complex, and revelation was performed using di-amino benzidine as chromogen.
For pre-embedding IHC, sections were incubated for 3 h in 20% glycerol and 20% saccharose PBS solution. The solution was then removed, and the sections were frozen-unfrozen 3 times over liquid nitrogen. Immunostaining with anti-LAMP1 antibodies was then performed as above except that no triton was used. Embedding was performed as described above for EM morphological analysis. The sections were analyzed without being contrasted.
Quantifications
Analysis of neuronal lysosomes in sections from the CA1 stratum pyramidale was done blind to genotype in three-, four-, and six-month-old APPxPS1-KI mice (4 mice per age), and in six-month-old PS1-KI or APP transgenic mice (3 mice per genotype). Additionally, a quantification was done in the CA3 stratum pyramidale in six-month-old APPxPS1-KI mice (3 mice). All neuronal cell bodies in the section were sampled (30–129 neuronal profiles per mouse, mean 68), and measurements of the surface of lysosomes accumulating fibrils were done on photomicrographs using the Image J software [25]. The fraction of neuronal profiles that contained such lysosomes and the mean value of the surface occupied by these organelles in neurons were then calculated for each mouse.
Analysis of lipofuscin granule numerical density and surface in neuronal cell bodies of the human brain biopsies was performed in sections from layers II-III of the right middle frontal gyrus and blind to pathology, as was done previously for the analysis of synapses [14]. Electron micrographs from all neuronal cell bodies in the section were taken for each case (17–43 neuronal profiles per case, mean 24), and measurements were done on photomicrographs using the Image J software [25]. The mean value per case was then calculated, and between-groups comparisons were performed using two-tailed, unpaired Student’s t-test. Statistical significance was set to a p value < 0.05.
RESULTS
Accumulation of Aβ42 fibrils in neuronal lysosomes in APPxPS1-KI mice
An intracellular accumulation of Aβ42 was previously observed in the CA1/2 pyramidal cell layer of the hippocampus in APPxPS1-KI mice [9]. In agreement, we observed an accumulation of 4G8 immunolabeling in the hippocampal pyramidal cell layer (Fig. 1A left) and in plaques (Fig. 1A right) of three-month-old APPxPS1-KI mice. To analyze the ultrastructural organization of Aβ accumulation in the soma of CA1 pyramidal neurons, we used post-embedding IHC in four-month-old APPxPS1-KI mice. We observed 4G8 positive, enlarged organelles that had a dark matrix with heterogeneous vesicles (Fig. 1B, E). A fibrillar organization was associated with the immunolabeling. Fibrils were densely packed and filled the organelles (Fig. 1B, E). A labelling of similar fibrils was observed in extracellular plaques (Fig. 1C). Comparable but smaller organelles in PS1-KI controls were not 4G8 positive and did not contain fibrils (Fig. 1D). In summary, Aβ accumulated as fibrils that filled enlarged organelles in neurons of APPxPS1-KI mice.
Accumulation of fibrils of Aβ in enlarged organelles in APPxPS1-KI mice. A) IHC using the 4G8 antibody labels Aβ both in the CA1 pyramidal cell layer (left) and in extracellular plaques (right). B-E) IHC using the 4G8 antibody and a secondary antibody coupled to 10 nm gold particles in neuronal cell bodies of the CA1 pyramidal cell layer in APPxPS1-KI (B, E) and PS1-KI (D) mice, and in a plaque (C). Organelles are delimited by dashed white lines and arrows show fibrils of Aβ. Note the accumulation of fibrils of Aβ in neuronal organelles in APPxPS1-KI mice. Scale bar in A is 20μm. Scale bar in B is 500 nm. Scale bars in C-E are 200 nm.
The dark matrix and heterogenous vesicular content of the organelles accumulating Aβ fibrils are characteristic of late endosomes/lysosomes. Late endosomes/lysosomes were labeled by an anti-LAMP1 IHC that showed their enlargement in the CA1 pyramidal cell layer of 3-month old APPxPS1-KI mice (Fig. 2A, B) [26]. No enlargement was observed in the adjacent CA3 pyramidal cell layer. Pre-embedding IHC using antibodies against LAMP1 showed the labeling of both small organelles (Fig. 2C) and enlarged lysosomes that accumulated undigested material (Fig. 2D) in the cell bodies of CA1 pyramidal neurons in APPxPS1-KI mice. Omission of the primary antibody yielded no labeling (Fig. 2E). These studies indicated that lysosomes indeed became enlarged in APPxPS1-KI mice.

Enlargement of lysosomes in CA1 pyramidal neurons in APPxPS1-KI mice. A, B) Light microscopy IHC using an antibody against LAMP1 in the CA1 pyramidal cell layer in PS1-KI (A) and APPxPS1-KI (B) mice. Note the enlarged dots in APPxPS1-KI mice (arrows). C-E) EM immunolabeling using the antibody against LAMP1 in the cell bodies of CA1 pyramidal neurons in APPxPS1-KI mice. Note the labeling of small organelles (arrows in C) or enlarged lysosomes (arrows in D), and the absence of labeling of organelles after primary antibody omission (arrows in E). m: mitochondria; n: nucleus. Scale bar in A is 50μm in A and B. Scale bars in C-E are 1μm.
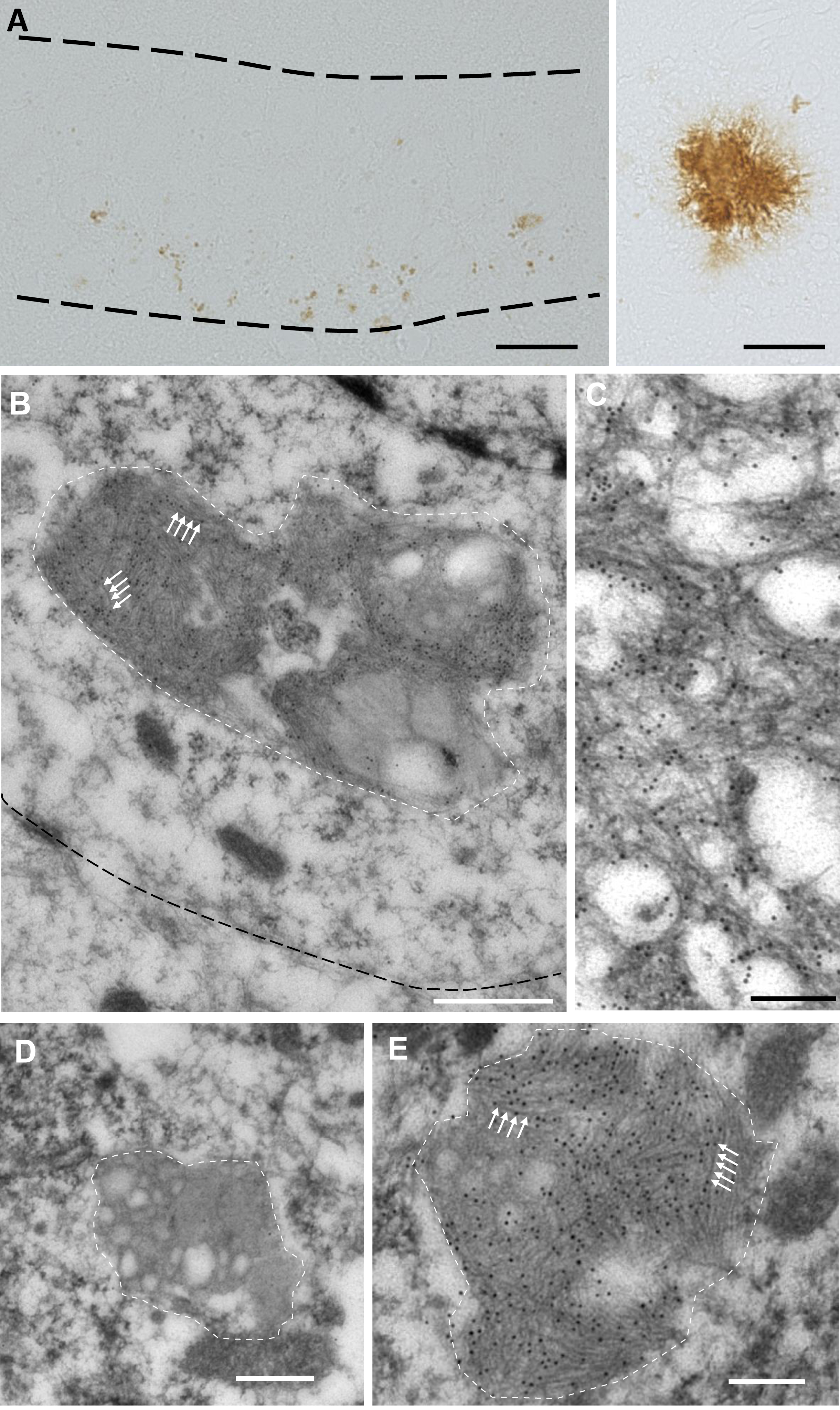
Because pre-embedding IHC may mask ultrastructural details, we used standard EM to analyze the alteration of lysosomal morphology in APPxPS1-KI mice. The ultrastructure of neuronal lysosomes as a function of age has been well described in mice [27]. Primary lysosomes with a smooth outline and a dark or gray content are first observed, followed by secondary lysosomes that contain lipid droplets of various size and fingerprint-like inclusions. Lipofuscin granules that increase in number with age are characterized by an irregular morphology and the accumulation of dense material in addition to the lipid droplets and inclusions. In agreement with this early study, the cell bodies of CA1 pyramidal neurons in APPxPS1-KI or PS1-KI mice contained primary lysosomes with a smooth outline and a dark or gray content (Fig. 3A), or secondary lysosomes with lipid droplets of various size and fingerprint-like inclusions (Fig. 3B, C). Additionally, CA1 pyramidal neurons in APPxPS1-KI mice had enlarged lysosomes due to the accumulation of a dense material (Fig. 3D, E), which consisted of fibrils (Fig. 3F, G). Organelles accumulating dense material were sometimes affixed to figures of autophagy (Fig. 3H). Their number and size did not change between 3 and 6 months of age (Fig. 3I, J). Strikingly, few or no of these enlarged lysosomes were observed in CA1 pyramidal neurons of APP transgenic or PS1-KI mice, or in CA3 pyramidal neurons of APPxPS1-KI mice at 6 months (Fig. 3J). Thus, the morphology of enlarged lysosomes accumulating fibrils was similar to that of lipofuscin granules.
Accumulation of lipofuscin granule-like organelles in CA1 pyramidal neurons in APPxPS1-KI mice. A-C) Examples of lysosomes in the cell bodies of CA1 pyramidal neurons in PS1-KI mice. D-G) Examples of enlarged lysosomes (lipofuscin granule-like organelles) containing undigested material in the cell bodies of CA1 pyramidal neurons in APPxPS1-KI mice. The insert in D and E is enlarged in F and G respectively. Arrowheads in F, G show fibrils accumulating in lipofuscin granule-like organelles. H) Figures of autophagy (membrane whorls) affixed to lipofuscin granule-like organelles containing undigested material. I) Quantification (mean + SEM) of the surface occupied by lipofuscin granule-like organelles per neuronal profile. J) Quantification (mean + SEM) of the fraction of neuronal profiles with lipofuscin granule-like organelles. Scale bars are 500 nm.
Accumulation of undigested material in neuronal lipofuscin granules in AD
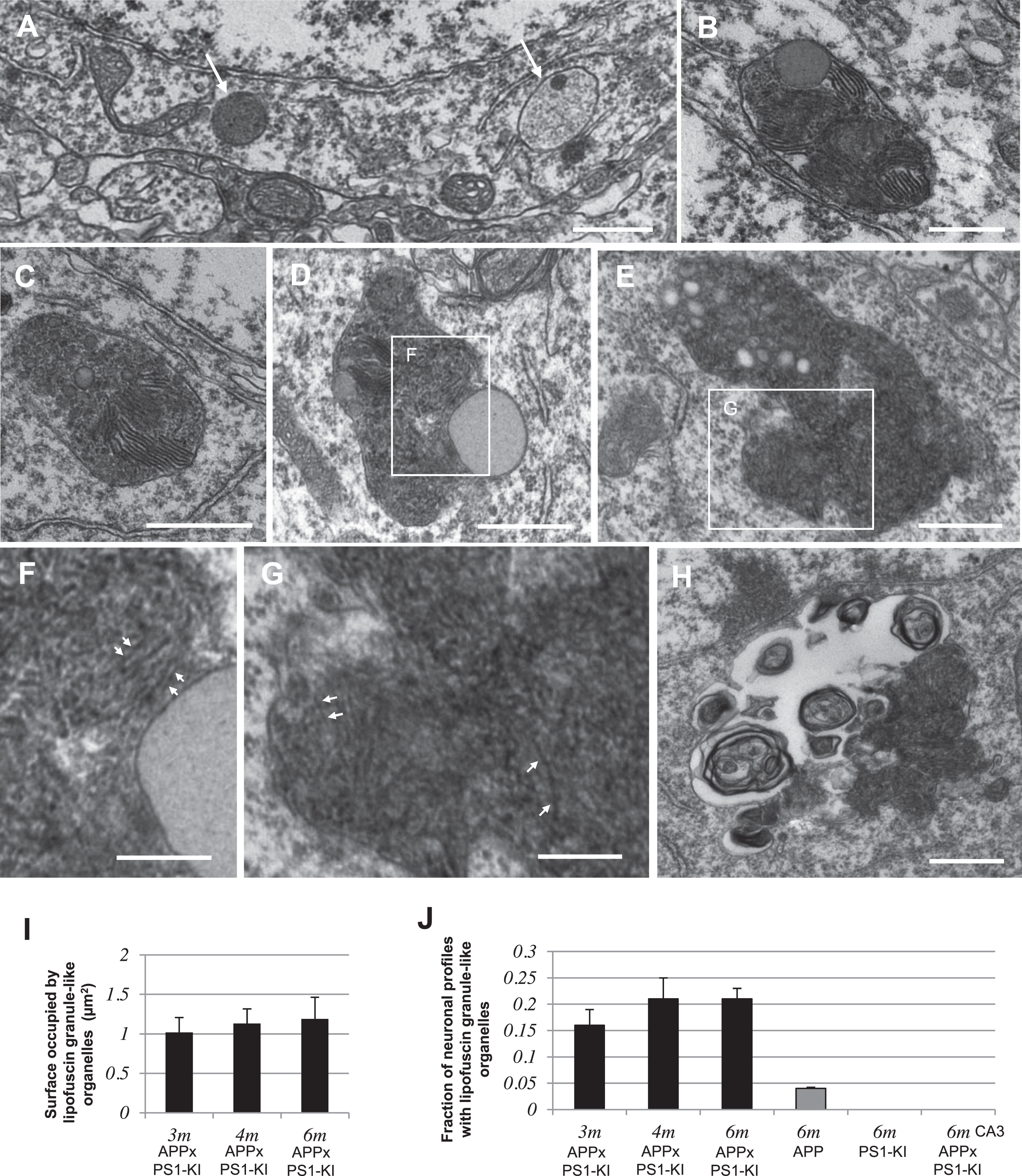
In order to determine if the lysosomal alterations observed in APPxPS1-KI mice could be relevant to the human pathology, we analyzed biopsies from AD and age-matched non-AD patients. The ultrastructure of layers II-III of the right middle frontal gyrus was well preserved, allowing the identification of neurons and the analysis of their organelles (Fig. 4A, B). In these neurons, lipofuscin granules were identified as dark organelles merged with lipid droplets. Lipofuscin granules showed an increased numerical density in neuronal profiles from AD patients when compared to age-matched non-AD patients (t(5) = 4.638, p = 0.0056; Fig. 4A-C). Additionally, lipofuscin granules occupied a larger area in neuronal profiles from AD patients (t(5) = 4.491, p = 0.0065; Fig. 4A, B, D). There was thus an accumulation of undigested material in lipofuscin granules in neurons from AD patients.
Accumulation of undigested material in lipofuscin granules in neurons from AD patients. A, B) Examples of neuronal cell bodies in the frontal cortex of non-AD (A) and AD (B) patients. Arrowheads show lipofuscin granules and asterisks indicate lipid droplets. C) Quantification (mean ± SEM) of the number of lipofuscin granule profiles per neuronal profile. D) Quantification (mean ± SEM) of the surface occupied by lipofuscin granule profiles per neuronal profile. **p < 0.01. Scale bar in A is 5μm for A and B.
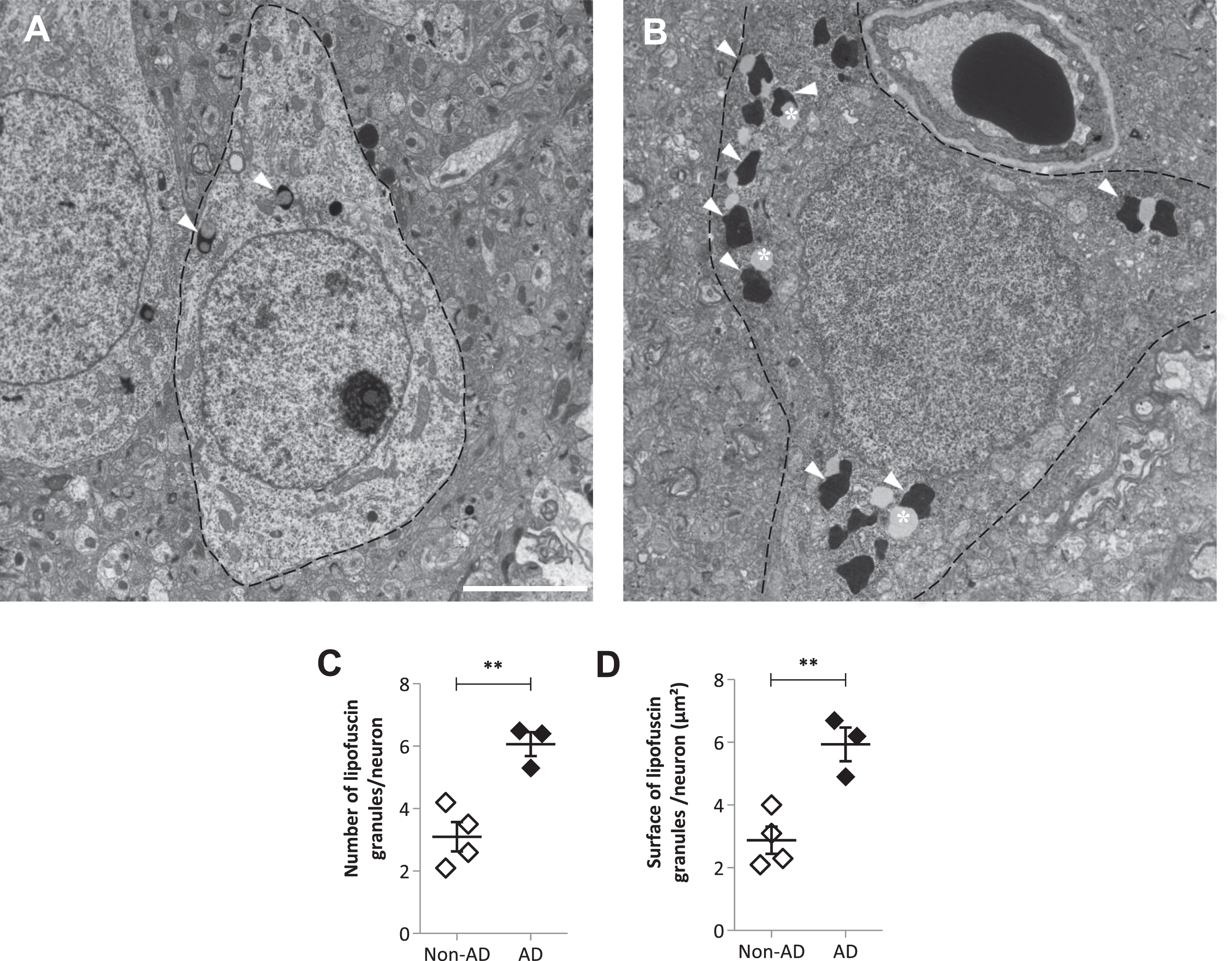
To determine if neuronal lipofuscin granules accumulated Aβ fibrils, we performed post-embedding IHC using the 4G8 antibody on postmortem samples from the frontal cortex or subiculum of AD patients. This technique gives to the antibodies access to cross sections of lipofuscin granules. No immunoreactivity was detected on lipofuscin granules (Fig. 5A, B), while Aβ-immunoreactivity was observed on fibrils in amyloid plaques on the same sections (Fig. 5C). It indicated that contrary to what was observed in the mouse model, the undigested material that accumulated in lipofuscin granules in AD did not contain detectable levels of Aβ fibrils.
Lipofuscin granules in neurons of AD patients did not contain detectable levels of Aβ fibrils. Immuno-labeling using the 4G8 primary antibody and a secondary antibody coupled to 10 nm gold particles, in a cell body (A, B) and in a plaque (C) in the frontal cortex of an AD patient. The insert in A is enlarged in B. Arrows show lipofuscin granules and asterisks indicate lipid droplets. n: nucleus. Scale bar in A is 1μm. Scale bars in B and C are 250 nm.
DISCUSSION
An intraneuronal accumulation of Aβ42 fibrils was previously observed specifically in areas with neuronal loss in transgenic mouse models of AD [9, 10], but their subcellular localization and relevance to the human pathology remained to be described. In this study, we found a similar accumulation of undigested material in enlarged lysosome-derived lipofuscin granules in neurons from APPxPS1-KI mice and AD patients. However, while the enlarged organelles accumulated Aβ fibrils in APPxPS1-KI mice, this particular composition was not detected in neurons from AD patients.
Complementary methods of EM and immuno-labeling allowed us to describe the intraneuronal accumulation of Aβ fibrils and associated organelle alteration in APPxPS1-KI transgenic mice. We found that Aβ fibrils accumulated in enlarged lysosomes resembling the lysosome-derived lipofuscin granules observed in neurons during aging [27]. This finding agrees with previous studies that found Aβ42 in enlarged lysosomes in transgenic mouse models of FAD [6, 28]. The intraneuronal accumulation of Aβ fibrils does not correlate with extracellular Aβ peptide deposition, but rather occurs in neurons with a high expression of the APP transgene [9]. The accumulation of Aβ fibrils in lysosomes can be explained by the fact that Aβ42 production occurs in the ELS [29]. Additionally, in vitro studies have shown that the accumulation of Aβ42 into the ELS can lead to its aggregation [30, 31]. Thus, the presence of fibrils likely results from a high concentration of Aβ42 in the ELS.
We observed a similar enlargement of lipofuscin granules in neurons from AD patients. The aging of neurons is characterized by an increased amount of lipofuscin granules [27, 32–34]. Lipofuscin granules accumulate undigestible material during aging [35]. Light microscopic analyses indicate either no change or an increase in the area occupied by these granules in neurons from AD patients compared with those from non-AD patients, depending on the brain area and cortical layer [36–38]. Yet these analyses are complicated by the fact that individual lipofuscin granules are not resolved when aggregated in clusters. Here, the use of EM allowed us to reach the resolution of individual granules. Although lipofuscin granules were enlarged, they did not accumulate fibrils. In brain tissue of AD patients, intracellular Aβ fibrils are only observed in microglial cells around plaques [16]. In contrast, the undigested material that accumulates progressively with age in lipofuscin granules is made of various components [39, 40]. As a decline in the intraneuronal labeling of Aβ could be noted with advancing amyloid pathology [5], the detection of Aβ in neurons of AD patients could be improved by immunoelectron microscopy methods preserving intraneuronal Aβ [41]. Nevertheless, in neurons of AD patients, accumulations of Aβ and organelle alterations have been observed at several steps along the ELS upstream the lipofuscin granules [3, 42–45]. Thus, an alteration of ELS traffic could be involved in the further accumulation of undigested material in lipofuscin granules.
Footnotes
ACKNOWLEDGMENTS
We are grateful to Prof. Jean-François Foncin for having established a collection of biopsies taken during neurosurgery (Profs. Le Beau and Philippon) and having provided it to us. We thank Pascal Barneoud and Laurent Pradier from Sanofi for sharing the APPxPS1-KI mouse line and for stimulating discussions. We also thank the ICM imaging facility icm.Quant. This work was partly carried out on the ICM HISTOMICS histology platform with the help of Annick Prigent and Celia Sayetta. All animal work was conducted at the ICM PHENOPARC Core Facility. We thank Nadège Sarrazin and Joanna Droesbeke from the PHENO.ICMice for their help. The NeuroCEB Neuropathology network includes: Franck Letournel (CHU Angers), Marie-Laure Martin-Négrier (CHU Bordeaux), Maxime Faisant (CHU Caen), Catherine Godfraind (CHU Clermont-Ferrand), Claude-Alain Maurage (CHU Lille), Vincent Deramecourt (CHU Lille), David Meyronnet (CHU Lyon), Nathalie Streichenberger (CHU Lyon), André Maues de Paula (CHU Marseille), Valérie Rigau (CHU Montpellier), Fanny Vandenbos-Burel (Nice), Charles Duyckaerts (CHU PS Paris), Danielle Seilhean (CHU PS, Paris), Susana Boluda (CHU PS, Paris), Isabelle Plu (CHU PS, Paris), Serge Milin (CHU Poitiers), Dan Christian Chiforeanu (CHU Rennes), Annie Laquerrière (CHU Rouen), Dr Béatrice Lannes (CHU Strasbourg).
This work was supported by the Fondation Alzhei-mer. The present work was also supported by funding from the program “Investissements d’avenir” ANR-10-IAIHU-06, ANR-11-INBS-0011-NeurATRIS: Translational Research Infrastructure for Biotherapies in Neurosciences.