
Abstract
Background:
Brain accumulation of amyloid-β is a hallmark event in Alzheimer’s disease (AD) whose underlying mechanisms are incompletely understood. Case-control genome-wide association studies have implicated numerous genetic variants in risk of clinically diagnosed AD dementia.
Objective:
To test for associations between case-control AD risk variants and amyloid PET burden in older adults, and to assess whether a polygenic measure encompassing these factors would account for a large proportion of the unexplained variance in amyloid PET levels in the wider population.
Methods:
We analyzed data from the Mayo Clinic Study of Aging (MCSA) and the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Global cortical amyloid PET burden was the primary outcome. The 38 gene variants from Wightman et al. (2021) were analyzed as predictors, with PRSice-2 used to assess the collective phenotypic variance explained.
Results:
Known AD risk variants in APOE, PICALM, CR1, and CLU were associated with amyloid PET levels. In aggregate, the AD risk variants were strongly associated with amyloid PET levels in the MCSA (p = 1.51×10–50) and ADNI (p = 3.21×10–64). However, in both cohorts the non-APOE variants uniquely contributed only modestly (MCSA = 2.1%, ADNI = 4.4%) to explaining variation in amyloid PET levels.
Conclusion:
Additional case-control AD risk variants added only modestly to APOE in accounting for individual variation in amyloid PET burden, results which were consistent across independent cohorts with distinct recruitment strategies and subject characteristics. Our findings suggest that advancing precision medicine for dementia may require integration of strategies complementing case-control approaches, including biomarker-specific genetic associations, gene-by-environment interactions, and markers of disease progression and heterogeneity.
INTRODUCTION
Amyloid accumulation in the brain is widely considered to be an early hallmark event in Alzheimer’s disease (AD) [1]. Although AD is a complex and heterogeneous disorder, reliable methods for individualized prediction of susceptibility to amyloid accumulation could guide early interventions to mitigate risk of future cognitive decline. Older age and presence of the APOE (apolipoprotein E) ɛ4 allele are the strongest known risk factors for brain amyloidosis [2], but are not fully explanatory.
Recent case-control genome-wide association studies (GWAS) have implicated additional genetic variants in risk of clinically probable AD dementia [3, 4], with the largest study to-date exceeding one million individuals and identifying 38 risk variants [5]. Here, we hypothesized that at least some of the genotypes from these AD risk variants would be associated with amyloid positron emission tomography (PET) burden (as an early hallmark of AD) in older adults. We tested this hypothesis using two large cohorts with PET imaging and GWAS data. We also hypothesized that a polygenic score encompassing the cumulative effects of these genetic factors [6], would account for a large proportion of the unexplained variance in amyloid PET levels, with a particular focus on a population-based sample to gauge potential utility for risk stratification in the wider population.
METHODS
Study participants
The primary sample for analysis was drawn from the Mayo Clinic Study of Aging (MCSA), a population-based prospective study of older adults residing in Olmsted County, Minnesota [7]. Individuals were identified for recruitment using the Rochester Epidemiology Project (REP) medical records linkage system [8, 9]. Clinical data through questionnaires and in-person history, multimodal neuroimaging, and laboratory tests were assessed at selected visits based on study protocols. Clinical diagnoses were made by a multidisciplinary consensus panel, incorporating all available information and standard definitions of cognitively unimpaired, mild cognitive impairment (MCI), and dementia [7, 10]. All MCSA individuals aged 50 years or older and having amyloid PET imaging and GWAS data were included in this study.
Data from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) was used for targeted validation and/or comparison of population-based findings with those from a sample recruited in a manner reflecting clinical trial cohorts. The ADNI is a longitudinal multicenter study to facilitate development of clinical, imaging, genetic, and biochemical biomarkers for the early detection and tracking of AD [11, 12]. Individuals were recruited from over 50 sites across the United States and Canada. Clinical diagnoses were made by a consensus panel as described previously [13]. Further information about the ADNI can be found at https://adni.loni.usc.edu/.
Neuroimaging and clinical data
In the MCSA, amyloid PET scans were performed with 11C-Pittsburgh compound B (PiB) and were analyzed using an in-house fully automated image processing pipeline as described elsewhere [14]. In the ADNI, amyloid PET was performed with 18F-florbetapir (AV-45) using acquisition and processing protocols as described at https://www.adni-info.org, and with summary measures downloaded from the ADNI database [15]. The target outcome was global cortical amyloid load from the baseline amyloid PET scan, reported as a standardized uptake value ratio (SUVR). For comparison to assess for wide differences across AD-relevant phenotypes, we also analyzed as secondary outcomes tau PET burden in a previously described subset of 754 MCSA participants [16] and clinical diagnosis in subsets of the MCSA (1483/1725) and ADNI (544/1068) samples. Tau PET was performed with 18F-flortaucipir (AV-1451), synthesized on site with precursor supplied by Avid Radiopharmaceuticals [17], and using SUVR within an AD signature composite region of interest as the target outcome [14]. Clinical diagnosis was restricted to cognitively unimpaired (MCSA = 1444, ADNI = 352) versus dementia (MCSA = 39, ADNI = 352) to match case-control designs.
Genetic data
For MCSA and ADNI participants, GWAS array data was acquired and filtered for standard quality control metrics as described elsewhere [18, 19]. Analyses were restricted to participants with non-Hispanic Caucasian ancestry [18, 19]. Genome-wide imputation was performed separately within each cohort (and for ADNI participants, separately within each batch by GWAS array and then merged) using the TOPMed Imputation Server and reference panel [20] which is based in Minimac4 [21]. Variants with low imputation quality (based on the Minimac-specific metric of r2 < 0.8) were filtered out [22]. Additional standard quality control filters were applied, with exclusion of variants having genotyping rate < 95%, Hardy-Weinberg equilibrium p < 1×10–6, or monomorphic genotype, and exclusion of samples with call rate < 98%, sex discordance with clinical data, or evidence of significant relatedness defined by PLINK identity-by-descent PI_HAT ≥0.25. This resulted in 24,118,699 variants (8,054,769 variants with MAF = minor allele frequency ≥1%) for 1727 individuals within the MCSA dataset and 16,502,548 variants (8,054,769 variants with MAF ≥1%) for 1661 individuals within the ADNI dataset. Specific to this study, the 38 AD risk variants from Wightman et al. [5] were extracted for further analysis. For post-hoc analyses we also extracted rs7412, the variant determining the APOE ɛ2 allele which was not part of the 38 susceptibility variants from Wightman et al. To account for potential confounding effects of population stratification, principal component eigenvectors were generated for use as covariates in the genetic analyses.
Statistical analyses
Single variant associations with amyloid PET
Genotype associations with amyloid PET burden were assessed with PLINK version 1.9 [23], utilizing linear regression under an additive genetic model and including age at scan, sex, and the first 5 genetic principal component eigenvectors as covariates. Individual variant associations were first analyzed within the MCSA and ADNI cohorts separately. Following this, METAL [24] was used for sample size weighted meta-analysis of the results across the two cohorts; an inverse variance meta-analysis approach was less suitable for this work due to the different amyloid PET tracers used in the MCSA (Pib) versus ADNI (AV-45) cohorts. To ensure no confounding effect related to APOE ɛ4 status, for variants displaying significant associations with amyloid levels in the meta-analysis we also repeated these protocols with the inclusion of APOE ɛ4 dose as an additional covariate.
Polygenic calculations for explaining variation in amyloid PET
We applied PRSice-2 [25] to assess the proportion of variance in amyloid PET burden explained by the aggregated effects on amyloid levels for the AD risk variants from Wightman et al. The top 38 variants from Wightman et al. (representing validated genome-wide significant associations with clinical AD dementia diagnosis from the largest such GWAS to-date) were used to define the set of input SNPs. The weights for each variant were based on that variant’s association with amyloid PET levels (i.e., its univariate linear regression coefficient) in the target dataset, using an additive genetic model and covarying for age, sex, and genetic principal components [26, 27]. The PRSice-2 algorithm uses a traditional clumping and thresholding (“C+T”) method. Specifically, the input variants are pruned to account for linkage disequilibrium, retaining only the top associated variant for any pairs with r2≥0.1 (which is the recommended stringent cutoff) [26]. Analyses were primarily performed within each cohort (MCSA versus ADNI) separately. For cross-validation we also repeated the analyses by utilizing the MCSA association profiles as the base for testing in ADNI (as the target), and vice-versa. Because the goal of this analysis was to assess the aggregate influence of a set of variants known to be associated with risk of clinically diagnosed AD dementia, we did not employ a variant-level p-value threshold for inclusion in the primary analyses. However, results were not substantially different when inclusion thresholds of p < 0.5 or p < 0.25 were used [26]. The proportion of variance explained (R2) was obtained from the PRSice-2 output, and the relative contribution of non-APOE variants to this measure was assessed by subtracting the R2 for APOE ɛ4 alone from the R2 attributed to the model including APOE ɛ4. As a post-hoc sensitivity analysis, we also repeated the calculations for amyloid PET levels in the MCSA after restricting the sample to cognitively unimpaired individuals (leveraging the large number of these participants in the MCSA) to assess for differential results.
Post-hoc analyses of complementary AD-relevant outcomes
To test whether the pattern of findings in our primary analyses were specific to amyloid PET levels (as compared with other AD-relevant outcomes), we applied a similar framework to assess the proportion of variance explained by the aggregated effects of the 38 AD risk variants on 1) tau PET burden in the MCSA and 2) clinical diagnosis in the MCSA and ADNI. For tau PET, the SUVR from the AD signature composite region of interest was used as the outcome. For tau PET, the polygenic variance explained was calculated using PRSice-2 based on the univariate linear regression coefficients for the variants from Wightman et al. with tau PET levels, including age, sex, genetic principal components, and global amyloid PET levels as covariates. For clinical diagnosis (cognitively unimpaired versus dementia), PRSice-2 was applied based on the logistic regression coefficients for the variants from Wightman et al., including age, sex, and genetic principal components as covariates. For the clinical diagnosis analyses, the Nagelkerke’s pseudo-R2 used to define variance explained for the binary phenotype.
Standard protocol approvals, registrations, and patient consents
All MCSA study protocols were approved by the Mayo Clinic and Olmsted Medical Center Institutional Review Boards. All ADNI study protocols were approved by each participating site’s Institutional Review Board. Written informed consent was obtained from all participants or their surrogates.
Data availability
Data from this study are available from the authors upon reasonable request.
RESULTS
Although similar age and sex distributions were observed in the MCSA (N = 1,725) and ADNI (N = 1,068) cohorts, other key variables included evident differences reflecting the distinct study designs and recruitment strategies (Table 1). While most MCSA participants were cognitively unimpaired at the time of amyloid PET imaging (83.7%), most ADNI participants had diagnosis of mild cognitive impairment or dementia (67%). The ADNI sample was also enriched for APOE ɛ4 carriers (44% versus 29% in the MCSA) and included a larger proportion of amyloid PET positive participants (54% versus 39% in the MCSA) based on published thresholds [17, 28].
Sample characteristics
Values displayed as mean (standard deviation) or number (percentage). CU, cognitively unimpaired; MCI, mild cognitive impairment; DEM, dementia; UNK/UNC, diagnosis unknown or unclassified amongst CU/MCI/DEM; SUVR, standardized uptake value ratio. aConsensus clinical diagnosis at the visit accompanying the baseline amyloid PET scan used for analysis. bDifferent amyloid PET tracers were used for the MCSA (11C-PiB) versus ADNI (18F-florbetapir) samples.
Nominal associations (p < 0.05) with amyloid burden were observed for 6 gene variants in the MCSA and 9 gene variants in the ADNI (Table 2). Given the relative concordance of variant-level findings across MCSA and ADNI, we performed meta-analysis of the 36 variants common to both samples. After Bonferroni correction to account for multiple comparisons, APOE, PICALM, CR1, and CLU displayed significant associations with amyloid levels in the meta-analysis (p < 0.05/36 = 1.39×10–3), with the direction of effect for each minor allele consistent with its impact on risk of clinically diagnosed AD dementia [5]. After including APOE ɛ4 dose as an additional covariate, the associations for CR1 (p = 1.17×10–4), PICALM (p = 2.38×10–4), and CLU (p = 6.45×10–4) all remained significant in the meta-analysis, indicating no confounding effect of APOE ɛ4 on these findings.
A polygenic score incorporating APOE and the additional AD risk variants was strongly associated with amyloid PET levels in the MCSA (p = 1.51×10–50). This measure included 32/38 candidate variants, with the same four variants (as in the MCSA) removed in the clumping and thresholding step, and with rs1761461 (LILRB2) and rs113020870 (AGRN) unavailable in this dataset. Although this aggregate measure accounted for 9.9% of the phenotypic variance, most of this fraction was explained by APOE ɛ4 (7.8%), with non-ɛ4 variants uniquely contributing modestly (2.1%). Results were similar when the analyses were restricted to the 1,443 MCSA participants who were cognitively unimpaired (thus supporting no confounding effect of clinical diagnosis), with the overall combination of risk alleles accounting for 8.0% of the phenotypic variance (p = 1.43×10–33) and with non-ɛ4 variants contributing modestly (1.8%) in comparison to APOE ɛ4 (6.2%).
Associations of Alzheimer’s dementia risk variants with amyloid PET
Blue shaded gene variants have significant associations on meta-analysis after Bonferroni correction for multiple comparisons (p < 0.05/36 = 1.39×10–3). aDenoted as chromosome:base pair (hg38 build). bAmyloid PET in the MCSA utilized 11C-Pittsburgh compound B (PiB). cAmyloid PET in the ADNI utilized 18F-florbetapir. dVariant not available for analysis in ADNI dataset.
A similar pattern was observed in the ADNI, where a polygenic score including APOE ɛ4 was strongly associated with amyloid PET burden (p =3.21×10–64, R2 = 23.5%). This measure included 32/38 candidate variants, with rs9369716 (CD2AP), rs3935067 (EPHA1-AS1), rs602602 (ADAM10), and rs2632516 (TSPOAP1-AS1) being removed in the clumping and thresholding step. Non-APOE variants uniquely accounted for only 4.4% of the phenotypic variance. Compared to findings from the MCSA, the relatively larger R2 for the polygenic measure in ADNI was due to the stronger effect of APOE ɛ4 in that cohort, likely reflecting the enrichment for APOE ɛ4 carriers in the ADNI which was recruited in a manner akin to clinical trial samples. Overall, age, sex, genetic principal components, and the polygenic score explained 31.1% of the variance in amyloid levels in the MCSA and 26.0% of the variance in amyloid levels in the ADNI, with age and APOE ɛ4 together accounting for nearly all of these totals in both cohorts.
Using a cross-validation approach (i.e., applying the MCSA summary statistics as the base for polygenic modeling in the ADNI, and applying the ADNI summary statistics as the base for polygenic modeling in the MCSA), we observed concordant results indicating only a modest added value in explaining variation in amyloid PET levels for non-APOE AD risk variants over and above APOE. Specifically, a polygenic score of 6 variants (yielding the maximum R2) within the MCSA (p = 2.95×10–21, R2 = 4.5%) was minimally better than APOE ɛ4 alone (p = 7.02×10–20, R2 = 4.2%). Similarly, a polygenic score of 3 variants (yielding the maximum R2) within the ADNI (p = 2.78×10–54, R2 = 19.8%) was minimally better than APOE ɛ4 alone (p = 2.69×10–52, R2 = 19.1%). In both cases, addition of more variants to the model did not improve fit.
The variant defining the APOE ɛ2 allele (rs7412) was not included in the 38 variants isolated from Wightman et al., and therefore was not included in our primary analyses. In post-hoc analyses, rs7412 displayed a modest protective association with amyloid PET levels in the MCSA (p = 8.52×10–6, β= –0.11, R2 = 0.9%) which remained significant after accounting for APOE ɛ4 dose (p = 1.76×10–3, β= –0.07, R2 = 0.4%).
In a comparison analysis of MCSA participants who also had tau PET imaging, amyloid PET burden (R2 = 16.2%, p = 1.43×10–34) and age (R2 = 11.2%, p = 1.41×10–3) were robustly associated with tau levels, while in the aggregate APOE ɛ4 and the other AD risk variants uniquely added only modestly to the variance explained (R2 = 2.6%, p = 2.34×10–7), suggesting that these AD risk variants do not have a disproportionately strong relationship with tau as opposed to amyloid accumulation. Comparable results were also observed in additional complementary analyses using clinical diagnosis (cognitively unimpaired versus dementia) as the outcome, with the polygenic measure including APOE ɛ4 explaining 15.8% of the phenotypic variance in the MCSA (p = 6.37×10–11) and 24.3% of the phenotypic variance in the ADNI (p = 1.08×10–19), including APOE ɛ4 as the strongest contributor in both cohorts.
DISCUSSION
As expected, age and APOE ɛ4 were robustly associated with amyloid PET burden in older adults. Although an aggregated polygenic measure based on the top risk variants for clinically probable AD dementia was also associated with amyloid levels, the added value of this measure over simply age and APOE together was modest, and all of these factors collectively still left a large majority of the variance in amyloid levels unexplained. These results were consistent across two independent cohorts with varying subject characteristics, including one cohort representing a large population-based sample of older adults.
There are several reasons to hypothesize that genetic factors may strongly account for susceptibility to amyloidosis. Twin studies support a high estimated heritability (0.60–0.80) [29] of AD, for which amyloidosis is an early disease hallmark [1]. A growing literature describes genetic associations with amyloid burden [30–33], and a recent twin study approach suggested that amyloidosis itself has at least moderate (0.41–0.52) heritability [34]. Our analyses confirmed associations of APOE ɛ4, APOE ɛ2, and known AD dementia susceptibility variants in PICALM, CR1, and CLU with amyloid deposition in older adults.
The second central aim of this study was to assess whether the aggregated effects of a large set of the strongest validated risk variants for clinically diagnosed AD dementia would account for a robust proportion of variance in amyloid PET levels. For this work, we utilized one application of the polygenic scoring framework, namely for extending association studies to calculate a collective variance explained by polygenic influences on a biologically relevant endophenotype [35]. Prior literature based around the ADNI cohort has supported that for amyloid deposition, APOE ɛ4 predominates over other common genetic variants linked to clinical AD dementia diagnosis. One study of an ADNI sample found no added value of an AD risk polygenic score (computed from case-control GWAS summary statistics) over APOE alone in predicting amyloid positivity [36]. Other analyses of the ADNI cohort identified associations of AD risk polygenic scores with brain amyloid levels, but with the aggregated measures adding in minor ways beyond APOE. Other studies of clinically derived cohorts have identified similar modest added value of polygenic scores beyond APOE for CSF AD biomarkers in autosomal dominant early-onset AD [40] and for plasma biomarkers in select populations of the ADNI sample [41]. Our study adds particular unique value by addressing this question in an amyloid PET sample larger than those of prior works and representing a population-based sample (distinct from those recruited in a manner similar to clinical trials). We also applied the results from the latest case-control GWAS of clinically diagnosed AD dementia to focus on well-validated prior hits. In summary, our study was designed to identify (if present) any robust collective effect on variance explained in amyloid deposition beyond APOE ɛ4 for other known case-control AD risk variants. Nevertheless, our findings support the conclusions from existing literature in this area: the genetic architecture of clinically probable AD dementia appears meaningfully different from the architecture underlying biologically defined measures of AD including amyloid deposition.
Through large sample sizes, AD case-control GWAS offer advantages in statistical power which may come at the expense of diagnostic specificity. Up to 10–20% of cases of clinically probable AD dementia do not meet criteria for biologically defined AD (i.e., they are not amyloid- and tau-positive) [42]. In addition, it is likely that a nontrivial proportion of individuals classified as non-demented controls may nevertheless have extant AD pathophysiology. That some AD case-control GWAS have implicated genes with known relationships to frontotemporal degenerative diseases (e.g., TMEM106B and GRN) could reflect common disease mechanisms but alternatively raises the spectre of a heterogeneous outcome measure yielding non-disease-specific results.
This work has limitations. Although the samples analyzed were robust for a PET-based study, they remain modest in comparison to those of other genomics studies and they lack in racial and ethnic diversity. It is also possible that the gene variants tested have relationships with alternative elements of AD pathophysiology not captured by amyloid or tau PET. A broader list of variants including those not meeting thresholds for genome-wide significance could theoretically account for further variance in amyloid levels, though likely with progressively diminishing returns and increasing likelihood of false positives. We also acknowledge that the GWAS hits from Wightman et al. may not pinpoint the true functional variants at these loci, and may not be all-inclusive in relation to other AD case-control GWAS [3, 4]. In addition, it should be mentioned that a subset of ADNI participants were included in the IGAP (International Genomics of Alzheimer’s Project) cohort that forms part of the much larger study by Wightman et al. (comprising more than one million individuals in total) and could in theory lead to a degree of overfitting. Reassuringly, the influence of any overlap with our study would be hypothesized to be minimal due to the distinct outcome measure (of amyloid PET levels) used here, the concordant results across the MCSA and ADNI, and prior work suggesting such modest overlap amongst ADNI is not likely to be material toward polygenic score assessment [36]. Further, although polygenic estimates of variance explained within a sample can be prone to overfitting and as a result should be interpreted with caution, it is reassuring that we observed a similar pattern of results in independent cohorts which were distinct in PET tracers and recruitment designs. Finally, there are other state-of-the-art approaches to polygenic score calculation, including those which use multi-parameter tuning to optimize LD clumping and thresholding [43] or which couple genetic effects across ancestral populations [44], and we acknowledge that these methodological differences could influence the results and conclusions from this line of work.
Within this context, it is important to note that risk prediction is not the sole purpose of genetic association studies including polygenic scoring methods, and that our findings specifically do not indicate a failure of the polygenic risk scoring approach which continues to show promise for complex diseases [6, 46]. Beyond risk prediction, implicated genetic factors point to potential disease mechanisms which can facilitate improved diagnostic and therapeutic strategies. Additional common and rare variants, haplotypes, epigenetic elements, and genetic interactions relevant for AD are yet to be discovered through complementary investigations. Nevertheless, our findings suggest that clinically applicable risk stratification in AD may require a multi-pronged approach (beyond solely case-control GWAS hits) integrating biology-specific omics associations, population heterogeneity, dynamic biomarker data, and environmental/lifestyle factors (Fig. 1).
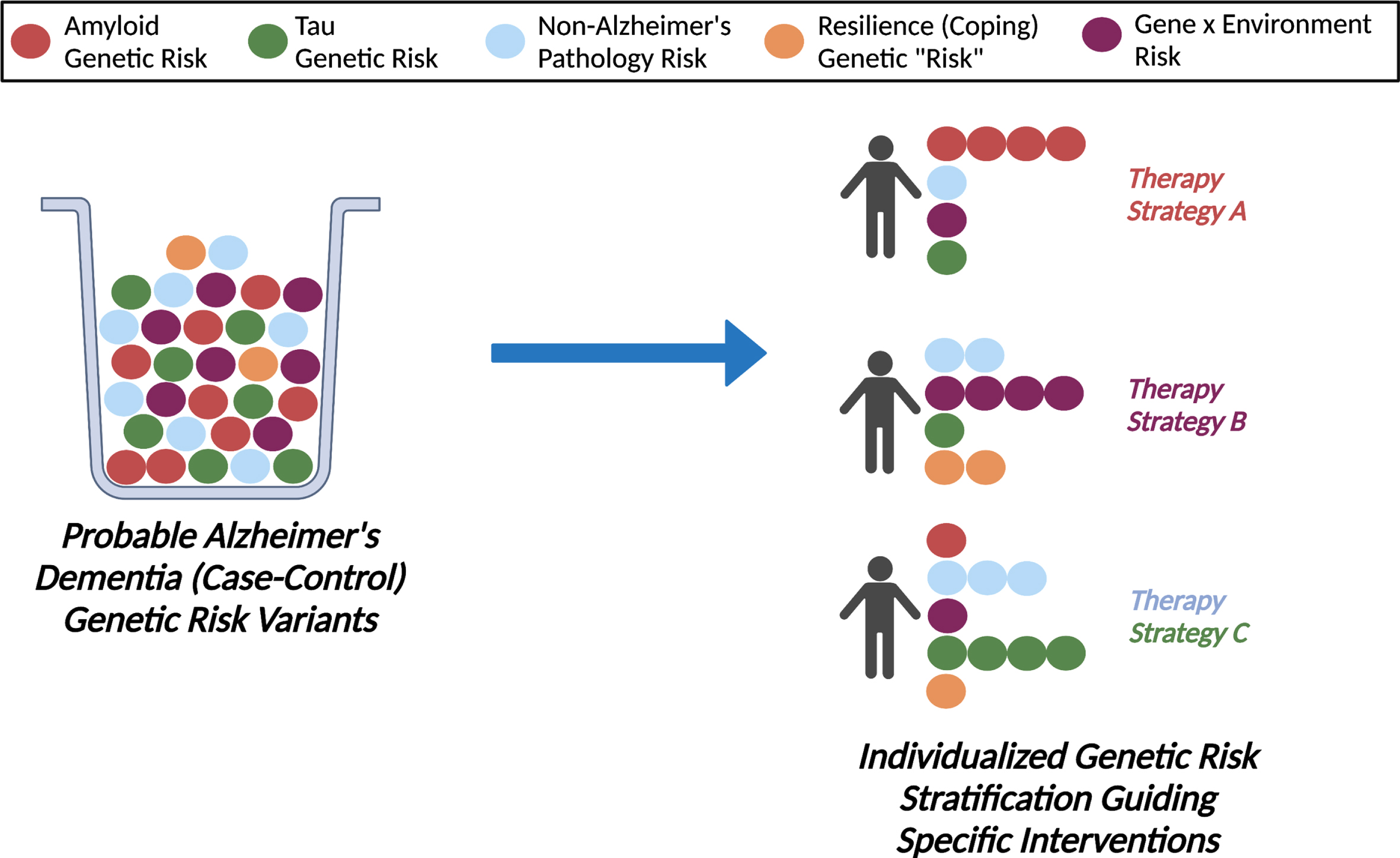
Enhancing Risk Stratification for Alzheimer’s Disease Via Multimodal Integration. We observed that a polygenic score of risk variants for clinically probable Alzheimer’s disease dementia added only modestly to APOE in explaining variation in amyloid levels (an early hallmark of Alzheimer’s disease) in a population-based sample of older adults, likely reflecting the underlying genetic heterogeneity of clinically diagnosed Alzheimer’s disease dementia. In the future, a more nuanced approach integrating biology-specific genetic associations, gene by environment interactions, and other biomarker measures of disease progression and heterogeneity needs to be considered to advance precision medicine for dementia care.
Footnotes
ACKNOWLEDGMENTS
The authors thank the study participants and staff in the Mayo Clinic Study of Aging, Mayo Alzheimer’s Disease Research Center, and Mayo Clinic Aging and Dementia Imaging Research laboratory.
This work was supported by NIH grants U01 AG006786 (PI: Petersen/Mielke/Jack), R01 NS097495 (PI: Vemuri), R01 AG56366 (PI: Vemuri), P50 AG016574 (PI: Petersen), P30 AG062677 (PI: Petersen), R37 AG011378 (PI: Jack), R01 AG041851 (PIs: Jack and Knopman), RF1 AG55151 (PI: Mielke), U54 NS100693 (PI: Ross), and R01 AG034676 (PI: Rocca); the GHR Foundation, the Alexander Family Alzheimer’s Disease Research Professorship of the Mayo Clinic, the Alzheimer’s Association, the Mayo Foundation for Medical Education and Research, the Liston Award, the Elsie and Marvin Dekelboum Family Foundation, the Schuler Foundation, and Opus Building NIH grant C06 RR018898.
We would like to greatly thank AVID Radiopharmaceuticals, Inc., for their support in supplying AV-1451 precursor, chemistry production advice and oversight, and FDA regulatory cross-filing permission and documentation needed for this work.
Data collection and sharing for the ADNI data utilized in this project was funded by the ADNI NIH grant U01 AG024904, other funding through the National Institute of Biomedical Imaging and Bioengineering, and private sector contributions from the following (facilitated by the Foundation for the National Institutes of Health with the grantee organization as the Northern California Institute for Research and Education): AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. The study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.