
Abstract
Background:
Varicella zoster virus (VZV) has been implicated in Alzheimer’s disease (AD), and vaccination against shingles, caused by VZV, has been found to decrease the risk of AD/dementia. VZV might reside latently in brain, and on reactivation might cause direct damage leading to AD, as proposed for herpes simplex virus type 1 (HSV-1), a virus strongly implicated in AD. Alternatively, shingles could induce neuroinflammation and thence, reactivation of HSV-1 in brain.
Objective:
To investigate these possibilities by comparing the effects of VZV and HSV-1 infection of cultured cells, and the action of VZV infection on cells quiescently infected with HSV-1.
Methods:
We infected human-induced neural stem cell (hiNSC) cultures with HSV-1 and/or VZV and sought the presence of AD-related phenotypes such as amyloid-β (Aβ) and P-tau accumulation, gliosis, and neuroinflammation.
Results:
Cells infected with VZV did not show the main AD characteristics, Aβ and P-tau accumulation, which HSV-1 does cause, but did show gliosis and increased levels of pro-inflammatory cytokines, suggesting that VZV’s action relating to AD/dementia is indirect. Strikingly, we found that VZV infection of cells quiescently infected with HSV-1 causes reactivation of HSV-1 and consequent AD-like changes, including Aβ and P-tau accumulation.
Conclusion:
Our results are consistent with the suggestion that shingles causes reactivation of HSV1 in brain and with the protective effects against AD of various vaccines, as well as the decrease in herpes labialis reported after certain types of vaccination. They support an indirect role for VZV in AD/dementia via reactivation of HSV-1 in brain.
Keywords
INTRODUCTION
Infectious diseases have long been known to cause a decrease in cognitive function which, in the case of herpes simplex virus type 1 (HSV-1) infection, is exacerbated by carriage of an APOE ɛ4 allele, and recent studies have shown that these diseases lead to a risk of Alzheimer’s disease (AD)/dementia [1–3]. (The term AD/dementia will be used here because in many studies the individual terms are not differentiated.) Several epidemiological studies have investigated the long-term effects of shingles, which is caused by reactivation of latent herpesvirus varicella zoster (VZV) in the periphery, in respect to the risk of dementia, and/or have examined the effect of vaccination against shingles on this risk. Chen et al. [4], Bae et al. [5], and Lophatananon et al. [6] implicated VZV in the disease in showing an increased risk after shingles, although the increase was very small. Importantly, Chen and later, Bae, discovered that prior antiviral treatment for shingles greatly reduced the risk, and Lophatananon et al. [6], Lehrer and Rheinstein [7], and Scherrer et al. [8] found that vaccination against shingles decreased the risk. Also, a recent cell biology study [9] implicated VZV further in detecting intranuclear formation of amyloid-β (Aβ) after infecting primary human spinal astrocytes cells with VZV. As far as we know, there have been no other previous studies investigating if VZV infection of cell cultures causes production of any AD-like characteristics. Whether or not the putative involvement of the virus in AD/dementia is direct or indirect—the latter perhaps via an effect on latent HSV-1 in brain—is unknown.
HSV-1 has been directly implicated in AD in that the viral DNA was discovered at a high prevalence in brain of elderly people, some thirty years ago [10]. Later it was found that in those who carried an APOE ɛ4 allele, HSV-1 in brain conferred a high risk of developing AD [11]. In a striking parallelism in the peripheral nervous system, APOE ɛ4 was shown to be a risk for cold sores (herpes labialis) [11], which are usually caused by HSV-1. Further support for a role for the virus came through the discovery of intrathecal antibodies to HSV-1, indicating that the virus had reactivated from latency in brain [12]. Also, it was found that in cell cultures, HSV-1 caused accumulation of Aβ [13] and abnormally phosphorylated tau (P-tau) [14], the main components of amyloid plaques and neurofibrillary tangles, which are the characteristic pathological features of AD, and that in brains of infected mice, HSV-1 caused deposition of Aβ [13]. Very significantly, in AD brains most of the viral DNA (72%) was found to be encaged in amyloid plaques [15]. Also, treatment of HSV-1-infected cells with various anti-HSV antivirals caused a substantial reduction in the accumulation of Aβ and P-tau ([16] et seq.)
Subsequently, over four hundred publications, using a variety of approaches, have provided further support for a major role for HSV-1 in AD. One of the most recent is our previous work using an in vitro 3D human brain model [17]. This study showed that HSV-1 infection of human-induced neural stem cells (hiNSCs) caused the formation of amyloid plaque-like formations, gliosis, neuroinflammation, and decreased functionality—significantly less electrophysiological activity, comparable to phenotypic changes observed in AD patients. These AD-like phenotypic changes occurred in the absence of any other intervention that might have caused such changes.
An obvious question that arises from these studies is whether or not VZV is present in latent form in brain and acts directly by causing cumulative damage during repeated reactivations, i.e., does it act in the way suggested for HSV-1 to explain the development of AD? We know of only two studies, using PCR, that have sought the viral DNA in autopsy brain specimens: in one case viral DNA was detected [18] but not in the other [19], despite the sensitivity of detection of VZV DNA being greater in the latter. The search for viral DNA in brain therefore needs to be repeated using current more sensitive detection methods. As well as this uncertainty about VZV presence in the brain, an argument against the direct involvement of VZV is the fact that it usually reactivates, causing shingles, only once, rarely doing so a second time: this single or once repeated VZV reactivation would not be consistent with the likely prerequisite for repeated viral reactivation to explain the lengthy progression of AD. Indirect VZV action could, in contrast, result from shingles causing inflammation which, in turn, would lead to neuroinflammation and thence to reactivation of HSV-1 in brain (inflammation is known to reactivate latent HSV-1, and in turn, reactivated HSV-1 causes inflammation as well as direct viral damage). Other causes of reactivation of HSV-1 include various types of stress, immunosuppression, UV light, and head injury (the latter explained by the fact that the axonal damage caused is known to be a reactivator of HSV-1) [20].
The possibility that peripheral infections might lead thus to HSV-1 reactivation was suggested almost 20 years ago by one of the present authors [21] as an explanation for the reduction in risk of dementia found by Verreault et al. [22] in subjects who had been vaccinated with the relevant vaccines to protect against influenza and poliomyelitis, and against bacterial infection causing diphtheria or tetanus. We proposed that vaccination reduced the number or severity of infections, thereby reducing the risk of HSV-1 reactivation and of subsequent development of AD/dementia. The seemingly inconsistent shingles results—that vaccination against shingles reduced greatly the risk of dementia [6, 23], yet shingles itself caused only a very small increase in risk [4–6]—could be explained if shingles causes HSV-1 reactivation in brain only if severe (perhaps in those who develop postherpetic neuralgia), whereas vaccination might well reduce mild as well as severe cases of shingles. If this is correct, it would be further support for an indirect effect of VZV on dementia.
We decided to determine if there is evidence for a direct action of VZV infection of cell cultures, similar to that of HSV1 infection, that might implicate VZV directly in AD. We therefore investigated if VZV, on infecting our hiNSCs cells and 3D brain model, causes the accumulation of Aβ and P-tau and other AD-like features, such as production of plaque-like formations (PLF), and gliosis (as shown by the presence of GFAP), as does HSV1 infection. We wanted also to investigate the proposal that latent HSV1 in brain is reactivated by inflammation induced by peripheral infection, by determining if VZV infection of cells harboring quiescent (resembling latent) HSV1 infection resulted in AD-like changes.
MATERIALS AND METHODS
Generation of hiNSCs
hiNSCs were generated as previously described [17]. Briefly, human foreskin fibroblasts were plated at a concentration of 105 cells in one gelatin-coated well of a 6-well plate, and cultured in fibroblast media (DMEM, 10% FBS, and 1% antibiotic-antimycotic). Concentrated aliquots of a polycistronic lentivirus expressing OCT4, KLF4, SOX2, and cMYC (Addgene #24603, a gift from Jose Cibelli) were used to infect cells in fibroblast medium using polybrene (Millipore) at a multiplicity of infection (MOI) = 1-2. Media were changed to hiNSC media: Knockout (KO) DMEM supplemented with 20% KO xeno-free serum replacement, 20 ng/mL recombinant bFGF, 1% Glutamax, 1% antibiotic-antimycotic, and 0.1 mM β-mercaptoethanol which also contained 1% KO growth factor cocktail (GFC) (Invitrogen). Four days later, cells were trypsinized and re-plated onto mouse embryonic fibroblast (MEF) feeder layers that had been previously inactivated by mitomycin C. hiNSC media (without KO-GFC) was subsequently changed every 1–3 days. At day 30 or later, colonies were mechanically picked and passaged onto fresh feeder MEF plates. Each picked colony was expanded to generate one hiNSC line. hiNSCs were enzymatically passaged as colonies using trypsin-like enzyme, TrypLE (Invitrogen), expanded, and subsequently frozen to make stocks. All generated lines tested negative for mycoplasma contamination.
hiNSC differentiation, HSV-1 and VZV infection, and Valacyclovir-HCl treatment
hiNSC colonies were trypsinized off MEF feeder layers using TrypLE (Invitrogen), then dissociated by pipetting. Cell suspensions were passaged through a 40μM cell strainer to remove larger aggregates. Dissociated hiNSCs were cultured on gelatin-coated plates in Neurobasal media supplemented with 2% B27 (Invitrogen), 1% Glutamax, and 1% antibiotic-antimycotic. HSV-1 McIntyre strain VR-539 and human herpesvirus 3 Varicella zoster virus Ellen strain VR-1367 were purchased from ATCC (Manassas, VA). For infections, we used purified HSV-1 and/or VZV to directly infect hiNSCs at an MOI of 0.0001 based on our previous study [17], which was calculated according to initial seeding density. For HSV-1, the purified virus preparation titration was 2×107 PFU/mL, which was adjusted to the desired MOI through serial dilution, 1μl per 2000 ml cell culture media. For VZV, the purified virus preparation titration was 4×104 PFU/mL, which was adjusted to desired MOI, 1μl per 4000μl cell culture medium. For both infections, purified virus was highly diluted with cell culture medium (ranging from 1:4000 to 1:2000000); thus, any potential effects from non-viral contaminants in the virus preparations would have been negligible. For mock infections, an equal volume of control culture medium from uninfected viral production cells was used (ATCC). All virus work was approved by Tufts Institutional Biosafety Committee. Valacyclovir-HCl (VCV) was reconstituted in dH2O and used at a concentration of 100μM for all subsequent experiments.
3D brain tissue model
A 3D brain tissue model was generated as previously described [24–26]. Briefly, silk protein sponges (pore size 500–600μm) were prepared from 6% (wt/vol) Bombyx mori-derived silk solution. Sponges were biopsy punched into 6 mm discs (2 mm in height), with 2 mm holes punched in the center to form donut shaped scaffolds. Scaffolds were autoclaved and coated with laminin (0.5 mg/ml) (Roche, Indianapolis, IN). Dissociated hiNSCs were seeded into the silk porous scaffolds at a density of 106 cells per scaffold and allowed to adhere overnight. The following day, collagen gels were made using type I rat tail collagen (Corning, Bedford, MA, USA) as previously described. 3D human brain tissue constructs were then cultured in neurobasal media (Invitrogen, Carlsbad, CA) supplemented with 2% B27 (Invitrogen, Carlsbad, CA), 0.5 mM Glutamax, and 1% antibiotic-antimycotic (Invitrogen, Carlsbad, CA) for 6 weeks to allow for mature network formation prior to HSV-1 infection, with medium changes every 3 days.
qRT-PCR
Total RNA was isolated using the RNeasy Mini kit (Qiagen). cDNA was generated using iScript (BioRad) according to the manufacturers’ protocols. Quantitative RT-PCR (qPCR) was performed using SYBR green and the CFX96 Real-Time PCR Detection System (BioRad) and normalized against the housekeeping gene GAPDH. All primer sequences are listed in Supplementary Table 1.
Immunofluorescence
Cells grown in monolayer tissue culture plates or in 3D scaffolds were fixed in 4% paraformaldehyde and washed with 1X phosphate-buffered saline (PBS). Samples were incubated with blocking buffer (PBS, 10% goat serum, and 0.1% Triton X-100). Primary antibodies were added to blocking buffer and incubated with samples overnight at 4°C. The next day, samples were washed several times with PBS, and incubated with a corresponding fluorescently-conjugated secondary antibody in blocking buffer for 1 h at room temperature. Nuclei were counterstained with DAPI (Invitrogen). All antibodies used in this study are listed in Supplementary Table 2.
Microscopy
Brightfield and fluorescent images were obtained using a Keyence BZ-X700 microscope and associated software.
Statistics
All data are expressed as mean±SD, and at least 3 biological replicates were analyzed per experiment. Individual experiments were repeated three times. Data with statistically significant differences were determined by 1-factor ANOVA with post-hoc Tukey test using the statistics software SYSTAT12 (Systat). A p-value less than 0.05 was considered significant.
RESULTS
VZV can infect hiNSCs
We previously showed that HSV-1 can infect hiNSCs and subsequently aimed to discover if hiNSCs were also infectable by VZV. We showed that VZV can infect hiNSCs, even at relatively low MOI (0.001) (Fig. 1), by demonstrating, using PCR, that several VZV-specific RNA were produced (additional VZV-specific markers were assayed also by qPCR - see Supplementary Figure 1). hiNSCs were treated with mock, or HSV-1 and/or VZV in monolayer cultures for four days. HSV-1 and VZV markers were detected via immunostaining (Fig. 1A) and qPCR (Fig. 1B,C).
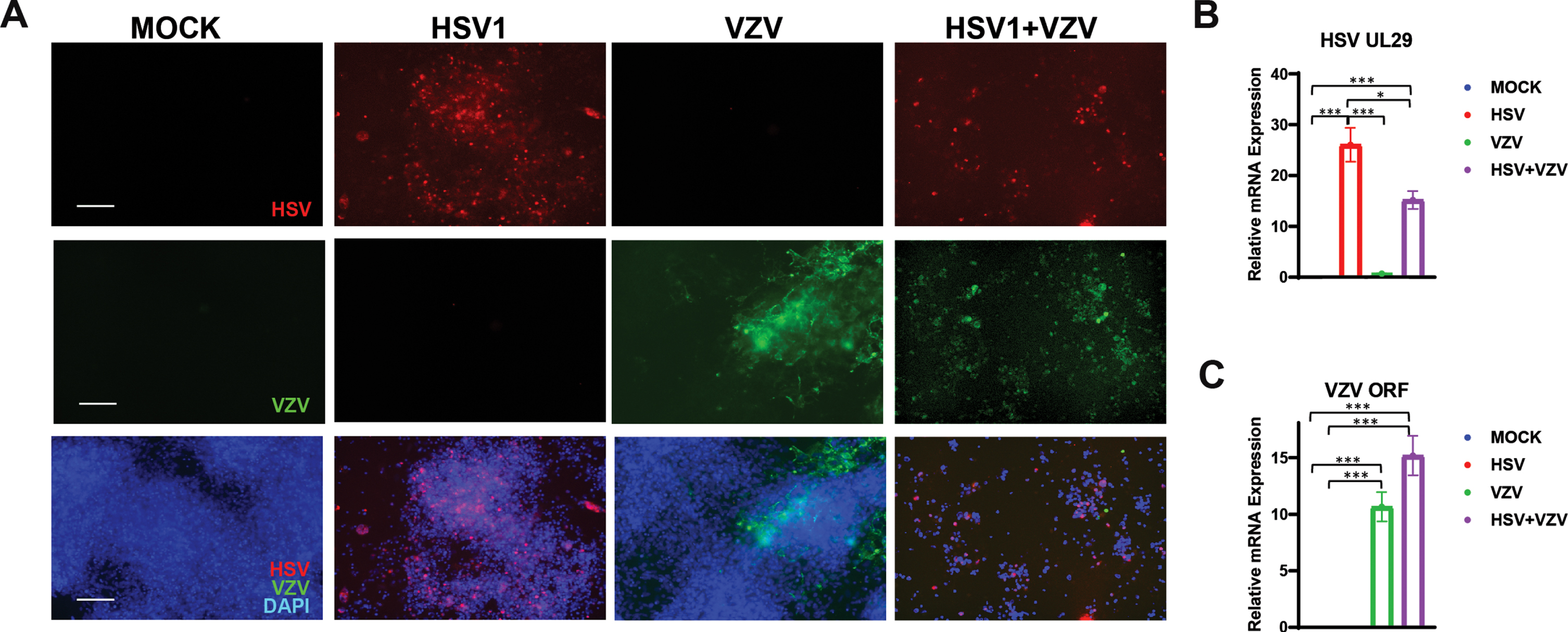
Varicella zoster virus (VZV) can infect human induced neural stem cells (hiNSCs). hiNSCs were treated with mock, HSV-1, and/or VZV in monolayer cultures for four days. HSV-1 and VZV markers were detected via immunostaining (A) and qPCR (B and C, respectively). Asterisks indicate statistically significant differences with error bars showing means±SD (*p≤0.05, **p≤0.01, and ***p≤0.001). Scale bars = 100μM.
HSV-1, but not VZV, induces Aβ and P-tau accumulation in hiNSCs
hiNSCs were treated with mock, HSV-1, and/or VZV in monolayer cultures for four days. Clearly, VZV, in marked contrast to HSV-1, did not cause the formation of the main features of AD—Aβ-positive PLFs (Fig. 2A) and P-tau (Fig. 2B), nor destruction of neurons (as shown by lack of difference of appearance of the neuronal marker TUJ1 between VZV-infected and mock-infected cells). However, when hiNSCs were co-infected with HSV-1 and VZV together, there was a significant increase in the size of the PLFs compared to those produced by HSV-1 infection alone, although the PLF numbers decreased (Supplementary Figure 2).
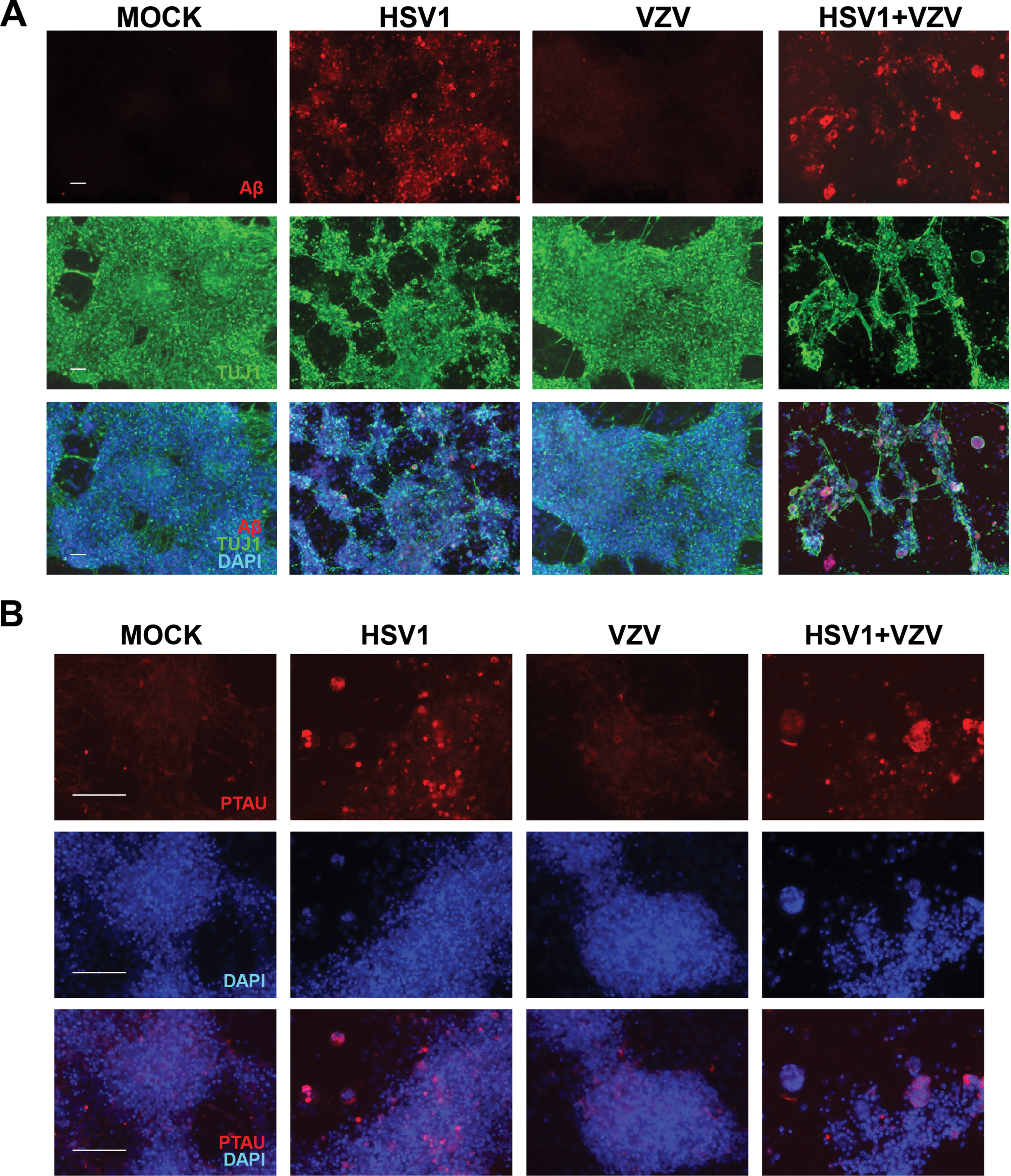
HSV-1 but not VZV induces amyloid-β and P-tau accumulation in hiNSCs. hiNSCs were treated with mock, HSV-1, and/or VZV in monolayer cultures for four days. A) Immunostaining against amyloid-β (Aβ, red) and pan-neuronal marker beta III tubulin (TUJ1, green). B) Immunostaining against phosphorylated tau (PTAU, red). Scale bars = 100μM.
HSV-1 and VZV independently cause gliosis and upregulation of pro-inflammatory cytokines
hiNSCs were treated with mock, HSV-1, and/or VZV in monolayer cultures for four days. Glial fibrillary acidic protein (GFAP) was upregulated in hiNSCs treated with HSV-1 or with VZV as demonstrated by immunostaining (Fig. 3A) and qPCR (Fig. 3B). Pro-inflammatory cytokines were also upregulated by either HSV-1 or VZV including IFN-γ, TNF-α, and IL-1β (Fig. 3C-E). Importantly, use of heat-inactivatedVZVdid not result in an upregulation of gliosis marker, GFAP (Supplementary Figure 3).
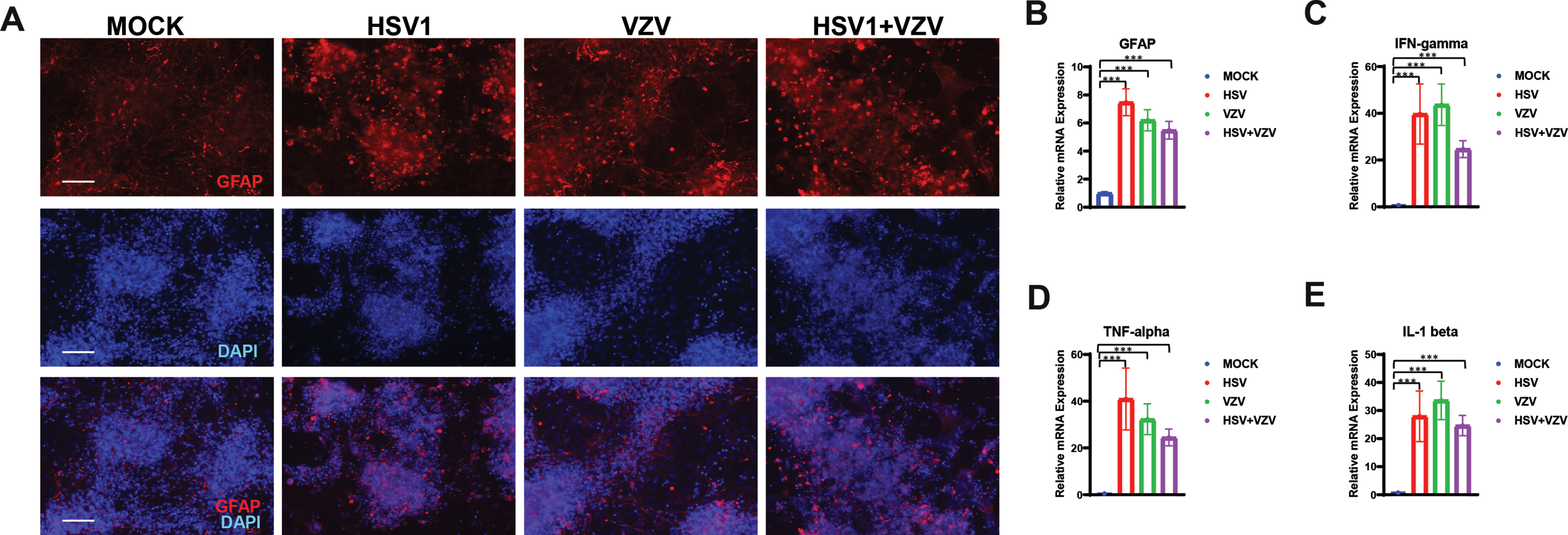
HSV-1 and VZV independently cause gliosis and upregulation of pro-inflammatory cytokines. hiNSCs were treated with mock, HSV-1 and/or VZV in monolayer cultures for four days. Glial fibrillary acidic protein (GFAP) was detected via immunostaining (A) and qPCR (B). Pro-inflammatory cytokines IFN-γ (C), TNF-α (D), and IL-1β (E) were analyzed by qPCR. Asterisks indicate statistically significant differences with error bars showing means±SD (*p≤0.05, **p≤0.01, and ***p≤0.001). Scale bars = 100μM.
Quiescent HSV-1 can be reactivated by VZV infection
Quiescence was achieved by treating hiNSCs acutely infected with HSV-1 for 24 h followed by 100μM of the antiviral agent VCV for 24 h; this resulted in the production of the latency-associated transcript (LAT) (Fig. 4A). This VCV-induced latency prevented active HSV-1 infection (Fig. 4B) and caused no accumulation of Aβ (Fig. 4C). However, treatment of quiescent HSV-1 hiNSC cultures with VZV resulted in upregulation of HSV-1 expression (Fig. 4B), accumulation of Aβ (Fig. 4C), and destruction of neurons, as shown by immunostaining.
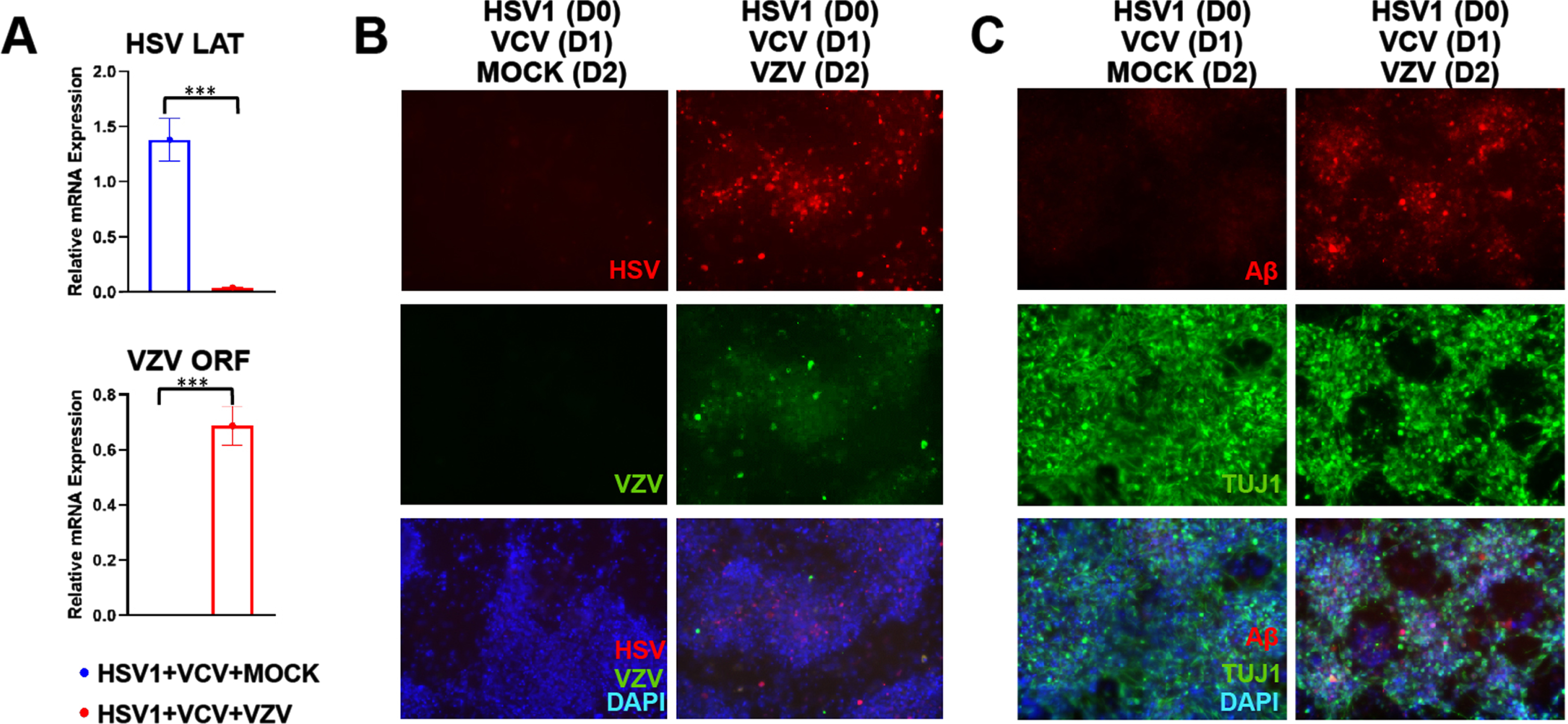
Quiescent HSV-1 can be reactivated by VZV infection. hiNSCs were treated with HSV-1 for 24 h followed by Valacyclovir HCl treatment for 24 h, then treatment with either mock or VZV in monolayer cultures for four days. A) VCV-induced quiescence of HSV-1 was demonstrated by expression of latency associated transcript (LAT) by qPCR. Subsequent VZV infection after HSV-1 latency was confirmed by qPCR. B) HSV (red) and VZV (green) marker expression were detected via immunostaining. C) Amyloid-β (Aβ, red) and pan-neuronal marker beta III tubulin (TUJ1, green) expression were detected via immunostaining. Asterisks indicate statistically significant differences with error bars showing means±SD (***p≤0.001). Scale bars = 100μM.
Bioengineered 3D human brain-like tissue constructs can be infected with VZV
Silk scaffolds were seeded with hiNSCs to allow for sufficient network formation. Cells were treated with mock or HSV-1 for 2 days, followed by 100μM Valacyclovir HCl treatment for 3 days. Human brain-like tissue constructs were then subjected to mock or VZV infection for 10 days. Immunostaining then showed that 3D construct could be infected with VZV (Fig. 5B).
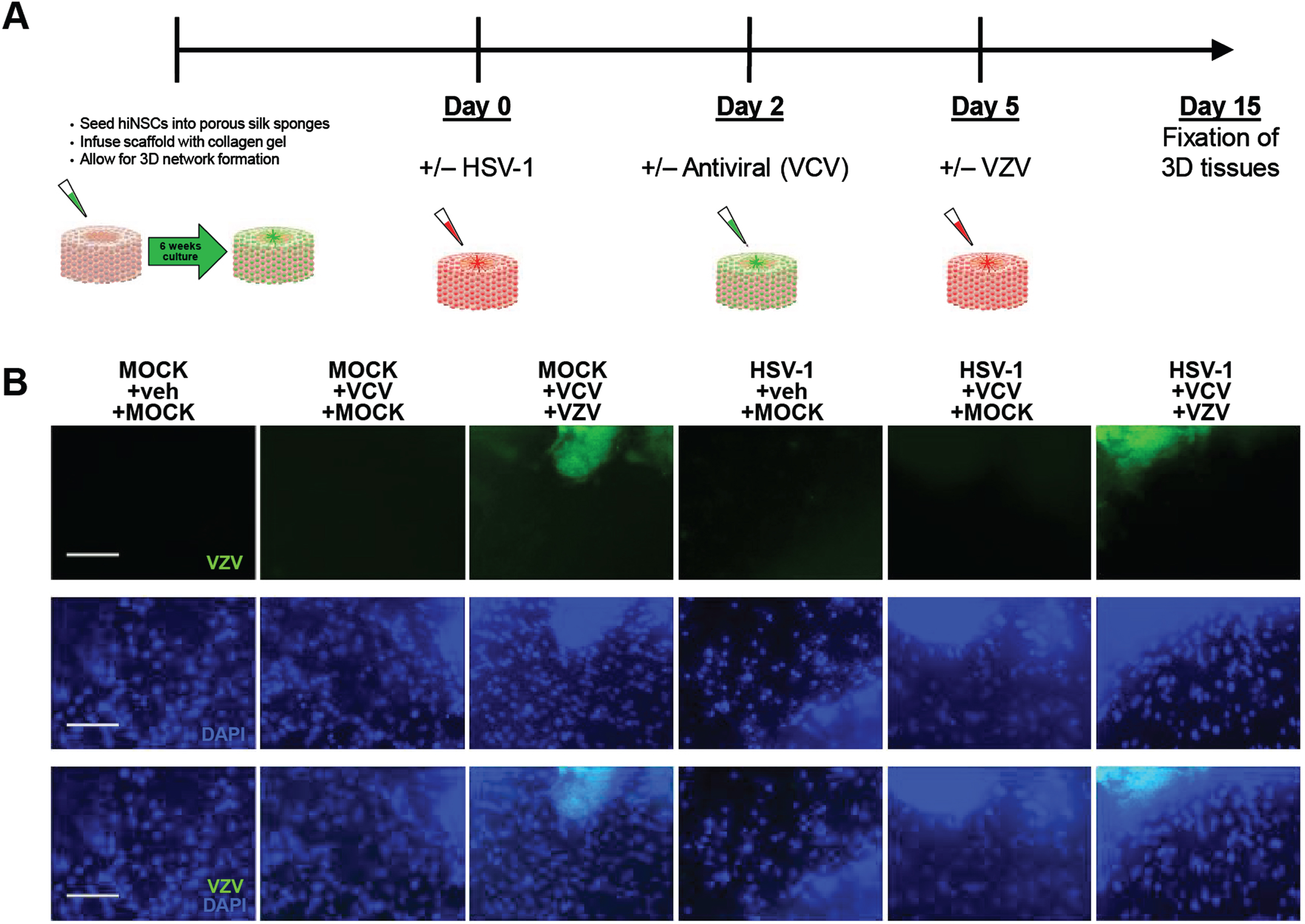
Bioengineered 3D human brain-like tissue constructs can be infected with VZV. A) Schematic diagram showing timeline of 3D human brain-like tissue construct generation and treatment. B) Immunostaining results showing expression of VZV marker (green) across all treatments. Scale bars = 100μM.
VZV infection reactivates quiescent HSV-1 to induce Aβ and P-t au accumulation in 3D human brain-like tissue constructs
Human brain-like tissue constructs with quiescent HSV-1 infection, then subjected to VZV infection, demonstrated a reactivation of HSV-1 infection as indicated by immunostaining (Fig. 6A). This HSV-1 reactivation resulted in accumulation of Aβ (Fig. 6A) and P-tau (Fig. 6B) in these 3D human brain-like tissue constructs, showing that VZV can trigger latent HSV-1 to become active, thereby inducing AD-like phenotypes.
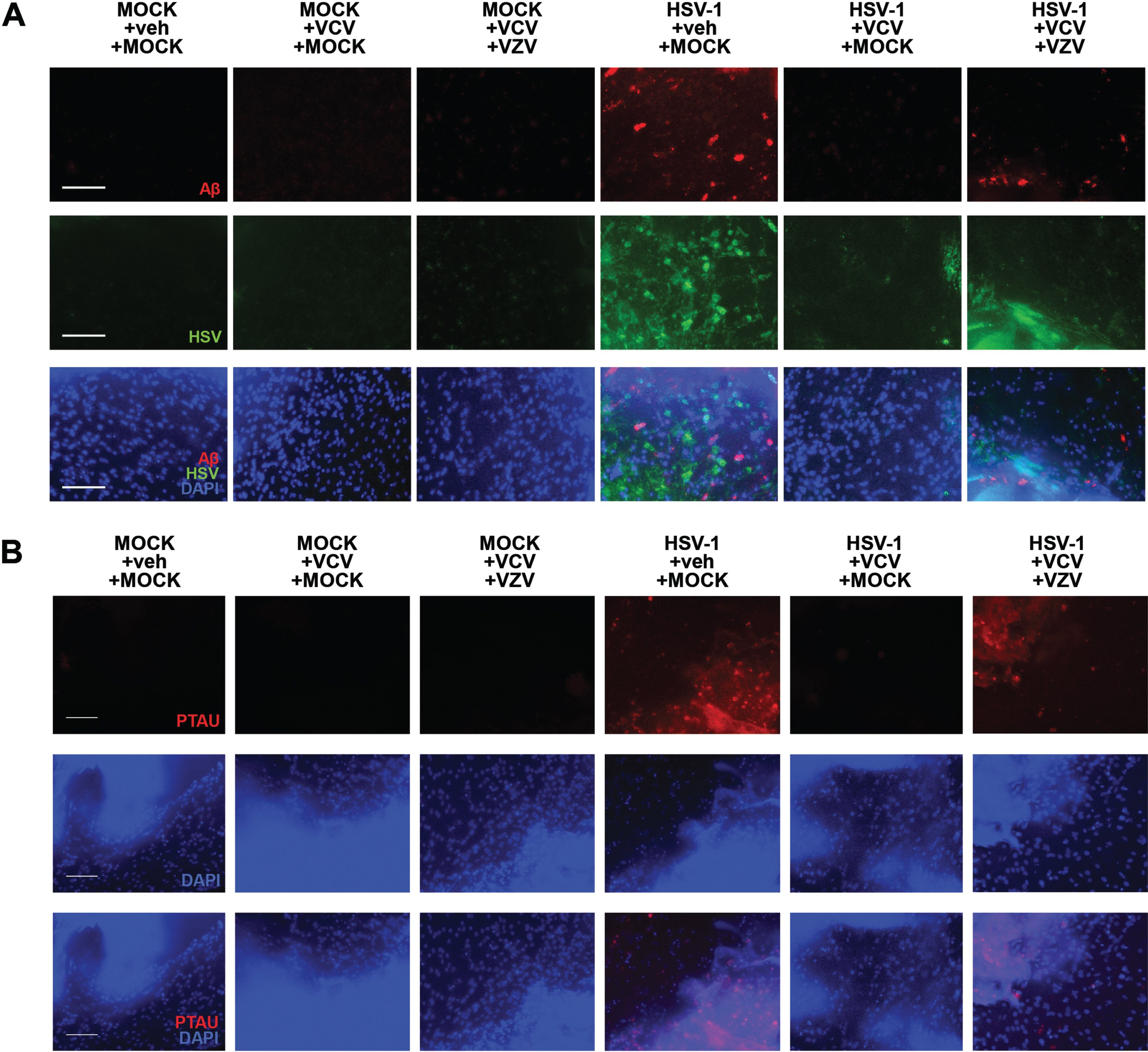
VZV infection reactivates quiescent HSV-1 to induce amyloid-β and P-Tau accumulation in 3D human brain-like tissue constructs. A) Immunostaining against amyloid-β (Aβ, red) and HSV marker (HSV, green). B) Immunostaining against phosphorylated Tau (PTAU, red). Scale bars = 100μM.
DISCUSSION
Our results show that VZV infection of cultured hiNSC cells does not lead to the formation of Aβ and P-tau, the main components respectively of AD plaques and neurofibrillary tangles. It therefore seems unlikely that VZV could be a direct cause of AD. These facts suggest instead that VZV has an indirect effect: reactivation of HSV-1.
Our other main finding is that the addition of VZV to hiNSC cells harboring quiescent HSV-1 causes a dramatic increase in Aβ and P-tau, in gliosis and in several pro-inflammatory cytokines, all features characteristic of the AD brain. What is striking is that reactivation occurred in our system despite the absence of a normal immune system. Interestingly and perhaps relevantly, it has been found that the HSV-1 protein ICP0, and certain other HSV proteins can trigger reactivation of HSV-1, HSV2, and pseudorabies virus in latently infected trigeminal ganglion cells in culture [27–29]. Presumably, the cytokines elicited by VZV infection cause the reactivation of quiescent HSV-1. This striking result would be totally consistent with the suggestion that in vivo, infections such as VZV cause an increase in neuroinflammation, leading to reactivation of latent HSV-1 and consequent damage in brain, so that repeated infections—causing recurrent reactivation—would lead eventually to the development of AD/dementia [21]. These proposed events would explain the fact that many studies have found that peripheral infections lead not only to cognitive decline but also to an increased risk of AD/dementia [3, 30]. In fact, two very recent publications have suggested that targeting infections might reduce the incidence of AD [31, 32]. The fact that infections often trigger herpes labialis, indicating that the virus has reactivated in the periphery, is consistent also with the proposed reactivation in the CNS.
Infection by SARS-CoV-2 is particularly relevant to the present aspect of infection and AD/dementia in that the neurological disturbances that occur in many COVID-19 patients continue long-term; these are of great concern because of the possibility that they might lead eventually to neurodegeneration, and specifically to AD/dementia. It is noteworthy and probably significant that a number of studies on COVID-19 have shown that HSV-1 and VZV are reactivated during or after SARS-CoV-2 virus infection [33–35], especially in older subjects and in those with weakened immune systems; and sometimes both are reactivated together [36]. In one study [37], reactivation of other herpesviruses—Epstein-Barr virus, cytomegalovirus, and human-herpes virus-6—was detected. Most authors though warn against assuming that COVID-19 causes an increased risk of reactivations; instead, some reactivations might merely be coincidental. These effects probably reflect the fact that many COVID-19 patients suffer protracted severe infection because of the absence of adequate treatments. Therefore, side effects such as herpes reactivation are more likely to occur and would be more noticeable than in the many other infectious diseases that are treatable and which, therefore, affect patients only relatively briefly.
Three of the studies on VZV and its relevance to AD/dementia examined the effect of vaccination against shingles on risk of dementia and all discovered that it reduces the risk considerably [6–8]. This too is totally consistent with the suggestion that the effect of various other vaccines against a variety of infections is protective in reducing reactivations of HSV-1 and hence, in reducing the incidence of AD/dementia. Indeed, further strong support for our hypothesis that the harmful effects of infections involve reactivation of HSV-1 and its subsequent damaging consequences is provided by several studies investigating certain types of vaccination, in particular Bacillus Calmette Guérin. These vaccinations have been found to lead to decreased frequency and severity of herpes labialis, and of genital herpes (caused usually by HSV2), as described in the reviews by Pittet and Curtis [38], and Adensanya [39]. Even though these observations apply to reactivation in the periphery, they are likely to apply also to reactivation in brain, but unfortunately, such activity in the brain is not at present detectable. This activity might be detectable in cerebrospinal fluid (CSF) if HSV-1 DNA were to be sought and found there, as the viral DNA is known to persist for a week or two after herpes simplex encephalitis [40]—assuming that HSV1 reactivations resemble very mild HSV-1 encephalitis; however, a search for the viral DNA would necessitate taking weekly samples of CSF from suspected cases—an unethical and impracticable procedure. Nonetheless, these studies and our current results support the likelihood that in brains of older humans, infection leads to HSV1 reactivation, and that various types of vaccination reduce the frequency and/or severity of reactivations and hence reduce the progression of damage leading to AD/dementia.
One of our aims now is to investigate the effects of other viruses, both herpesviruses and non-herpesviruses, as well as other common infectious pathogens, on cells of various types quiescently infected with HSV-1, to find if they too cause reactivation of the HSV-1. The results should help to elucidate the pathways whereby infections increase the risk of AD/dementia and the ways this disease could be combatted by appropriate antivirals for treatment, or just possibly, for prevention [6, 42].