
Abstract
Background:
Urokinase-type plasminogen activator (uPA) is a serine proteinase found in excitatory synapses located in the II/III and V cortical layers. The synaptic release of uPA promotes the formation of synaptic contacts and the repair of synapses damaged by various forms of injury, and its abundance is decreased in the synapse of Alzheimer’s disease (AD) patients. Inactivation of the Wingless/Int1 (Wnt)-β-catenin pathway plays a central role in the pathogenesis of AD. Soluble amyloid-β (Aβ) prevents the phosphorylation of the low-density lipoprotein receptor-related protein-6 (LRP6), and the resultant inactivation of the Wnt-β-catenin pathway prompts the amyloidogenic processing of the amyloid-β protein precursor (AβPP) and causes synaptic loss.
Objective:
To study the role of neuronal uPA in the pathogenesis of AD.
Methods:
We used in vitro cultures of murine cerebral cortical neurons, a murine neuroblastoma cell line transfected with the APP-695 Swedish mutation (N2asw), and mice deficient on either plasminogen, or uPA, or its receptor (uPAR).
Results:
We show that uPA activates the Wnt-β-catenin pathway in cerebral cortical neurons by triggering the phosphorylation of LRP6 via a plasmin-independent mechanism that does not require binding of Wnt ligands (Wnts). Our data indicate that uPA-induced activation of the Wnt-β-catenin pathway protects the synapse from the harmful effects of soluble Aβ and prevents the amyloidogenic processing of AβPP by inhibiting the expression of β-secretase 1 (BACE1) and the ensuing generation of Aβ40 and Aβ42 peptides.
Conclusion:
uPA protects the synapse and antagonizes the inhibitory effect of soluble Aβ on the Wnt-β-catenin pathway by providing an alternative pathway for LRP6 phosphorylation and β-catenin stabilization.
INTRODUCTION
Loss of synaptic structure and function is a strong predictor of disease progression in Alzheimer’s disease (AD) patients [1]. More specifically, the harmful effects of soluble amyloid-β (Aβ) on the presynaptic release of neurotransmitters [2], structure of dendritic spines [3], development of synaptic plasticity [4], and proper functioning of neural networks [5] have been robustly correlated with the severity of cognitive decline in these individuals [1, 7]. Importantly, the deleterious effects of soluble Aβ on the synapse precede neuronal death [8, 9], and thus they are potentially amenable to therapeutic interventions before progression to irreversible brain damage.
The Wnt-β-catenin pathway plays a pivotal role in synaptogenesis and preservation of synaptic structure and function [10, 11]. When this pathway is inactive, glycogen synthase kinase-3β (GSK3β) targets β-Catenin to proteasomal degradation by eliciting its phosphorylation at Ser33/Ser37/Thr41. In contrast, binding of Wnt ligands (Wnts) to Frizzleds (Fz) receptors and its co-receptor the low-density lipoprotein receptor-related protein-6 (LRP6) on the cell membrane prompts the phosphorylation of the intracellular domain of LRP6 at Ser1490 (pLRP6). In turn, pLRP6 recruits and inactivates GSK3β, rendering it unable to phosphorylate β-Catenin and thus allowing its cytoplasmic accumulation and subsequent nuclear translocation. Once in the nucleus, β-Catenin induces the expression of Wnt-target genes by binding to the transcription factor T cell factor (TCF)/lymphoid enhancer factor (LEF) [10]. Soluble Aβ prevents the binding of Wnts to LRP6 and Fz receptors [12], and the resultant inactivation of the Wnt-β-catenin pathway causes synaptic loss [13–15], and triggers the amyloidogenic processing of amyloid-β protein precursor (AβPP) and tau phosphorylation [16]. In line with these observations, restoration of Wnt-β-catenin pathway function prevents soluble Aβ-induced synaptic dysfunction and cognitive decline in animal models of AD [13].
Urokinase-type plasminogen activator (uPA) is a serine proteinase that upon binding to its receptor (uPAR) generates plasmin and activates cell signaling pathways [17]. In the developing brain, uPA induces neurogenesis and the formation of dendrites and axonal branches [18]. In the mature brain, this proteinase is found in synapses of excitatory neurons located in the II/III and V cortical layers [19]; and its release promotes the formation of synaptic contacts [20–22] and the repair of synapses damaged by various forms of injury [19–26]. Our earlier studies indicate that the abundance of uPA is decreased in the synapse of AD patients [19]. Importantly, although it has been proposed that uPA has a protective effect in the brain of AD patients by its ability to induce plasmin-mediated cleavage of extracellular Aβ containing plaques [27], recent studies have revealed that uPA protects the synapse from the harmful effects of soluble Aβ by a mechanism that does not require plasmin generation [19].
Here we report that binding of uPA to uPAR activates the Wnt-β-Catenin pathway in cerebral cortical neurons by triggering the phosphorylation of LRP6 via a Wnts-independent mechanism that does not require plasmin generation. Our data indicate that the resultant cytoplasmic accumulation of β-Catenin protects the synapse from the harmful effects of soluble Aβ. Remarkably, we found that by providing an alternative pathway for LRP6 phosphorylation, uPA also antagonizes the inhibitory effect of soluble Aβ on the Wnt-β-Catenin pathway, and that the ensuing activation of β-Catenin signaling prevents the amyloidogenic processing of AβPP by inhibiting the transcription of β-secretase-1 (BACE1). This is a new role for uPA in the central nervous system with potential implications for the development of therapeutic strategies aimed to protect the synapse of AD patients.
MATERIALS AND METHODS
Animals and reagents
We used 6-month-old male wild-type (Wt), and uPA (uPA-/-) and plasminogen deficient (Plg-/-) mice on a C57BL/6J background. The Institutional Animal Care and Use Committee (IACUC) of Emory University, Atlanta, GA, approved the animal experiments. A murine neuroblastoma cell line stably transfected with the APP-695 Swedish mutation (K595 N/M596 L) and herein referred to as N2asw, was a kind gift from Dr. G. Thinakaran (University of South Florida). Other reagents were recombinant murine uPA and its amino terminal fragment (ATF, devoid of proteolytic activity; Molecular Innovations, Novi, MI; Cat # MATF and MUPA, respectively), recombinant murine WIF-1 and Wnt3a (R & D Systems; Cat # 133-WF-050 and 1324-WN-010, respectively), dynabeads protein G immunoprecipitation kit, ultrapure agarose, Pierce BCA protein assay kit, sample buffer 5X, bovine serum albumin, penicillin/streptomycin, ProLong gold antifade, phalloidin DyLight 554, Hoechst, and ELISA kits for murine Aβ 1–40 and 1–42 peptides (ThermoFisher, Grand Island, NY; Cat # 10007D, 16500 - 500, 23225, 1859594, J64655, 15140-122, P36930, 21834, H3570, KMB3181 and KMB3441, respectively), triton X-100 and poly-l-lysine (Millipore, Burlington, MA; Cat # 9400 and P1399, respectively), oligomers of β-amyloid 1-42 (rPeptide, Watkinsville, GA; Cat # A - 1163), XAV-939 (Tocris Bioscience, Minneapolis, MN; Cat # 3748), Rneasy mini kit (Qiagen, Germantown, MD; Cat # 74106), protease inhibitors complete tablets EASYpack (Roche, Indianapolis, IN; Cat # 46931320001), RIPA buffer (TEKNOVA, Hillister, CA; Cat # R3792), iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA; Cat # 1708891), and the following antibodies: goat anti-mouse uPAR (R & D Systems; Cat # AF534), rabbit polyclonal LRP6 and mouse anti-β-actin (ThermoFisher; Cat # PA5-89161 and A1978, respectively), mouse anti-β-Catenin (BD Biosciences, San Jose, CA; Cat # 610153), monoclonal β-Catenin non-phosphorylated at Ser37/Thr41 (active β-Catenin), monoclonal anti-LRP6, and goat anti-mouse IgG HRP conjugate (Millipore; Cat # 05-665, MABS341, AP127P, respectively), mouse bassoon (Enzon, Farmingdale, NY; Cat # ENZ- ABS666-0200), rabbit anti-histone-H3, rabbit anit-PSD-95 and rabbit anti-phospho-LRP6 (Ser1490; Cell Signaling, Denver, MA; Cat # 4499, 2507 and 2568, respectively), sheep anti-uPAR (Molecular Innovations; Cat # SASMUPAR-306), IRDye 800CW donkey anti-rabbit, IRDye 800CW donkey anti-mouse, IRDye 680RD donkey anti-mouse, and IRDye 680RD donkey anti-rabbit (Li-Cor, Lincoln, NE; Cat # 926-32213, 929-32212, 926-68072, and 927-50000, respectively), and donkey anti-mouse Alexa Fluor 594, donkey anti-rabbit Alexa 594, goat anti-mouse Alexa Fluor 488, goat anti-rabbit Alexa Fluor 488, donkey anti-goat Alexa Fluor 488, and donkey anti-sheep IgG HRP conjugated (ThermoFisher; Cat # A21203, A21207, A11017, A11008, A11055, and A16041, respectively). PCR primers for Actb (forward 5’-ACTGGGACGACATGGAGAAG-3’, reverse 5’-GGGGTGTTGAAGGTCTCAAA-3’) and BACE1 (forward 5’-CTTTGTGGAGATGGTGGAC-3’, reverse 5’-GTTACTACTGCCCGTGTC-3’) were purchased from Integrated DNA Technologies (Coralville, IA).
Murine neuronal cultures
We cultured Wt cerebral cortical neurons from E16-18 mice as described [28]. Briefly, the cerebral cortex was dissected, transferred into Hanks’ balanced salt solution containing 100 units/ml penicillin, 100μg/ml streptomycin, and 10 mM HEPES, and incubated in trypsin containing 0.02% DNase at 37°C for 15 min. Tissue was triturated and the supernatant was re-suspended in GS21-supplemented neurobasal medium containing 2 mM l-glutamine, and plated onto 0.1 mg/ml poly-l-lysine-coated wells.
Preparation of nuclear extracts
Wt neurons incubated for 60 min with 5 nM of uPA or its ATF, or vehicle (control) were washed and centrifuged at 1,600 x g for 10 min. The pellet was resuspended in a hypotonic buffer containing 10 mM Hepes pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT, and protease and phosphatase inhibitors, incubated in ice for 10 min, homogenized with a Pyrex homogenizer with 1 ml volume capacity and centrifuged for 10 min at 3,400 x g. The pellet was resuspended in a buffer containing 20 mM Hepes pH 7.9, 20% glycerol, 0.42 mM KCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, and protease and phosphatase inhibitors before centrifugation for 30 min at 21,000 x g. Supernatants containing nuclear extracts were used for western blot analysis. To test the purity of these preparations cytoplasmic and nuclear extracts were immunoblotted with antibodies against histone-3 and GAPDH (data not shown).
Preparation of Aβ142 oligomers
To prepare Aβ142 oligomers [29], 80μM Aβ142 monomers lyophilized in 1,1,1,3,3,3-Hexafluoro-2-Propanol were incubated overnight after resuspension to a 5 mM concentration in DMSO. Experiments were performed with final concentration of 100 nM.
Western blot analyses
The abundance of β-Catenin was studied in Wt cerebral cortical neurons treated for 30 min with 5 nM of uPA or a comparable volume of vehicle (control), or 24 h with 100 nM of soluble Aβ142 oligomers in the presence of three doses of 5 nM of uPA or vehicle (control) administered at time 0, 12, and 23 h, or in nuclear extracts prepared from Wt cerebral cortical neurons treated during 60 min with 5 nM of uPA or its ATF, or in N2asw cells incubated during 15 min with 5 nM of uPA. The abundance of active β-Catenin was studied in Wt cerebral cortical neurons treated during 30 min with 5 nM of uPA. The abundance of LRP6 phosphorylated at Ser1490 (pLRP6) was studied in Wt cerebral cortical neurons incubated 15 min with 5 nM of uPA. Cells were then homogenized with RIPA buffer. Protein was loaded onto 4–15% gels (Bio-Rad) and transferred into a nitrocellulose membrane (Bio-Rad). Then, membranes were incubated with antibodies against either total β-catenin (1:2,000), or active β-catenin (1:1,000), or LRP6 phosphorylated at Ser1490 (1:1,000), or β-actin (1:50,000), or histone-3 (1:1,000). Membranes were then washed and incubated with either one of the following secondary antibodies prepared at a 1:10,000 dilution: IRDye 800CW donkey anti-rabbit, or IRDye 800CW donkey anti-mouse, or IRDye 680RD donkey anti-mouse, or IRDye 680RD donkey anti-rabbit, or IRDye 800CW goat anti-mouse IgG1. LI-COR Odyssey Fc reader and IMAGE STUDIO version 5.2 were used to detect and measure infrared signal (Li-Cor). Values were expressed as a ratio to β-actin and normalized to intensity detected in extracts prepared from untreated cells.
Polymerase chain reaction
RNA was extracted from Wt cerebral cortical neurons and N2asw cells treated 24 h with 5 nM of uPA or vehicle (control), and from Wt cerebral cortical neurons incubated during 24 h with vehicle (control) or 1μM of XAV-939, followed by 24 h of treatment with either 5 nM of uPA or vehicle (control). A subgroup of neurons was treated during 24 h with 5 nM of uPA’s ATF. cDNA was prepared using iScript cDNA Synthesis kit, and a MultiGene OptiMax Thermal Cycler [72°C×2 min; 30 cycles of 98°C×was used 10 s, 60°C×30 s, 72°C×20 s, and 72°C×10 min (Labnet International, Edison, NJ)] was used to detect a PCR product to detect either BACE1 or Actb with the primers described above in Animals and reagents. The LI-COR Odyssey Fc reader and IMAGE STUDIO ver 5.2 was used to detect and measure the amplified PCR product.
Immunocytochemistry
To study the effect of uPA on the synapse, Wt cerebral cortical neurons were treated during 24 h with 1μM of XAV-939 or vehicle (control), and then incubated for 24 h with 100 nM of soluble Aβ oligomers or vehicle (control), alone or in the presence of three doses of 5 nM of uPA administered at 0, 12, and 23 h. To study the effect of uPA on LRP6 phosphorylation at Ser1490, Wt cerebral cortical neurons were incubated 15 min with 5 nM of uPA or vehicle (control); or 30 min with 10μg/ml of WIF1 or vehicle (control) followed by 15 min of treatment with with 5 nM of uPA or 80μg/ml of Wnt3a; or 24 h with vehicle or 100 nM of soluble Aβ oligomers, followed by 15 min of treatment with vehicle, or 80μg/ml of Wnt3a, or 5 nM of uPA; or 15 min with vehicle (control), or 5 nM of uPA, or 6μg/ml of anti-uPAR antibodies, or a combination of either uPA or Wnt3a and anti-uPAR antibodies. To study the effect of uPA on the nuclear translocation of β-Catenin Wt cerebral cortical neurons were treated 60 min with vehicle (control) or 5 nM of uPA. To study the effect of uPA on BACE1 expression, Wt cerebral cortical neurons were treated 24 h with 5 nM of uPA or vehicle (control). At the end of each time point cells were fixed for 10 min with 4% paraformaldehyde, washed, permeabilized, blocked with 0.1% triton X-100 and 3% BSA in TBS, and incubated overnight at 4°C with either one of the following antibodies at a 1:100 dilution: mouse anti-bassoon, and/or rabbit anti-PSD-95, and/or rabbit anti-phospho-LRP6, and/or mouse anti- β-Catenin. Cells were then washed and incubated with either phalloidin DyLight 488 (1:1000) and/or Hoechst (1:5000), and either one of the following antibodies at a 1:500 dilution: donkey anti-mouse Alexa Fluor 594, donkey anti-rabbit Alexa 594, goat anti-mouse Alexa Fluor 488, goat anti-rabbit Alexa Fluor 488, or donkey anti-goat Alexa Fluor 488. Confocal images were obtained using a Fluoview FV10i automated confocal laser-scanning microscope (Olympus, Center Valley, PA) with a 60x lens UPLSAPO60XO NA 1.35, 1X pinhole (50 um), 512×512 pixels, speed 4.4 s/frame, scanning device 2 galvano-meter mirrors, XY resolution 300 nm, immersion oil IMMOIL F30CC. Pictures were deconvoluted using 64 iterations of CellSens dimension 1.17 software (Olympus). To quantify the nuclear translocation of β-Catenin, we used the CellSens dimension 1.17 software to delineate the nucleus in the Hoechst channel. Then, the area immunoreactive to anti β-Catenin antibodies was quantified, and values were expressed as percentage of nuclear β-Catenin immunoreactivity in control cells. The ImageJ puncta analyzer was used to quantify pLRP6- and bassoon/PSD-95-positive puncta in 50μm extensions of neurons in each experimental group.
ELISA
Brain cortices were harvested and processed following manufacturer’s instructions. Briefly, 100 mg of tissue was homogenized 8 times with 100μl of 5M guanidine-HCl/50 mM Tris at pH 8 and mixed at room temperature for 3 h. The lysate was diluted 10-fold with cold PBS plus protease inhibitors cocktail and centrifuged at 4°C 16,000 x g for 20 min. The supernatant was collected and diluted two times using the kit dilution buffer. N2asw cells were maintained in culture media containing 1:1 DMEM and Opti-MEM, 5% fetal bovine serum, 100 units/ml penicillin/streptomycin, 200 mM L-glutamine, and 0.2 mg/ml G418. Upon reaching 80-90% confluence, cells were plated onto 35 mm wells and treated for 24 h with either vehicle control or 5 nM of uPA. Then, 100μl of culture medium was used to measure the concentration of Aβ oligomers following manufacturer’s instructions. 100μl of brains or cells, or standards were incubated for 2 h in the antibody-coated plate. Wells were then washed four times with the kit wash buffer and incubated with the kit detector antibody for 1 h at room temperature. The plate was then incubated with anti-IgG HRP for 30 min at room temperature, and then washed again and treated during 30 min with 100μl of the kit chromogen buffer. The absorbance was read at 450 nm after addition of 100μl of the kit stop solution. Protein concentration was measured in cortices and cells, and values were expressed as pg of Aβ40 or Aβ42 per mg (tissue) or μg (cells) of protein.
Statistics
Statistical analysis was performed with student’s t-test and one- or two-way ANOVA with corrections, as deemed appropriate and described in each figure legend. p-values of < 0.05 were considered as significant.
RESULTS
UPA protects the synapse from the harmful effects of soluble Aβ by abrogating its inhibitory effect on the accumulation of β-catenin
First, we decided to study the effect of uPA on the expression of β-Catenin. We found that 30 min of treatment with 5 nM of uPA increases the abundance of β-Catenin in wild-type (Wt) cerebral cortical neurons (Fig. 1A, B). Because soluble Aβ decreases the abundance of β-Catenin [30], we studied its expression in Wt cerebral cortical neurons incubated during 24 h with 100 nM of soluble Aβ oligomers, alone or in the presence of 5 nM of uPA. Importantly, our previous studies indicate that with this experimental paradigm soluble Aβ does not induce neuronal death [19]. These experiments revealed that soluble Aβ decreases the abundance of β-Catenin in cerebral cortical neurons, and that this effect is abrogated by treatment with uPA (Fig. 1 C, D). To determine if uPA also has an effect of endogenously generated soluble Aβ on the abundance of β-Catenin, we studied its expression in a murine neuroblastoma cell line stably transfected with the APP-695 Swedish mutation (N2asw) that by rendering AβPP a more optimal substrate for BACE-1 increases the production of Aβ40 and Aβ42 [31]. Our data show that the low baseline level of β-Catenin in N2asw cells is increased after 15 min of treatment with 5 nM of uPA (Fig. 1E, F).
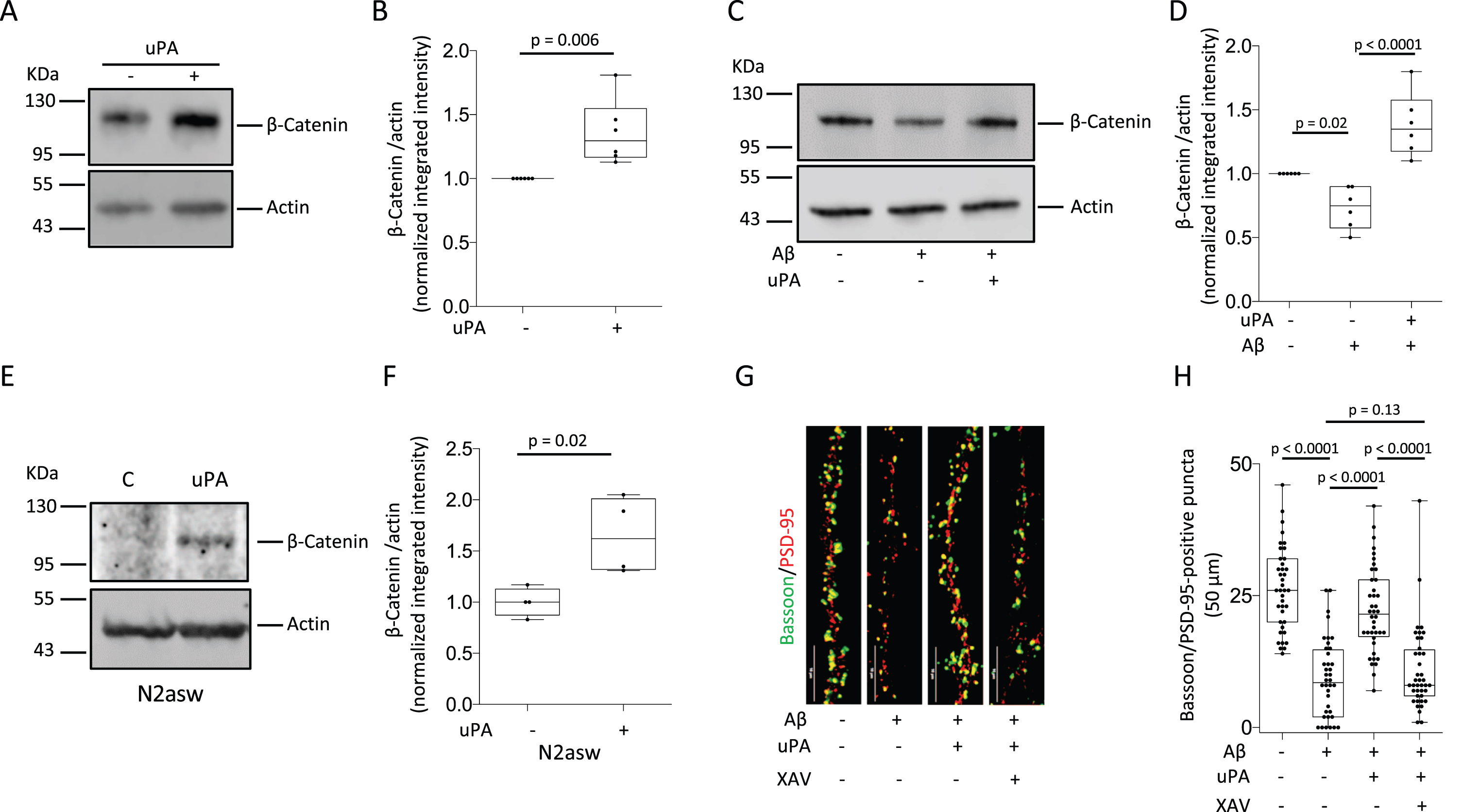
UPA protects the synapse from the harmful effects of soluble Aβ by reversing its inhibitory effect on the accumulation of β-Catenin. A, B) Representative western blot analysis (A) and quantification of the intensity of the band (B) of β-Catenin expression in wild-type (Wt) cerebral cortical neurons incubated 30 min with 5 nM of uPA or a comparable volume of vehicle (control). n = 6 observations per experimental group. Statistical analysis: two-tailed student’s t-test. C, D) Representative western blot analysis (C) and quantification of the intensity of the band (D) of β-Catenin abundance in Wt cerebral cortical neurons incubated 24 h with 100 nM of soluble Aβ oligomers or a comparable volume of vehicle (control), alone or in the presence of 5 nM of uPA. n = 6 observations per experimental group. Statistical analysis: one-way ANOVA with Holm-Sidak’s multiple comparisons test. E, F) Representative western blot analysis (E) and quantification of the intensity of the band (F) of β-Catenin abundance in N2asw cells incubated 15 min with 5 nM of uPA or a comparable volume of vehicle (control). n = 4 per experimental condition. Statistical analysis: two-tailed student’s t-test. G) Representative confocal micrographs at 60 X magnification of bassoon (green)- and PSD-95 (red)- immunoreactive puncta in extensions of Wt neurons treated during 24 h with 1μM of XAV-939 or vehicle (control), and then incubated for 24 h with 100 nM of soluble Aβ oligomers or vehicle (control), alone or in the presence of 5 nM of uPA. Scale bar: 10μm. H) Number of bassoon/PSD-95-positive puncta in extensions of 40 Wt neurons exposed to the experimental conditions described in G. Statistical analysis: one-way ANOVA with Holm-Sidak’s multiple comparisons test.
β-Catenin plays a pivotal role in the formation of synaptic contacts and our previous studies indicate that uPA protects the synapse from the harmful effects soluble Aβ [19]. Thus, to determine if β-Catenin mediates the protective effect of uPA on the synapse, we used confocal microscopy to quantify the number of intact synaptic contacts (bassoon/PSD-95-positive puncta) in extensions of Wt cerebral cortical neurons incubated during 24 h with soluble Aβ oligomers in the presence of 5 nM of uPA, alone or in combination with 1μM of XAV-939 (promotes β-Catenin degradation [32, 33]). We found that the effect of soluble Aβ on the number of intact synaptic contacts (a decrease from 26.41±7.8 in control cells to 9±7.46 in neurons incubated with soluble Aβ; p < 0.0001) was attenuated by treatment with uPA (22.28±8.2; p < 0.0001). However, the protective effect of uPA on the synapse was abrogated when the accumulation of β-Catenin was prevented with XAV-939 (10.85±7.83; p = 0.13 compared to control cells and p < 0.0001 compared to cells treated with uPA alone. n = 40 per experimental group; Fig. 1 G, H).
UPA/uPAR signaling antagonizes the inhibitory effect of Aβ on LRP6 by providing a Wnt-independent alternative pathway for LRP6 phosphorylation
When the Wnt β-Catenin pathway is inactive, GSK3β targets β-Catenin to proteasomal degradation by triggering its phosphorylation at Ser33/Ser37/Thr41 [10]. Thus, to investigate if uPA promotes the accumulation of β-Catenin by preventing its phosphorylation by GSK3β, we used an antibody that does not recognize phosphorylated β-Catenin, to study the expression of β-Catenin non-phosphorylated at Ser33/Ser37/Thr41 (also known as active β-Catenin, is not degraded by the proteasome) in Wt cerebral cortical neurons treated during 30 min with 5 nM of uPA or vehicle (control). These experiments revealed that uPA prevents GSK3β-induced β-Catenin phosphorylation, as denoted by an increase in the abundance of active β-Catenin in uPA-treated neurons (Fig. 2A, B).
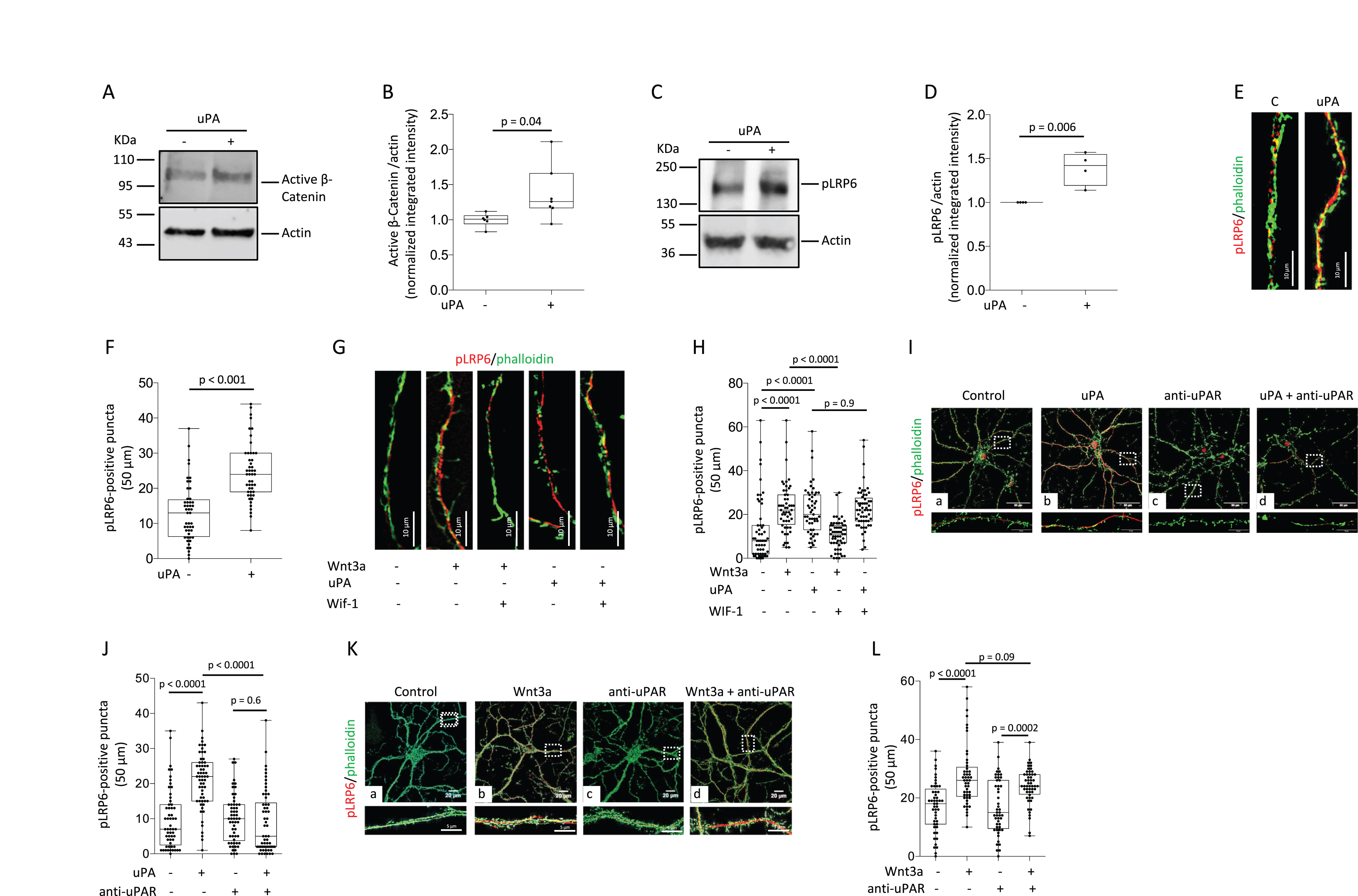
UPA binding to uPAR induces LRP6 phosphorylation via a Wnt-independent mechanism. A, B) Representative western blot analysis (A) and quantification of the intensity of the band (B) of the abundance of active β-Catenin in Wt cerebral cortical neurons incubated 30 min with 5 nM of uPA or vehicle (control). n = 6 per experimental condition. Statistical analysis: two-tailed student’s t-test. C, D) Representative western blot analysis (C) and quantification of the intensity of the band (D) of the abundance LRP6 phosphorylated at S1490 (pLRP6) in Wt cerebral cortical neurons incubated 15 min with 5 nM of uPA or a comparable volume of vehicle (control). n = 4 per experimental condition. Statistical analysis: two-tailed student’s t-test. E) Representative confocal micrographs at 60X magnification of pLRP6 (red) and phalloidin (green) staining in extensions of Wt cerebral cortical neurons incubated 15 min with 5 nM of uPA or a comparable volume of vehicle (control). Scale bar: 10μm. F) Number of pLRP6-positive puncta in extensions of 52 Wt cerebral cortical neurons exposed to the experimental conditions described in E. Statistical analysis: two-tailed student’s t-test. G) Representative confocal micrographs at 60X magnification of puncta immunoreactive to anti-pLRP6 antibodies (red) in extensions of Wt cerebral cortical neurons treated during 30 min with 10μg/ml of WIF-1 or vehicle (control), and then incubated 15 min with 5 nM of uPA or 80μg/ml of Wnt3a. Green is phalloidin. Scale bar: 10μm. H) Number of pLRP6-positive puncta in extensions of Wt cerebral cortical neurons exposed to the experimental conditions described in G. n = 52 - 58 cells per experimental condition. Statistical analysis: one-way ANOVA with Holm-Sidak’s multiple comparison’s test. I) Representative confocal micrographs at 60X magnification of puncta immunoreactive to pLRP6 antibodies (red) in extensions of Wt cerebral cortical neurons incubated during 15 min with vehicle (control; a), or 5 nM of uPA (b), or 6μg/ml of anti-uPAR antibodies (c), or a combination of uPA and anti-uPAR antibodies (d). Scale bars: 50μm. Lower panels correspond to a 1.5 magnification of the area depicted by the corresponding white squares. Green corresponds to phalloidin. J) Number of pLRP6-positive puncta in extensions of Wt cerebral cortical neurons exposed to the experimental conditions described in I. n = 57 - 60 cells per experimental condition. Lower panels correspond to a 1.5 magnification of the area depicted by the corresponding white squares. Statistical analysis: two-way ANOVA with Holm-Sidak’s multiple comparisons test. K) Representative confocal micrographs at 60X magnification of pLRP6-positive puncta (red) in extensions of Wt cerebral cortical neurons incubated during 15 min with vehicle (control; a), or 80μg/ml of Wnt3a (b), or 6μg/ml of anti-uPAR antibodies (c), or a combination of Wnt3a and anti-uPAR antibodies (d). Lower panels correspond to a 1.5 magnification of the area depicted by the corresponding white squares. Green corresponds to phalloidin. Scale bars: 50μm. L) Number of pLRP6-positive puncta in extensions of Wt cerebral cortical neurons exposed to the experimental conditions described in K. n = 49 –52 cells per experimental condition. Statistical analysis: two-way ANOVA with Holm-Sidak’s multiple comparisons test.
Because LRP6 phosphorylation at Ser1490 (pLRP6) triggered by binding of Wnts to LRP6 on the cell membrane prompts the recruitment and inactivation GSK3β, we then decided to investigate the effect of uPA on the abundance of pLRP6. We found that 15 min of incubation with 5 nM of uPA induces the phosphorylation of LRP6 in Wt cerebral cortical neurons (Fig. 2 C, D), and in line with these observations, our confocal microscopy studies show that the number of pLRP6-positive puncta in extensions of Wt cerebral cortical neurons increases from 13±1.81 in control cells to 24.63±1.2 in uPA-treated neurons (p < 0.0001; n = 48; Fig. 2E, F).
To determine if uPA increases the abundance of pLRP6 by promoting the binding of Wnts to LRP6 on the cell membrane, we studied the abundance of pLRP6 in extensions of Wt cerebral cortical neurons treated with either vehicle (control), or Wnt3a (Wnt3a binding to LRP6 triggers its phosphorylation in cerebral cortical neurons [34]), or uPA, or with the Wnt inhibitor WIF-1 in combination with either Wnt3a or uPA. We found that the number of pLRP6-positive puncta increases from 12.32±1.9 in control cells (n = 57) to 22.84±1.27 (n = 57) and 22.96±1.57 (n = 52), in Wnt3a- and uPA-treated cells, respectively (p < 0.0001 when control cells are compared to either Wnt3a- or uPA-treated neurons; Fig. 2 G, H). However, while WIF-1 abrogates the effect of Wnt3a (10.7±0.9; p < 0.0001 compared to cells treated with Wnt3a alone), it does not have an effect on uPA-treated neurons (22±2.3; p = 0.96 compared to cells treated with uPA alone, n = 51). To determine if uPAR mediates the effect of uPA and Wnt3a, we performed similar observations in the presence of anti-uPAR neutralizing antibodies. Our data indicate that uPAR mediates the effect of uPA (Fig. 2I, J) but not Wnt3a (Fig. 2K, L) on the abundance of pLRP6.
Because soluble Aβ inhibits Wnt-β-catenin pathway signaling by preventing LRP6 phosphorylation [35], we decided to investigate if uPA is capable to antagonize this effect. We used confocal microscopy to quantify the abundance of pLRP6 in extensions of Wt cerebral cortical neurons incubated during 24 h with 100 nM of soluble Aβ oligomers, and then treated during 15 min with 5 nM of uPA or 80μg/ml of Wnt3a. We found that the number of pLRP6-positive puncta decreases from 20.11±10.07 in control cells (n = 80) to 10.68±4.68 in neurons incubated with soluble Aβ (p < 0.0001). Significantly, this effect was antagonized by treatment with uPA (20.31±10.88; p < 0.0001, n = 74) but not Wnt3a (12.09±9.49; p = 0.6, n = 65; Fig. 3A, B).
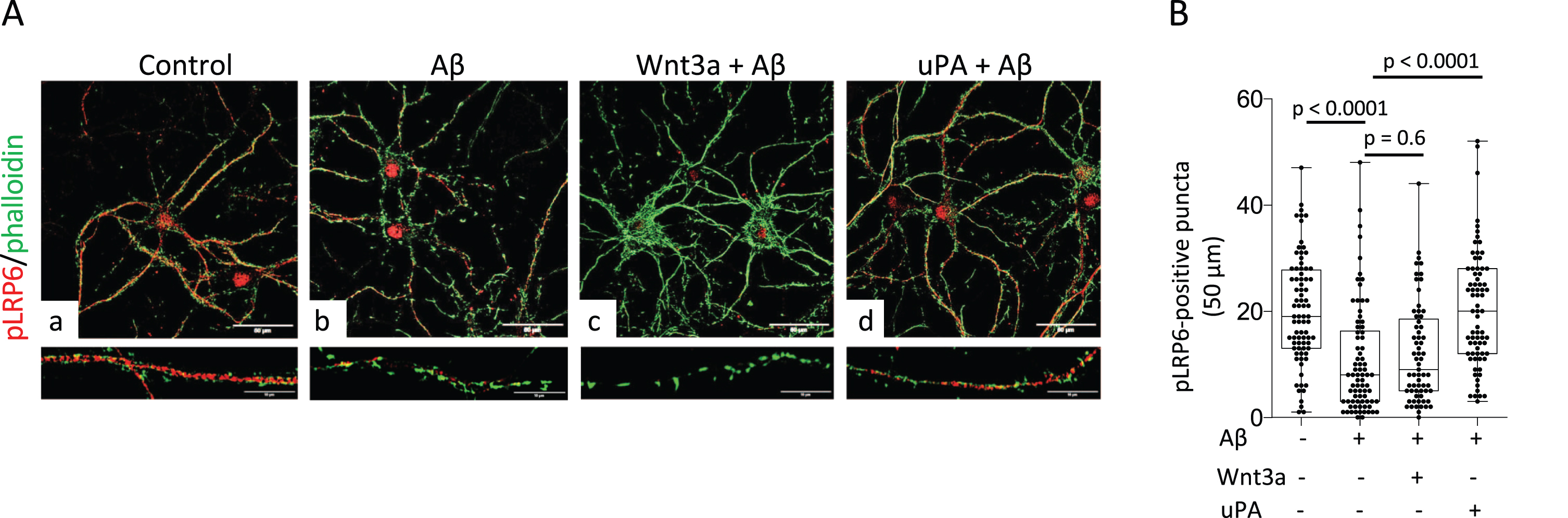
UPA reverses the inhibitory effect of soluble Aβ on LRP6 phosphorylation. A) Representative confocal micrographs at 60X magnification of Wt cerebral cortical neurons incubated during 24 h with vehicle (control; panel a), or 100 nM of soluble Aβ oligomers (b, c, and d), followed by 15 min of treatment with vehicle (control; b), or 80μg/ml of Wnt3a (c), or 5 nM of uPA (d). Lower panels correspond to a 1.5 magnification of the area depicted by the corresponding white squares. Scale bars: 50μm. B) Number of pLRP6-positive puncta in extensions of Wt cerebral cortical neurons exposed to the experimental conditions described in A. n = 80 (control cells), 78 (cells treated with Aβ and vehicle-control), 65 (cells treated with Aβ and Wnt3a), and 74 (cells treated with Aβ and uPA). Statistical analysis: one-way ANOVA with Holm-Sidak’s multiple comparison’s test.
UPA induces the nuclear translocation of β-Catenin via a plasminogen-independent mechanism
Because the cytoplasmic accumulation of β-Catenin is followed by its nuclear translocation [10], we used confocal microscopy to quantify the area of the nucleus immunoreactive to anti-β-Catenin antibodies in Wt cerebral cortical neurons treated during 60 min with 5 nM of uPA or a comparable volume of vehicle (control). Our data indicate that compared to control-treated cells, uPA induces a 23.71±5.1% increase in the area of the nucleus immunoreactive to anti-β-Catenin antibodies (Fig. 4A, B). To further characterize these observations, we studied the abundance of β-Catenin in nuclear extracts prepared from Wt cerebral cortical neurons treated during 60 min with 5 nM of either uPA, or its amino terminal fragment [ATF (Ser1 -Lys135): this fragment of uPA binds to uPAR as its contains its epidermal growth factor-like (EGF) domain, but is devoid of proteolytic activity because lacks the serine protease domain], or vehicle (control). We found that the cytoplasmic accumulation of β-Catenin induced by uPA treatment is followed by its nuclear translocation (Fig. 4 C, D) via a mechanism that does not require plasmin generation (Fig. 4E, F).
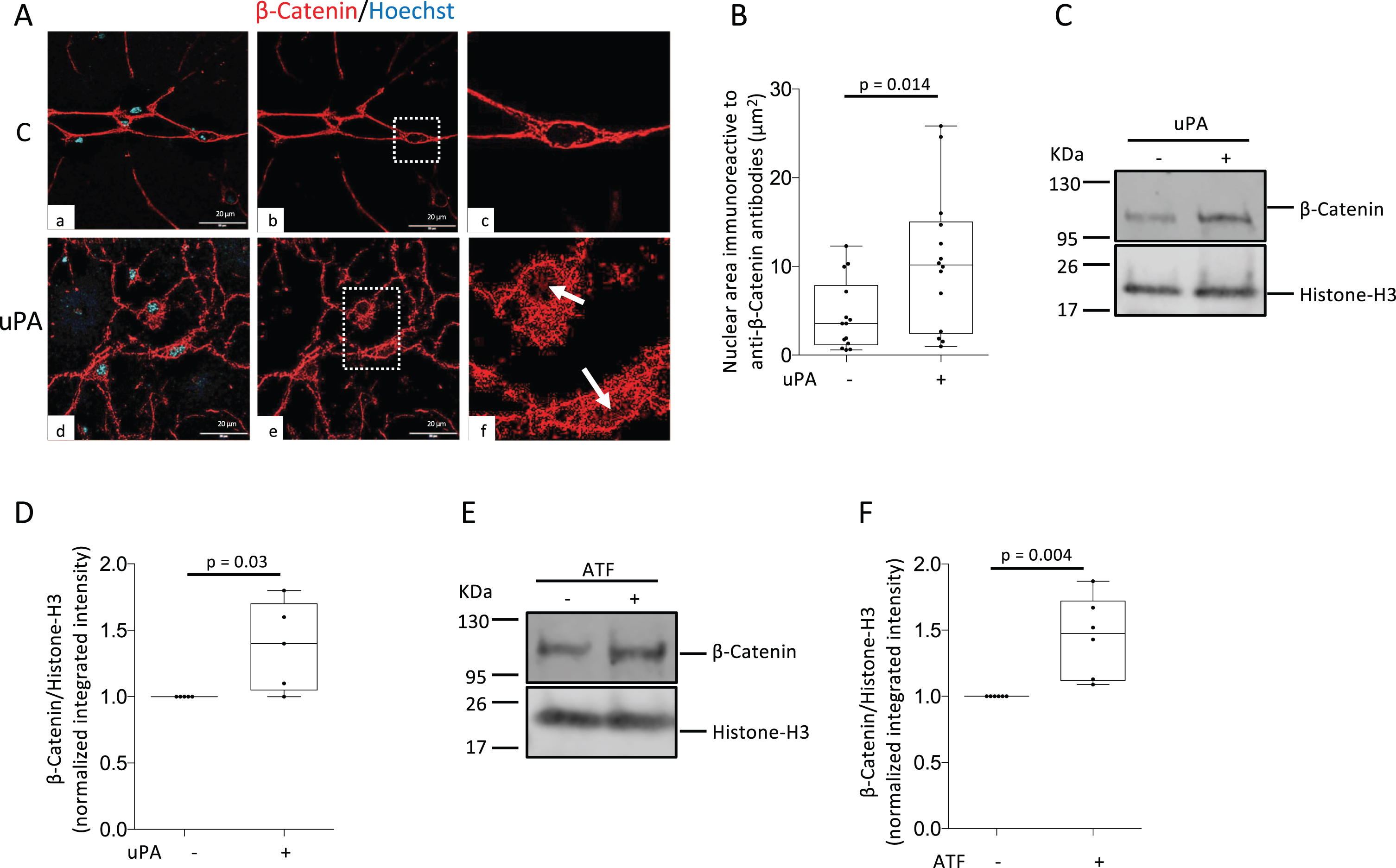
UPA induces the nuclear translocation of β-Catenin via a plasminogen-independent mechanism. A) Representative confocal micrographs of cerebral cortical neurons stained with anti-β-Catenin antibodies (red) and Hoechst (blue) following 60 min of treatment with vehicle (control: C; panels a-c) or 5 nM of uPA (panels d-f). Magnification in panels a, b, d, and e: 60 X. Panels c and f correspond to a 1.5 X magnification of the areas denoted by the white squares in b and e. Arrows in f denote examples of β-Catenin immunoreactivity in the nucleus of uPA-treated neurons. Scale bars: 20μm. B) β-Catenin-immunoreactive area in the nucleus of Wt neurons exposed to the experimental conditions described in A. n = 14 neurons from three different cultures per experimental group. Statistical analysis: two-tailed student’s t-test. C-F) Representative western blot analyses (C, E) and quantification of the intensity of the band (D, F) of β-Catenin expression in nuclear extracts prepared from Wt cerebral cortical neurons following 60 min of treatment with 5 nM of either uPA (C, D) or its amino terminal fragment (ATF: E, F), or with a comparable volume of vehicle (control). n = 5 in D and 6 in F. Statistical analysis in D and F: two-tailed student’s t-test.
UPA-triggered β-Catenin signaling inhibits the amyloidogenic processing of the AβPP
Because the nuclear translocation of β-Catenin represses BACE1 transcription by promoting binding of TCF-4 to its promoter [31], we studied the expression of BACE1 mRNA in Wt cerebral cortical neurons incubated during 24 h with 5 nM of uPA, alone or in the presence of XAV-939, which inhibits Wnt-β-Catenin signaling by promoting the degradation of β-Catenin. We found that uPA effectively decreases the abundance of BACE1 mRNA and that this effect is abrogated by XAV-939 (Fig. 5A, B). In line with these observations, our confocal microscopy studies show that the number of BACE-1-positive puncta in extensions from Wt cerebral cortical neurons decreases from 23.98±7.93 in control cells to 13.25±2.5 in neurons incubated with uPA (n = 48 per experimental group. p < 0.0001; Fig. 5 C, D). To study if the effect of uPA requires its proteolytic activity, we studied the expression of BACE1 mRNA in cerebral cortical neurons treated with 5 nM of uPA’s ATF (binds to uPAR but is devoid or proteolytic activity as it lacks uPA’s serine protease domain). These experiments revealed that the effect of uPA on BACE1 mRNA does not require the conversion of plasminogen to plasmin (Fig. 5E, F). Because BACE1 plays a pivotal role in the amyloidogenic processing of AβPP [36], we quantified BACE1 mRNA and the concentration of Aβ40 and Aβ42 in N2asw cells and their culture medium, respectively, following 24 h of incubation with 5 nM of uPA or vehicle (control). These experiments revealed that uPA effectively decreases the abundance of BACE1 mRNA (Fig. 5 G, H) and the generation of Aβ40 and Aβ42 (Fig. 5I) in cells with enhanced amyloidogenic processing of AβPP [31]. To determine the translational relevance of these observations, we quantified the concentrations of Aβ40 and Aβ42 in the cerebral cortex of 6-month-old Wt, uPA-/- and Plg-/- mice. In line with our previous observations, we found that genetic deficiency of uPA is associated with increased generation of Aβ40 and Aβ42 in the cerebral cortex via a plasminogen-independent mechanism (Fig. 5J).
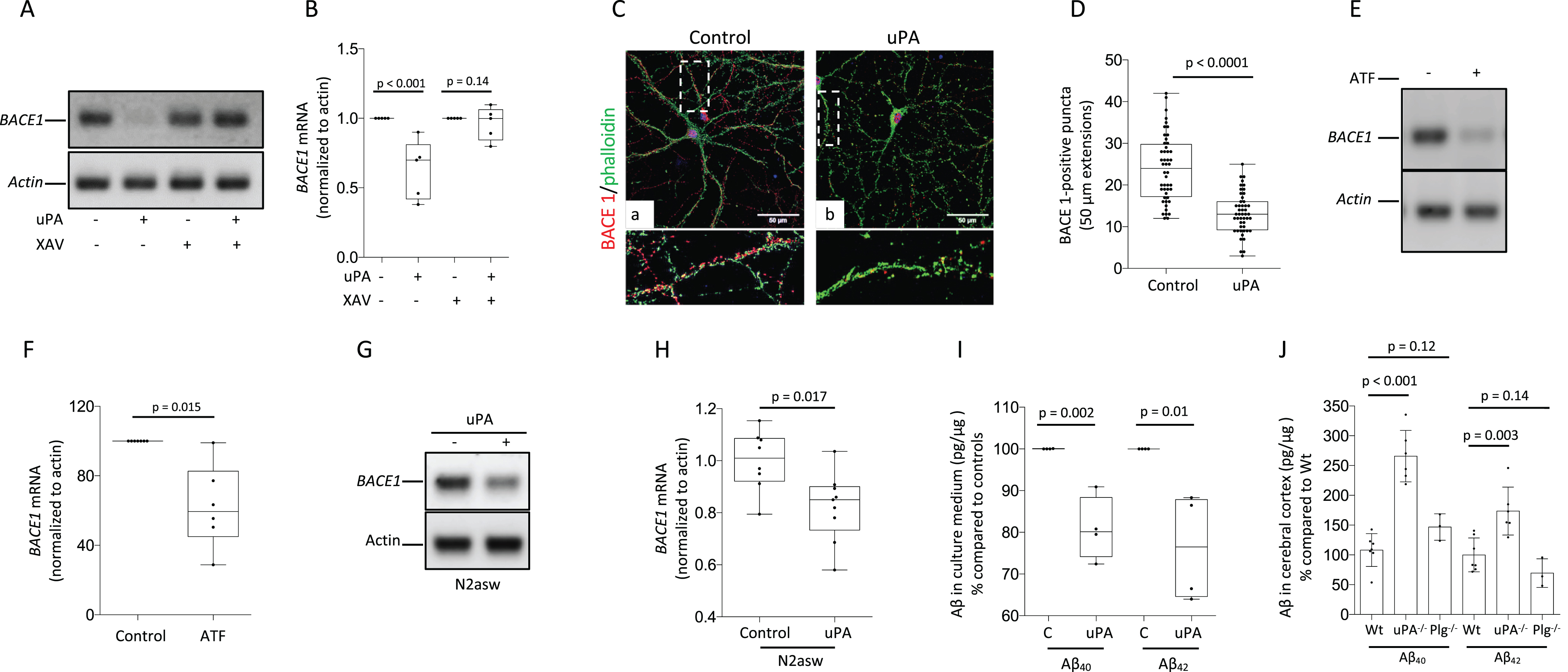
UPA-induced activation of the Wnt-β-Catenin pathway inhibits the expression of BACE-1 and the amyloidogenic processing of AβPP. A, B) Representative PCR analysis (A) and quantification of BACE1 mRNA normalized to actin and to BACE1 mRNA in control cells (B) in Wt cerebral cortical neurons incubated during 24 h with vehicle (control) or 1μM of XAV-939, followed by 24 h of treatment with either 5 nM of uPA or vehicle (control). n = 5 per experimental condition. Statistical analysis: two-way ANOVA with Holm-Sidak’s multiple comparison’s test. C) Representative confocal micrographs at 60X magnification of BACE 1 (red) and phalloidin (green) expression in Wt cerebral cortical neurons treated 24 h with 5 nM of uPA or vehicle (control). Lower panels correspond to 1.5 magnifications of the areas depicted by the white squares. Scale bar: 50μm. D) Number of BACE1-positive puncta in extensions of 48 Wt cerebral cortical neurons exposed to the experimental conditions described in C. Statistical analysis: two-tailed student’s t-test. E, F) Representative PCR analysis (E) and quantification of BACE1 mRNA normalized to actin and to BACE1 mRNA in control cells (F) in Wt cerebral cortical neurons incubated during 24 h with vehicle (control) or 5 nM of uPA’s ATF. n = 6 per experimental condition. Statistical analysis: two-tailed student’s t-test. G, H) Representative PCR analysis of BACE1 mRNA (G) and quantification of the intensity of the band (H) in N2asw cells treated during 24 h with 5 nM of uPA or a comparable volume of vehicle (control). n = 4 per experimental group. Statistical analysis: two-tailed student’s t-test. I) Aβ40 and Aβ42 in the culture medium of N2asw cells treated during 24 h with 5 nM of uPA or a comparable volume of vehicle (control). n = 4 per experimental group. Statistical analysis: two-tailed student’s t-test. J) Aβ40 and Aβ42 in the cerebral cortex of 6-month-old Wt (n = 7), uPA -/- (n = 6) and Plg-/- mice (n = 3). Statistical analyses: one-way ANOVA with Holm-Sidak’s multiple comparisons test.
DISCUSSION
Binding of the serine proteinase urokinase to its receptor (uPAR) on the cell surface catalyzes the conversion of plasminogen into plasmin [37, 38]. Despite the fact that for a long time it was thought that the only role of uPA was to induce cell migration by triggering plasmin-mediated degradation of the extracellular matrix [39], it was soon evident that uPA/uPAR binding also activates cell signaling pathways that promote cell survival, proliferation, and invasion [40, 41]. Here we show that by triggering Wnts-independent phosphorylation of LRP6 in cerebral cortical neurons, uPA is capable to antagonize the inhibitory effect of soluble Aβ on the Wnt-β-Catenin pathway. Furthermore, we report that uPA-induced activation of the Wnt-β-Catenin pathway protects the synapse from the harmful effect of soluble Aβ and inhibits the amyloidogenic processing of AβPP.
Different lines of experimental evidence have linked uPA to the pathogenesis of AD. Hence, it has been shown that the abundance of uPA is decreased in the synapse of cerebral cortical neurons of AD patients [19], that nucleotide polymorphisms of the uPA encoding gene (PLAU) are associated with late onset AD [42–44], and that the activity of uPA’s inhibitor (plasminogen activator inhibitor-1) is increased in the brain of AD patients [45–47]. It has been reported that uPA plays a role in the pathogenesis of AD by inducing plasmin-mediated cleavage of insoluble Aβ-containing extracellular plaques [27]; and our data indicate that uPA-induced plasmin-independent activation of the Wnt-β-Catenin protects cerebral cortical neurons from the harmful effects of soluble Aβ and prevents the amyloidogenic processing of AβPP. Together, these data indicate that uPA has both a plasmin-mediated effect on the cleavage of insoluble Aβ-containing extracellular plaques and a plasmin-independent protective effect on soluble Aβ-induced inhibition on β-Catenin signaling. Remarkably, we found that uPA has a rapid effect on the abundance of β-Catenin, which agrees with previous reports on the effect of uPA treatment [48] and mechanical strain [49] on the cytoplasmic accumulation of β-Catenin in cerebral cortical astrocytes and CIMC-4 cells harvested from mouse calvariae, respectively.
Based on these observations, we postulate that the reported decreased abundance of uPA in the synapse of AD patients [19] allows soluble Aβ to silence the Wnt-β-Catenin pathway, thus triggering a sequence of events that result in synaptic loss. In line with this hypothesis, our data indicate that by providing an alternative pathway for LRP6 phosphorylation treatment with uPA antagonizes the inhibitory effect of soluble Aβ on the Wnt-β-Catenin pathway, that uPA protects the synapse from the harmful effects of soluble Aβ, and that this protective effect requires the cytoplasmic accumulation and nuclear translocation of β-Catenin. The translational relevance of these observations is underscored by a wealth of neuropathological studies indicating that by their ability to impair NMDAR-dependent long term potentiation [50], enhance long-term depression [4], and cause mitochondrial damage [51] and synaptic loss [7], soluble Aβ contributes to the development of cognitive decline in AD patients [52–54]. Thus, our data suggest that restoring the synaptic abundance of uPA may be a potential therapeutic strategy to prevent disease progression in AD.
LRP6 is an essential coreceptor for the activation of the Wnt-β-Catenin pathway [34]. More specifically, binding of Wnt ligands to Fz receptors and LRP6 on the cell membrane triggers the oligomerization of LRP6 in signalosomes [55], which then serve as adaptors for the recruitment of cytoplasmic kinases that phosphorylate the intracellular domain of LRP6 at Ser1490 [56]. This sequence of events plays a central role in the pathogenesis of AD. Indeed, soluble Aβ inhibits Wnts-induced LRP6 phosphorylation [12], two single-nucleotide polymorphisms and an alternative splice variant of LRP6 have been associated with increased risk of developing AD [57], and deficient LRP6 signaling accelerates AβPP processing to Aβ, impairs synaptic plasticity [58], and causes age-dependent synaptic loss and memory deficits [59]. We found that uPA and Wnt ligands (Wnts) activate LRP6 by different mechanisms. More specifically, our data show that while binding of uPA to uPAR induces LRP6 phosphorylation, Wnts trigger LRP6 phosphorylation by a uPAR-independent pathway. More importantly, the studies presented here indicate that uPA but not Wnts antagonize the inhibitory effect of soluble Aβ on LRP6 phosphorylation. These findings have important translational implications because they show that uPA is capable to antagonize soluble Aβ-induced inhibition of Wnt-β-Catenin signaling thus preventing its harmful effects on synaptic structure and function. Once phosphorylated, LRP6 recruits GSK3β and inhibits its effect on β-Catenin, thus enabling its cytoplasmic accumulation and subsequent nuclear translocation [60]. In line with these observations, our data indicate that uPA promotes the cytoplasmic accumulation and nuclear translocation of β-Catenin. Importantly, the effect of uPA on the nuclear translocation of β-Catenin does not require the conversion of plasminogen into plasmin, as it is also observed following treatment with uPA’s ATF (binds to uPAR but is devoid of proteolytic activity).
Aβ peptides are generated by sequential proteolytic cleavage of AβPP [61]. More specifically, α-secretase cleaves AβPP at the Aβ sequence, thus preventing the formation of Aβ peptides [36]. However, AβPP molecules that are not cleaved by α-secretase are cleaved by β-secretase 1 (BACE1) and γ-secretase to generate Aβ peptides, most of which are 40 and 42 amino acids in length (Aβ40 and Aβ42, respectively) [61, 62]. LRP6 deletion and the ensuing inactivation of the Wnt- β-Catenin pathway accelerate the generation of Aβ peptides [58], which in turn have a harmful effect on axonal boutons, dendritic spines [63, 64], synaptic transmission [65], and network activity [7]. In agreement with these data, LRP6 phosphorylation and Wnt-β-catenin pathway activation abrogate the amyloidogenic processing of AβPP [16, 66]. More specifically, LRP6 phosphorylation inhibits the effect of GSK3β on the cytoplasmic accumulation and nuclear translocation of β-Catenin. In turn, binding of β-Catenin to TCF4 in the nucleus represses the transcription of BACE1. In line with these observations, Wnt-β-Catenin inhibition increases BACE1 expression and activity [31].
Our work indicates that uPA decreases the abundance of BACE1 mRNA and protein in cerebral cortical neurons, and that this effect is mediated by β-Catenin signaling as it is abrogated by prevention of its cytoplasmic accumulation with XAV-939. Additionally, we found that treatment with uPA decreases the abundance of BACE1 mRNA and the production of Aβ peptides in a cell line with the APP-695 Swedish mutation that increases the generation of Aβ40 and Aβ42 by rendering AβPP a more optimal substrate for BACE 1 [31]. Remarkably, our data indicates that the effect of uPA on BACE1 mRNA expression does not require its proteolytic activity, as it was also observed following treatment with uPA’s ATF fragment. Here is important to keep in mind that BACE1 not only mediates the amyloidogenic processing of AβPP, but also plays a central role in several physiological processes such as axon myelination and guidance, regulation of voltage-gated sodium channel function and hippocampal astrogenesis [67]. Thus, future research should be performed to determine if by repressing BACE1 transcription, uPA/uPAR signaling also has an effect on these processes. Independently of these considerations, the in vivo relevance of our observations is indicated by our experiments revealing increased Aβ40 and Aβ42 production in the cerebral cortex of aged uPA-/- but not Wt or Plg-/- mice, supporting our observation that uPA inhibits the amyloidogenic processing of AβPP via a mechanism that does not require plasmin generation.
In conclusion, the data presented here show that uPA-induced, uPAR-mediated, Wnts-independent phosphorylation of LRP6 provides an alternative pathway for LRP6 phosphorylation that antagonizes the inhibitory effect of Aβ on the Wnt-β-Catenin pathway (Fig. 6). These results indicate that the ensuing activation of β-Catenin signaling pathway not only protects the synapse from the harmful effects of soluble Aβ but also inhibits the amyloidogenic processing of AβPP. This is a novel role for uPA in the central nervous system with potential translational implications for the treatment of AD patients.
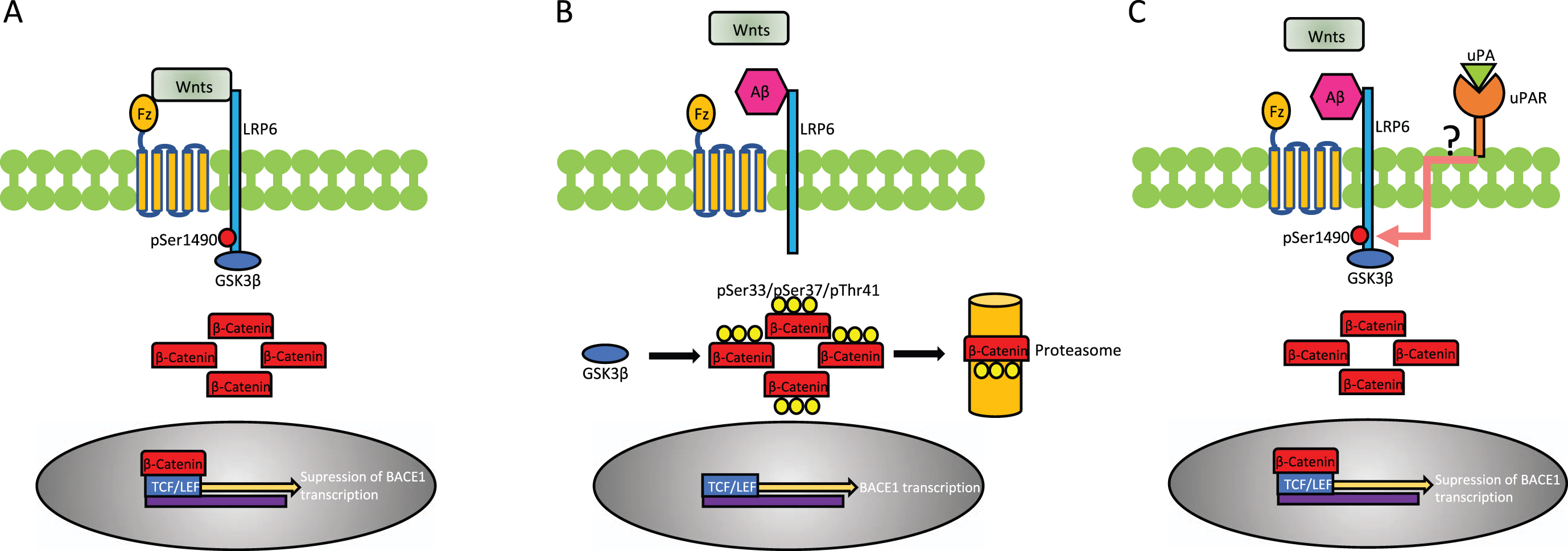
Schematic representation of the proposed mechanism whereby uPA/uPAR binding antagonizes the inhibitory effect of soluble Aβ on the Wnt-β-catenin pathway. A) Under physiological conditions, binding of Wnt ligands (Wnts) to Frizzleds (Fz) receptors and the low-density lipoprotein receptor-related protein-6 (LRP6) triggers the phosphorylation of the intracellular domain of LRP6 at Ser1490 (pLRP6). In turn, pLRP6 recruits and inactivates GSK3β, rendering it unable to phosphorylate β-Catenin and thus allowing its cytoplasmic accumulation and subsequent nuclear translocation where after binding to the transcription factor T cell factor (TCF)/lymphoid enhancer factor (LEF) represses the transcription of BACE1. B) Soluble Aβ prevents the binding of Wnts to LRP6 and Fz receptors, thus averting LRP6 phosphorylation. This allows GSK3β to target β-Catenin for proteasomal degradation by phosphorylating it at Ser33/Ser37/Thr41. These sequence of events permit the transcription of BACE1. C) UPA binding to uPAR provides an alternative Wnt-independent pathway for LRP6 phosphorylation, via an as yet unidentified coreceptor (question mark), hence allowing β-Catenin to accumulate, translocate to the nucleus and repress BACE1 transcription, despite binding of soluble Aβ to LRP6 and Fz receptors.