
Abstract
Background:
Aging is characterized by systemic alterations and forms an important risk factor for Alzheimer’s disease (AD). Recently, it has been indicated that blood-borne factors present in the systemic milieu contribute to the aging process. Exposing young mice to aged blood plasma results in impaired neurogenesis and synaptic plasticity in the dentate gyrus, as well as impaired cognition. Vice versa, treating aged mice with young blood plasma rescues impairments associated with aging.
Objective:
Whether blood-borne factors are sufficient to drive impairments outside the dentate gyrus, how they impact neurophysiology, and how the functional outcome compares to impairments found in mouse models for AD is still unclear.
Methods:
Here, we treated adult mice with blood plasma from aged mice and assessed neurophysiological parameters in the hippocampal CA1.
Results:
Mice treated with aged blood plasma show significantly impaired levels of long-term potentiation (LTP), similar to those present in APP/PS1 mice. These impaired levels of LTP in plasma-treated mice are associated with alterations in basic properties of glutamatergic transmission and the enhanced activity of voltage-gated Ca2+ channels.
Conclusion:
Together, the data presented in this study show that blood-borne factors are sufficient to drive neurophysiological impairments in the hippocampal CA1.
INTRODUCTION
Aging is characterized by cognitive decline and is a major risk factor for many neurodegenerative diseases, including Alzheimer’s disease (AD) [1]. As the population ages, cognitive decline is expected to become an increasing socio-economic problem. Understanding the biological basis of cognitive decline is therefore of critical importance. Cognition comprises a large variety of processes, ranging from short-term memory to verbal knowledge [2]. Studies indicate that aging individuals generally show impaired working and spatial memory, short-term recall, and information processing [3, 4]. Brain regions involved in encoding these cognitive processes include the hippocampus and the prefrontal cortex [5, 6]. The activity of these brain regions decreases with age, as shown by decreased functional MRI activity when aged individuals perform memory-related tasks [7, 8].
The prefrontal cortex and hippocampus display variable degrees of altered dendritic morphology, impaired neuronal activity, and reduced synaptic plasticity in the aging brain [9, 10]. Subregions of the hippocampus that are most affected include the dentate gyrus (DG) and the area cornu ammonis 1 (CA1). Early reports indicate that the DG suffers from impaired neurogenesis, increasing levels of neuronal loss, and altered dendritic morphology with age [11, 12]. The CA1 was initially assumed to be largely resistant to age-related neuronal death and alterations in spine density [13]. Yet, there are indications that dendritic branching in the hippocampal CA1 is negatively affected by aging [14]. Similarly, the DG and CA1 experience a substantial decline in synaptic plasticity in AD [15, 16], associated with alterations in morphological complexity and a reduction in the postsynaptic spine density [17, 18]. CA1 pyramidal neurons are additionally subjected to several age-related neurophysiological impairments in Ca2+ homeostasis. The increased expression [19] and function [20] of voltage-gated Ca2+ channels (VGCCs) are well-documented in aged CA1 pyramidal neurons. This particularly concerns the expression and function of high-voltage-activated (HVA) L-type currents [19–22]. Yet, also the increased activity of low-voltage-activated T-type currents has been implicated in the aging basal forebrain [23]. Aged CA1 pyramidal neurons are furthermore characterized by unchanged or reduced Ca2+ clearance mechanisms [24–26]. These changes directly impact Ca2+ conductance [20, 27] and the intracellular [Ca2+] during aging and neuronal activity [28, 29]. This contributes to an enhanced after-hyperpolarizing potential (AHP) [27, 30] and reduced N-methyl-D-aspartate (NDMA) receptor functioning [31], affecting neuronal excitability [32], synaptic plasticity [29, 33], and ultimately cognitive function [34]. Additional neurophysiological properties, such as resting membrane potential, membrane resistance, and action potential threshold generally remain unchanged [9, 36]. A similar shift in Ca2+ homeostasis has been demonstrated in mouse models for AD [37–40].
Additional evidence for altered Ca2+ homeostasis in the aging hippocampus is derived from studies that indicate the decreased presence of cytosolic calcium-binding (buffering) proteins [41], and age-related impairments in Ca2+ extrusion through Na+/Ca2+ exchanger and Ca2+-ATPase dysfunction [42]. Transcriptional studies further reveal the altered expression of genes that are mediated by the intracellular [Ca2+]. These include the immediate-early genes Narp, Arc, and cAMP-responsive element-binding protein (CREB), all crucial for the induction of long-term synaptic plasticity [43–45]. Since Ca2+ acts as a second messenger and is essential for many biological processes, including neurogenesis, cell proliferation and apoptosis, and modulation of neuronal excitability [46], prolonged variations in the intracellular [Ca2+] will have detrimental consequences for healthy brain functioning. Indeed, the hippocampal CA1 demonstrates age-related impairments in long-term potentiation (LTP) and an increasing susceptibility to the induction of long-term depression [47–49], two important correlates of memory processes [50].
The causes of these alterations in gene expression, VGCC current density, and subsequent changes in Ca2+ homeostasis in the aging milieu are still unclear. Recently, it was suggested that blood-borne factors are involved in the induction of age-related impairments in the brain [51, 52]. It was demonstrated that exposure of young mice to blood plasma of aged mice decreases hippocampal neurogenesis, reduces LTP in the DG, and impairs contextual and spatial memory performance [51]. Vice versa, exposure to young blood plasma improves LTP in the DG and cognitive performance in aged mice [52]. These improvements are associated with increased levels of phosphorylated CREB, which activity depends on intracellular Ca2+ homeostasis [53]. Similarly, exposure to young blood plasma rescued the expression of synaptophysin and the calcium-binding protein calbindin in the DG of AD model mice [54]. Additional transcriptomic analyses revealed a significant enrichment of genes encoding proteins involved in calcium ion binding, indicating that blood-borne factors can act on signaling pathways related to Ca2+ homeostasis.
Whether blood-borne factors are sufficient to drive impairments outside the DG and how they impact neurophysiology is still unclear. To answer these questions, we systemically injected 5-month-old (M) C57BL/6 wild-type (WT) mice with blood plasma obtained from aged 18M-22M C57BL/6 WT mice. We then compared a selection of neurophysiological parameters in the hippocampal CA1, including the capability to induce synaptic plasticity, pre- and post-synaptic functioning, and VGCC activity. We additionally assessed the capability to induce synaptic plasticity in the hippocampal CA1 of APP/PS1 mice to see how plasma-induced alterations compare to impairments that exist in a mouse model for AD. The hippocampal CA1 is suitable for assessing plasma-induced neurophysiological impairments, as the hippocampus is actively involved in learning and memory, and CA1 pyramidal neurons display age-related changes in the intracellular [Ca2+] upon neuronal activity [28]. Overall, the results presented here indicate neurophysiological changes in the hippocampal CA1 following systemic treatment with aged blood plasma and we propose altered VGCC activity as a potential mechanism of action.
METHODS
Animals
Experiments were performed using adult (5M - 6M) and aged (18M–22M) male and female C57BL/6 mice (Charles River [strain C57BL/6; strain code 027; https://www.criver.com/) and hemizygous double-transgenic APPswe/PS1dE9 (APP/PS1) male and female mice [55] (The Jackson Laboratory, strain B6.Cg-Tg(APPswe, PSEN1dE9)85Dbo/Mmjax; stock number 34832; https://jaxmice.jax.org/). APP/PS1 mice were backcrossed onto a C57BL/6 background and were maintained as hemizygotes by crossing with C57BL/6 mice. APP/PS1 mice express a humanized form of the amyloid-β protein precursor containing the K595N/M596L Swedish mutation, and the presenilin 1 (PS1) splicing enzyme with exon-9 deleted (PS1dE9) [55, 56]. All animals were bred on location, handled regularly and group housed under standard laboratory conditions (12-h light/dark cycle, 21°C, 50% humidity) in the presence of nesting material. Food and water were provided ad libitum. Experiments were performed across three separate cohorts and in accordance with protocols and guidelines approved by the Institutional Animal Care and Use Committee (IvD Utrecht) operating under standards set by EU Directive 2010/63/EU.
Plasma collection and injection
Following the administration of an overdose Euthanimal®, aged blood was acquired from 18M to 22M C57BL/6 mice by cardiac puncture using ethylenediaminetetraacetic acid- (EDTA, 0.5M) coated syringes and then collected in EDTA-coated Eppendorf tubes. Plasma was obtained by centrifugation at 1000 g. All samples were stored at –80°C until further use. To remove EDTA, plasma samples were pooled and dialyzed using 3 mL 3.5-kDa Slide-A-Lyzer dialysis cassettes (cat. no. 66110, ThermoFisher Scientific) in phosphate-buffered saline (PBS, pH 7.4, cat. no. 10010031, Gibco®). Dialyzed plasma samples were then administered to 5M WT animals. Animals received 8 systemic injections with plasma (150μl) intravenously via the tail vein over a period of 28 d (2 injections/week), starting at 5M±1 week. During administration, animals were restrained using a Broome-style rodent restrainer (Plas Labstrademark 551BSRR). Control and APP/PS1 animals received 8 systemic injections of PBS (150μl) over a similar period. As a control, we used PBS instead of age-matched plasma due to ethical considerations to minimize the number of animals needed. Previous studies indicate that saline treatment and exposure to aged-matched plasma have a similar effect [57]. The day after the final injection, at 6M±1 week, animals were used for electrophysiological recordings.
Extracellular recordings
Slice preparation
Extracellular field potential recordings were performed on submerged coronal slices (350μm) containing the hippocampus. Mice were decapitated following anesthesia with isoflurane. Brains were rapidly dissected after which slices were cut on a Leica VT1000S vibratome using slicing artificial cerebrospinal fluid (aCSF) containing (in mM, 300 mOsm, pH adjusted 7.35) 139 Choline Cl, 3.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 6 MgSO4, 10 Glucose, 25 NaHCO3. Slices were subsequently incubated in recording aCSF containing (in mM, 300 mOsm, pH adjusted 7.35) 120 NaCl, 3.5 KCl, 1.25 NaH2PO4, 2.5 CaCl2, 1.3 MgSO4, 10 Glucose, 25 NaHCO3 at 32°C for±10 min. Slices were then left to recover at room temperature (RT) in recording aCSF for at least 1 h. All recordings were performed in recording aCSF at 32°C.
Field excitatory postsynaptic potentials (f-EPSPs)
To evoke a postsynaptic response, the Schaffer collaterals were stimulated with a concentric bipolar tungsten stimulus electrode (World Precision Instruments, Berlin, Germany). Extracellular f-EPSPs were recorded by placing glass electrodes (2 - 3 MΩ) containing recording aCSF in the stratum radiatum of the CA1 hippocampal area. 0.1 ms biphasic stimuli were applied at 0.033 Hz using a DS4 current generator (Digitimer, Welwyn Garden City, UK). Stabilization of synaptic responses was monitored over a period of±30 min followed by an input-output (IO) curve. The half-maximal stimulus intensity (
Whole-cell recordings
Slice preparation
Coronal slices (250μm) containing the hippocampus were cut on a Leica VT1000S vibratome using slicing aCSF containing (in mM, 300 mOsm, pH adjusted 7.35) 92 Choline Cl, 2.5 KCl, 1.2 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7 MgSO4, 20 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), 25 Glucose, 20 N-methyl-D-glucamine (NMDG), 10 Sodium Ascorbate, 2 Thiourea, 3 Sodium Pyruvate, 3.1 N-acetyl-L-cysteine. Slices were then incubated in slicing aCSF to recover from slicing at 36°C for 10 min. Slices were put in incubation aCSF at RT containing (in mM, 300 mOsm, pH adjusted 7.35) 92 NaCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 2 CaCl2, 2 MgSO4, 20 HEPES, 25 Glucose, 3 Sodium Ascorbate, 2 Thiourea, and 3 Sodium Pyruvate for at least 1 h or until further use. All whole-cell recordings were performed on pyramidal neurons located in the hippocampal CA1 using recording aCSF containing (in mM, 300 mOsm, pH adjusted 7.35) 124 NaCl, 2.5 KCl, 1 NaH2PO4, 26 NaHCO3, 2.5 CaCl2, 1.3 MgSO4, 5 HEPES, and 11 Glucose at 32°C. CA1 pyramidal cells were identified by their typical shape, their location in the stratum pyramidale, and their membrane capacitance (Cm) as indicated by amplifier readings. Cells were patched at –70 mV resting membrane potential in voltage-clamp configuration. Immediately after break-in, cells were allowed to rest for 5 min for the recording to stabilize.
(m)EPSCs recordings
To assess miniature excitatory postsynaptic currents (mEPSCs), tetrodotoxin (TTX, 1μM, cat. no. L8503, Latoxan) was added to the recording aCSF to block Na+ channels and prevent the initiation of action potentials. When recording evoked NMDA and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) responses, (–)-bicuculline methobromide (BIC, 0.5μM, cat no. sc-200436, Santa Cruz Biotechnology) was additionally added to prevent γ-aminobutyric-acid-type-A (GABAa) receptor-mediated currents. Cells were patched using borosilicate glass (Science Products) electrodes (3–4 MΩ) containing (in mM, 300 mOsm, pH adjusted 7.3) 139 Cs methanesulfonate, 5 CsCl, 2 MgCl2, 0.2 EGTA, 10 HEPES, 10 Creatine Phosphate, 4 Na2-ATP, and Na3-GTP. AMPA-mediated excitatory postsynaptic currents (EPSCs) and mEPSCs were recorded at –70 mV membrane potential. NMDA-mediated currents were recorded at +40 mV membrane potential. EPSCs were evoked by a 0.1 ms biphasic stimulus pulse administered similarly as described above.
Ca2+ currents
To record Ca2+ currents, recording aCSF additionally contained TTX (1μM, cat. no. L8503, Latoxan) to block Na+ channels and tetraethylammonium-Cl (10 mM, cat no. T2265, Sigma), 4-aminopyridine (5 mM, cat no. A78403, Sigma), and CsCl (5 mM, cat no. C4036, Sigma) to block K+ channels. Cells were patched using borosilicate glass (Science Products) electrodes (3–4 MΩ) containing (in mM, 300 mOsm, pH adjusted 7.3) 141 Cs methanesulfonate, 10 HEPES, 5 BAPTA, 2 Mg-ATP, and 0.1 Na3-GTP. Cells were voltage-clamped at –70 mV and series resistance (Rs) was compensated for approximately 70%. Ca2+ currents were evoked by inducing 200 ms depolarizing pulses in 10 mV steps varying from –80 mV to 20 mV [59]. Before the 200 ms pulse, a 3 s pre-potential step at –100 mV was applied to remove all Ca2+ channel steady-state inactivation. A period of 10 s was allowed between successive pulses to promote full extrusion of Ca2+ which had entered upon depolarization.
For all whole-cell patch-clamp recordings, a single cell per brain slice was patched. All obtained recordings were amplified and low-pass filtered at 5 kHz using an Axopatch 200B amplifier (Axon). Recordings were digitized and stored using Clampex 10.6 software (Axon). Recordings were excluded from analysis when the series resistance fluctuated >15% while recording.
All recordings (i.e., f-EPSP and whole-cell) were performed under visual guidance using an upright microscope (SliceScope Pro 6000, Scientifica) equipped with oblique illumination and a 4x (f-EPSP) and 40x (whole-cell) water immersion objective. Recording solutions were kept at their indicated temperature using an automatic temperature controller (TC-324B, Warner Instrument Corporation) combined with a peristaltic flow pump (Watson-Marlow, 120 S/DM2) at a±2 ml flow rate. All solutions mentioned above were continuously perfused with 95% O2 and 5% CO2.
Data processing and analysis
All obtained recordings were analyzed using Clampfit 10.6 (Axon). Post-processing of the data was performed using a combination of Microsoft Excel (2016) and custom-written MatLab (version 2019b) scripts. Cm used for group comparison was calculated offline based on Rs and tau time constants. When analyzing extracellular f-EPSPs recordings, we quantified the slope (10% –90%) of the postsynaptic response to minimize the effect of peak contamination. The slopes were measured after which relative changes were estimated by normalizing the post-tetanic data against baseline. The NMDA/AMPA ratio at
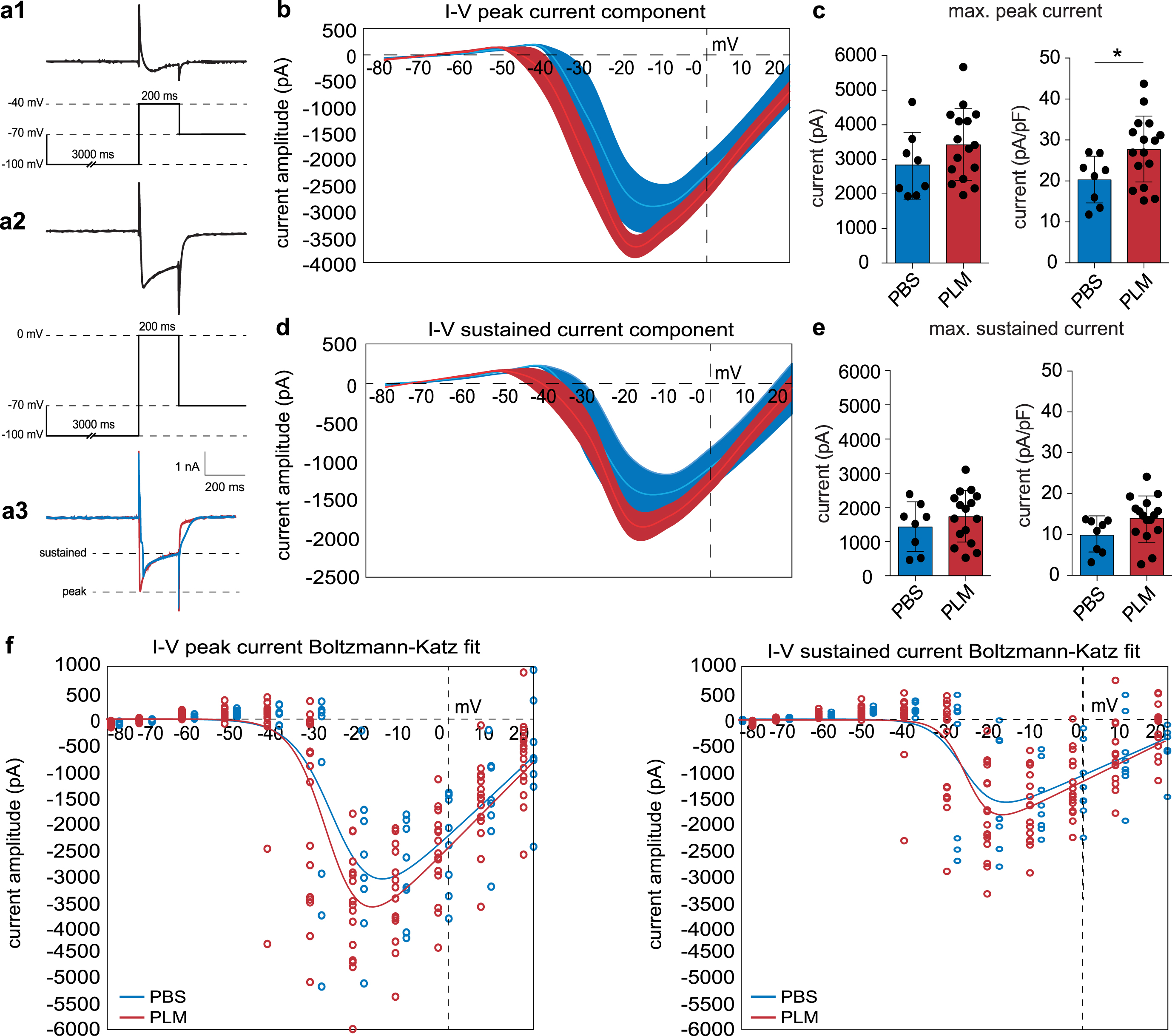
Treatment with aged blood plasma enhances VGCC currents in CA1 pyramidal neurons. a) Experimental approach. Ca2+ currents were isolated in the presence of 1μM TTX, 10 mM tetraethylammonium-Cl, 5 mM 4-aminopyridine and 5 mM CsCl. Ca2+ currents were evoked by administrating 10 mV depolarizing steps (–80 mV–20 mV) subsequent to a 3 s hyperpolarizing step (–100 mV). a1 shows a typical response at –40 mV. a2 shows a typical response at 0 mV. a3 shows representative Ca2+ traces at –15 mV for PBS and PLM in blue and red, respectively. The peak and sustained component were quantified at every voltage step and used for further analyses. b) The I–V relation of the peak component displaying the Ca2+ current amplitude (pA) as a function of the membrane potential (mV) at every depolarizing step, for both PBS (blue) and PLM (red) conditions. c) Quantification of the maximum peak current (at –15 mV) indicating both the absolute current (left, two-tailed Student’s t-test, PBS (2836.00±959.20 pA, nc = 8, N = 4) versus PLM (3421.00±1032.00 pA, nc = 16, N = 6), p = 0.1943) and the current compensated for cell capacitance (right, two-tailed Student’s t-test, PBS (20.27±5.86 pA/pF, nc = 8, N = 4) versus PLM (27.68±8.30 pA/pF, nc = 16, N = 6), p = 0.0348). d) The I–V relation of the sustained component displaying the Ca2+ current amplitude (pA) as a function of the membrane potential (mV) at every depolarizing step, for both PBS (blue) and PLM (red) conditions. e) Quantification of the maximum sustained current (at –15 mV) indicating both the absolute current (left, two-tailed Student’s t-test, PBS (1428.00±709.00 pA, nc = 8, N = 4) versus PLM (1730.00±751.00 pA, nc = 16, N = 6), p = 0.3560) and the current compensated for cell capacitance (right, two-tailed Student’s t-test, PBS (9.86±4.15 pA/pF, nc = 8, N = 4) versus PLM (13.99±5.51 pA/pF, nc = 16, N = 6), p = 0.0754). f) Graphs displaying the Bolzmann-Katz model fit (solid lines) to the recorded peak and sustained current component values from PBS and PLM mice. Individual values from PBS and PLM mice to which the model was fitted are represented by the blue and red dots, respectively. For each fit the standard error and 95% confidence interval were determined and are presented in Table 2.
Statistics
Data are presented as mean values±standard deviation (SD). Statistical comparisons between two groups were performed using a two-tailed (un)paired Student’s t-test. Analyses on more than two groups were done using a one-way analysis of variance (ANOVA). Whenever a F-test or Brown-Forsythe test for equal variances indicated significantly different SDs, non-parametric equivalents were used. Statistical significance for the data on Ca2+ current kinetics was tested by performing an independent two-tailed two-sample Student’s t-test using the obtained estimates and SDs (Table 2). The t-score was determined according to:
Fitting the current-voltage relation for the peak and sustained current components to the Boltzmann-Katz equation obtained estimates for the slope (k) and half-maximum activation potential (V1/2)
CA1 pyramidal neuron VGCC kinetics in PLM mice did not significantly differ from PBS conditions. The standard error and 95% confidence interval are indicated for each estimate. None of the differences between groups were statistically significant. kpeak, slope of the peak component; V1/2peak, half-maximum activation potential of the peak component; ksustained, slope of the sustained component; V1/2sustained, half-maximum activation potential of the sustained component; CI, confidence interval. PBS, nc = 8, N = 4; PLM, nc = 16, N = 6.
RESULTS
Systemic injection with aged blood plasma impairs LTP in the hippocampal CA1
To assess whether blood-borne factors are sufficient to drive neurophysiological impairments, WT mice were injected with aged blood plasma, starting at 5M (Fig. 1a). We measured synaptic plasticity in the hippocampal CA1 by recording f-EPSPs in this area (Fig. 1b) and compared LTP between plasma-treated (PLM) and PBS-treated (PBS) WT mice (Fig. 1c). The initial comparison of LTP levels between PLM and PBS mice revealed some differences. Whereas post-tetanic potentiation was comparable between groups, differences became apparent in the early phase of LTP and became more pronounced when LTP levels stabilized (Fig. 1c). PLM mice showed significantly reduced levels of LTP compared to PBS mice (Fig. 1d, one-way ANOVA, F(2, 34) = 8.178, p = 0.0013; Bonferroni’s, PBS (148.60±36.81, ns = 11, N = 7) versus PLM (117.30±26.75, ns = 14, N = 5), p = 0.0437). As aging is a major risk factor for AD and AD is associated with significantly impaired levels of LTP [1], we were interested in how LTP levels in PLM mice compare to those seen in the APP/PS1 mouse model at a similar age (Fig. 1a-c). We compared LTP between PBS mice, PLM mice, and 6M APP/PS1 mice and the data showed that both LTP levels in PLM and 6M APP/PS1 mice differ significantly from the WT PBS condition (Fig. 1d, one-way ANOVA, F(2, 34) = 8.178, p = 0.0013; Bonferroni’s, PBS versus APP/PS1 (98.11±26.88, ns = 10, N = 5), p = 0.0009; PBS versus PLM, p = 0.0437). Comparison of 6M APP/PS1 and PLM mice revealed no significant difference (Fig. 1d, one-way ANOVA, F(2, 34) = 8.178, p = 0.0013; Bonferroni’s, PLM versus APP/PS1, p = 0.3404). Further within-group comparison revealed significant LTP levels in PBS and PLM mice, as opposed to those measured in 6M APP/PS1 mice (two-tailed paired Student’s t-test, PBS (148.60±36.81), p = 0.0014; PLM (117.30±26.75), p = 0.0306; APP/PS1 (98.11±26.88), p = 0.8116).
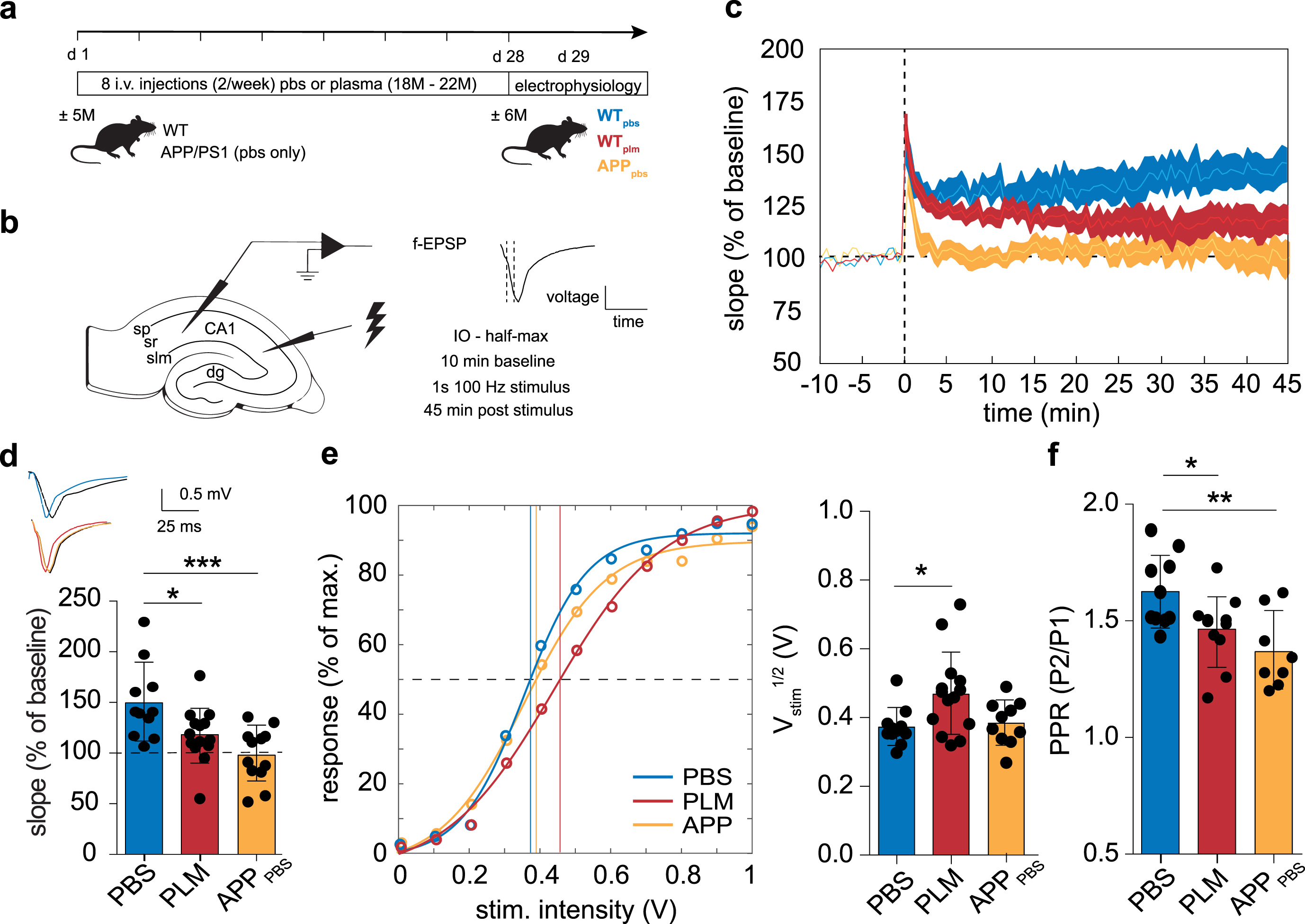
Treatment with aged blood plasma results in impaired LTP in the hippocampal CA1. a) Experimental overview. WT mice received 8 systemic injections with either PBS or aged blood plasma over a 4-week period, starting at the age of 5M. In the same period and at the same age, APP/PS1 mice received 8 systemic injections with PBS. b) Schematic approach of measuring f-EPSPs in the hippocampal CA1. LTP was induced by running a 1 s - 100 Hz stimulation protocol following a 10-min baseline recording. Stimuli were administered at Vstim1/2. LTP was assessed over a 45-min period. c, d) PLM and APP/PS1 mice display significantly impaired LTP compared to PBS mice (one-way ANOVA, p = 0.0013; Bonferroni’s, PBS (148.60±36.81 %, ns = 11, N = 7) versus PLM (117.30±26.75 %, ns = 14, N = 5), p = 0.0292; PBS versus APP/PS1 (98.11±26.88 %, ns = 10, N = 5), p = 0.0006). LTP traces of the 45 min recording (c) and quantification of the last 5 min (d) are shown. e) The Vstim1/2 was determined from the IO curve and is significantly increased in PLM mice (one-way ANOVA, p = 0.0161; Bonferroni’s, PBS (0.369±0.055 V, ns = 11, N = 5) versus PLM (0.465±0.118 V, ns = 14, N = 6), p = 0.0226; PBS versus APP/PS1 (0.374±0.065 V, ns = 10, N = 5), p > 0.9999). The graph displays the Bolzmann-Katz model fit (solid lines) to the IO data normalized to the maximum response from PBS, PLM, and APP/PS1 mice. The circles represent the group averages the model was fitted to. The bar graph shows the quantification of the Vstim1/2. f) Presynaptic release probability was assessed by recording extracellular paired-pulse recordings with a 100 ms interval. PLM and APP/PS1 mice show a significantly decreased paired pulse ratio (Fig. 1f, one-way ANOVA, p = 0.0054; Bonferroni’s, PBS (1.62±0.16, ns = 10, N = 5) versus PLM (1.46±0.15, ns = 11, N = 4), p = 0.0497; PBS versus APP/PS1 (1.37±0.16, ns = 8, N = 4), p = 0.0035). IO, input-output; CA1, cornu ammonis area 1; sp, stratum pyramidale; sr, stratum radiatum; slm, stratum lacunosum moleculare; dg, dentate gyrus.
Further comparison of f-EPSPs showed that post-synaptic baseline responses were similar in all groups. Additional analysis, however, indicates that the
Although not to the same extent as seen in 6M APP/PS1 mice, these data show that LTP and neuronal transmission are significantly affected in the hippocampal CA1 following systemic treatment with aged blood plasma. These changes involve alterations in presynaptic neurotransmitter release probability, a reduced postsynaptic response to a given presynaptic stimulation, and reduced levels of LTP.
Plasma treatment enhances spontaneous presynaptic glutamate release in the hippocampal CA1
We next aimed to further unravel plasma-induced alterations in synaptic transmission. Impaired LTP is associated with reduced NMDA receptor function [31, 61]. Moreover, aging is characterized by changes in presynaptic glutamate release and altered AMPA receptor conductivity [62–64]. To assess whether plasma treatment has similar consequences, we quantified the basic properties of glutamatergic transmission in CA1 pyramidal neurons. To this end, we recorded mEPSCs and postsynaptic AMPA and NMDA responses of CA1 pyramidal neurons in PBS and PLM mice using whole-cell patch-clamp recordings (Fig. 2a, c). We observed no significant changes in the NMDA/AMPA ratio (Fig. 2b, two-tailed unpaired Student’s t-test, PBS (0.036±0.019, nc = 8, N = 3) versus PLM (0.042±0.032, nc = 10, N = 4), p = 0.6185), which demonstrates similar postsynaptic AMPA and NMDA receptor function between PBS and PLM conditions. Accordingly, analysis of the mEPSC amplitude did not reveal significant differences between groups (Fig. 2e, two-tailed unpaired Student’s t-test, PBS (24.49±4.95, nc = 7, N = 4) versus PLM (30.95±10.75, nc = 6, N = 3), p = 0.1803). Further quantification, however, indicated increased spontaneous presynaptic glutamate release, as PLM mice displayed a significant increase in mEPSC frequency (Fig. 2c, d, two-tailed unpaired Student’s t-test, PBS (1.07±0.36, nc = 7, N = 4) versus PLM (1.56±0.35, nc = 6, N = 3), p = 0.0298).
Together, these data indicate that systemic treatment with aged blood plasma specifically alters spontaneous presynaptic glutamate release in the hippocampal CA1.
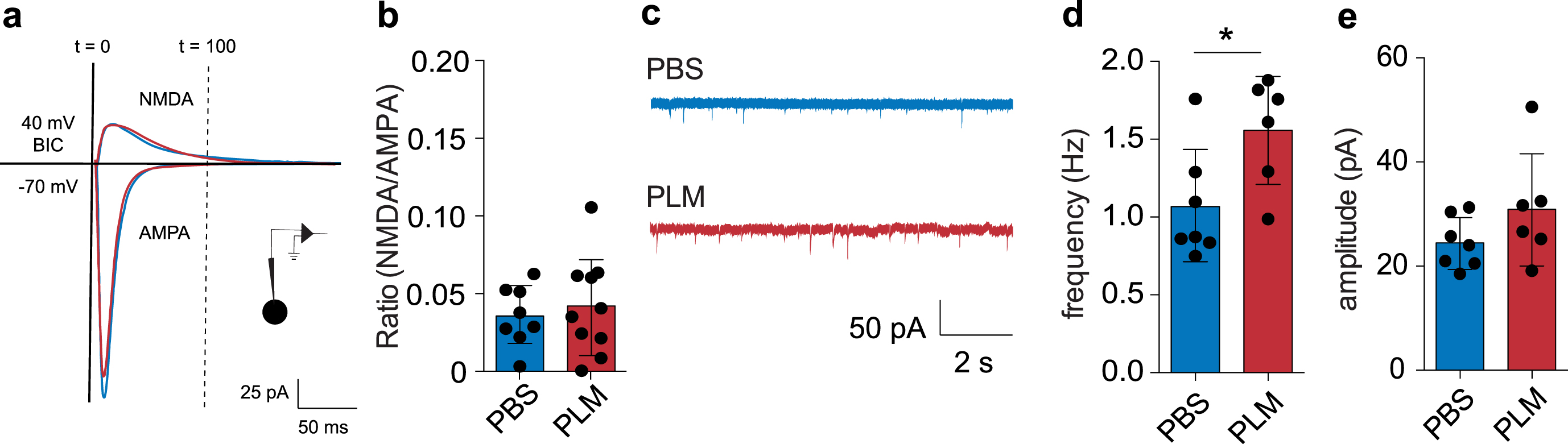
Treatment with aged blood plasma affects basic features of glutamatergic transmission in hippocampal CA1 pyramidal neurons. a) Schematic representation of experimental approach and quantification of NMDA/AMPA ratio. The responses indicate representative traces for PBS and PLM conditions. b) PBS and PLM mice show a similar NMDA/AMPA ratio (two-tailed unpaired Student’s t-test, PBS (0.036±0.019, nc = 8, N = 4) versus PLM (0.042±0.032, nc = 10, N = 4), p = 0.6185). c) Example traces displaying mEPSCs in PBS and PLM mice. d) Quantification indicating the mEPSC frequency in PBS and PLM mice (two-tailed unpaired Student’s t-test, PBS (1.07±0.36 Hz, nc = 7, N = 4) versus PLM (1.56±0.35 Hz, nc = 6, N = 3), p = 0.0298). e) Quantification indicating the mEPSC amplitude in PBS and PLM mice (two-tailed unpaired Student’s t-test, PBS (24.49±4.95 pA, nc = 7, N = 4) versus PLM (30.95±10.75 pA, nc = 6, N = 3), p = 0.1803).
Plasma-treated CA1 pyramidal neurons display increased conductance of voltage-gated Ca2+ channels
Neurotransmitter release and LTP are strongly linked to Ca2+ homeostasis [33, 66]. Many biological processes impact Ca2+ homeostasis, including the altered expression and activity of VGCCs [19, 28]. To find out whether peripheral factors in aged blood plasma impact VGCC activity, we recorded and assessed Ca2+ currents from CA1 pyramidal neurons of PBS and PLM mice. Ca2+ currents were isolated and analyzed as shown in Fig. 3a. For all cells, passive membrane properties were monitored while recording (Table 1). Cm (two-tailed unpaired Student’s t-test, PBS (146.90±51.17, nc = 11, N = 5) versus PLM (134.20±45.43, nc = 18, N = 6), p = 0.4920), membrane resistance (Rm) (two-tailed unpaired Student’s t-test, PBS (185.50±45.01, nc = 11, N = 5) versus PLM (183.00±73.09, nc = 18, N = 6), p = 0.9202), and Rs (two-tailed unpaired Student’s t-test, PBS (16.66±4.45, nc = 11, N = 5) versus PLM (15.08±4.18, nc = 18, N = 6), p = 0.3416) were not significantly different between PBS and PLM conditions and were comparable to what has been shown previously [67, 68]. Analysis of the recorded currents revealed an increase in the maximum peak Ca2+ current amplitude in PLM conditions (Fig. 3b, c). This increase in Ca2+ influx was significant when compensated for Cm (Fig. 3c, two-tailed Student’s t-test, PBS (20.27±5.86, nc = 8, N = 4) versus PLM (27.68±8.34, nc = 16, N = 6), p = 0.0348). No significant difference was present when comparing the sustained Ca2+ current component compensated for Cm (Fig. 3d, e, two-tailed Student’s t-test, PBS (9.86±4.15, nc = 8, N = 4) versus PLM (13.99±5.51, nc = 16, N = 6), p = 0.0754). Corresponding to the increase in peak Ca2+ current amplitude, CA1 pyramidal neurons of PLM mice displayed an enhanced VGCC chord conductance for the peak Ca2+ current component (Table 1). This increase in chord conductance was significant for the peak Ca2+ current component after compensating for Cm (Table 1, two-tailed Student’s t-test, PBS (0.45±0.13, nc = 8, N = 4) versus PLM (0.62±0.19, nc = 16, N = 6), p = 0.0348). To determine whether these increases in Ca2+ current amplitude and specific chord conductance are associated with altered VGCC kinetics, we subsequently estimated V1/2 for the peak and the sustained Ca2+ current components by fitting the I–V relation to a modified Boltzmann-Katz equation (Fig. 3f) [69, 70]. This approach additionally yielded a slope estimate for the I–V relation to further describe VGCC kinetics. Our analysis showed that the V1/2 of the peak (Fig. 3f Table 2, independent two-sample Student’s t-test, PBS (–25.46±1.96, nc = 8, N = 4) versus PLM (–27.48±1.02, nc = 16, N = 6), p = 0.3727) and sustained (Fig. 3f, Table 2, independent two-sample Student’s t-test, PBS (–27.16±2.40, nc = 8, N = 4) versus PLM (–26.24±1.39, nc = 16, N = 6), p = 0.7439) Ca2+ current components did not significantly differ between PBS and PLM mice. Accordingly, the peak (Fig. 3f, Table 2, independent two-sample Student’s t-test, PBS (4.88±1.37, nc = 8, N = 4) versus PLM (4.16±0.82, nc = 16, N = 6), p = 0.6574) and sustained (Fig. 3f, Table 2, independent two-sample Student’s t-test, PBS (3.79±1.90, nc = 8, N = 4) versus PLM (2.96±0.88, nc = 16, N = 6), p = 0.6965) slope estimates were not significantly different between both conditions (Fig. 3f, Table 2).
General membrane and VGCC properties of patch-clamped CA1 pyramidal cells from PBS and PLM mice
CA1 pyramidal cells in PLM mice generally show membrane characteristics similar to PBS conditions (Cm, Rs, Rm - data: PBS, nc = 11, N = 5; PLM, nc = 18, N = 6). The peak chord conductance of VGCCs is significantly increased in PLM mice and associated with a significantly increased peak current amplitude. Cm, capacitance; Rs, series resistance; Rm, membrane resistance; Ipeak _ max, maximum peak current; Ipeak _ max/Cm, normalized maximum peak current; Isustained _ max, maximum sustained current; Isustained _ max/Cm, normalized maximum sustained current; Gpeak _ max, peak chord conductance; Gpeak _ max/Cm, normalized peak chord conductance; Gsustained _ max, sustained chord conductance; Gsustained _ max/Cm, normalized chord conductance. PBS, nc = 8, N = 4; PLM, nc = 16, N = 6.
Together, these data indicate that systemic treatment with aged blood plasma significantly increases the chord conductance of VGCCs in CA1 pyramidal neurons. This increased chord conductance is characterized by enhanced peak and sustained Ca2+ currents in PLM mice without significantly affecting VGCC kinetics.
DISCUSSION
Aging is characterized by cognitive decline, including progressive memory impairments [71]. These cognitive changes reflect impairments in neuronal activity and synaptic plasticity in various brain regions, including the hippocampal CA1 and DG. Recently, it was suggested that blood-borne factors are involved in the induction of age-related impairments in the DG [51, 52]. Nevertheless, how these blood-borne factors induce neurophysiological impairments and to what extent these impairments reach beyond the DG remains unclear. The data presented here indicate that the systemic treatment of adult WT mice enhances presynaptic glutamate release and VGCC activity and conductivity in the hippocampal CA1. These enhancements are, however, associated with reduced levels of LTP.
First, we showed that PLM mice display significantly impaired levels of LTP in the hippocampal CA1, similar to those present in APP/PS1 mice. These impairments arose in the early phase of LTP and became significant when LTP levels stabilized. This is in accordance with earlier studies in which impaired LTP in the DG followed exposure to aged blood plasma [52]. We here show that this effect is not exclusive to the DG, indicating that peripheral factors present in aged blood plasma affect general hippocampal physiology. Impaired synaptic transmission and plasticity are overall characteristic for the aging and AD rodent hippocampus [47–49, 72–74]. There are, however, contrasting studies that indicate no substantial effect of aging on synaptic plasticity [75, 76]. This disparity likely results from variations in experimental conditions, as the hippocampal CA1 is particularly sensitive to the applied stimulation protocol [9]. Studies using peri-threshold stimulation intensities regularly report impaired synaptic plasticity [49, 72], as opposed to studies using supra-threshold stimulation [75, 76]. As such, the peri-threshold stimulus protocol we used in our approach made it more likely to reveal differences in the synaptic plasticity induction capacity. Generally, plasticity impairments in the aging hippocampal CA1 are associated with a decrease in synaptic strength [9]. Indeed, our data indicate that the neuronal network in the hippocampal CA1 in PLM mice is affected and leads to a reduced postsynaptic response to a given presynaptic stimulation, while maintaining a response threshold similar to the PBS condition. This effect was not present in APP/PS1 mice, indicating that the reduced capability to induce synaptic plasticity might result from an underlying defect different from that in PLM mice. Indeed, AD mouse models regularly show a significant reduction in postsynaptic spine density [15]. Yet, the similar cell capacitances in CA1 pyramidal neurons of PBS and PLM mice indicate that significant alterations in cellular and dendritic morphology are most likely not present in PLM mice. Thus, the impaired postsynaptic response of CA1 pyramidal neurons in PLM mice likely arises from alterations in synaptic strength and our data indicate that systemic treatment with aged blood plasma is sufficient to trigger these alterations. Moreover, we show that the impaired capability to induce synaptic plasticity in PLM mice resembles the functional outcome present in APP/PS1 mice.
Our results further show a decreased paired-pulse ratio and an associated increase in spontaneous presynaptic glutamate release onto CA1 pyramidal neurons in PLM mice, suggesting that the presynaptic release probability increases following the exposure to peripheral factors present in aged blood plasma. Both increases in tonic glutamate concentration and enhanced presynaptic glutamate release have been associated with impaired levels of LTP in the hippocampal CA1 [77, 78]. Moreover, enhanced tonic glutamate concentrations are characteristic for the aging rodent brain [63]. In some cases, this was associated with increased spontaneous and evoked presynaptic glutamate release [79–81]. Comparison of the NMDA/AMPA ratio revealed no apparent changes in postsynaptic AMPA and NMDA receptor function between PBS and PLM mice. This is in contrast with earlier studies that show a strong association between aging and reduced NMDA receptor conductivity and expression in the hippocampal CA1 [31, 82]. Treatment with aged serum, however, did not significantly alter the NMDA/AMPA ratio in cultured human neurons, suggesting that peripheral factors affect NDMA receptor conductivity differently from an endogenous aging milieu [83]. Studies on age-related changes on AMPA receptor expression and function are inconclusive. Early studies did not reveal differences in AMPA receptor expression or activity [62, 85], and additional studies showed contrasting results [64, 87]. Similar to our data, a recent study on cultured human neurons revealed no effect of aged serum on evoked AMPA responses [83]. Together, this suggests that the LTP impairment observed in our data is associated with changes in presynaptic glutamate release, rather than with alterations in postsynaptic AMPA or NMDA conductivity. These changes in presynaptic glutamate release are potentially triggered by peripheral factors present in aged blood plasma. Indeed, cultured human neurons revealed a comparable increase in release probability upon the prolonged exposure to aged serum [83].
CA1 pyramidal neurons are further subjected to several age-related changes in Ca2+ homeostasis, including the increased expression [19] and function [20] of VGCCs [21, 22]. Our data indicate a similar effect in PLM mice, as the Ca2+ current amplitude of the peak component and the VGCC chord conductance are significantly increased compared to the PBS condition, indicating an enhanced influx of Ca2+ immediately after VGCC activation. Our data further show that VGCC kinetics of CA1 pyramidal neurons of PLM mice does not differ significantly from the PBS condition, indicating that treatment with aged blood plasma does not impact the rate of Ca2+ influx after VGCC activation. This makes it unlikely that changes in VGCC subunit composition contribute to the enhanced Ca2+ currents [88].
Increased Ca2+ currents potentially affect the intracellular [Ca2+] and make it less likely for significant fluctuations in Ca2+ to occur, thereby affecting many physiological processes [46]. Enhanced VGCC currents do, however, not necessarily affect the [Ca2+] intracellularly, especially because neurons have multiple mechanisms for Ca2+ buffering that are potentially capable of rectifying increases in VGCC current density. There are indeed indications that aging is associated with increased Ca2+ buffering in both cortical and CA1 pyramidal neurons [89–91]. Yet, the increased Ca2+ buffering capacity of aged CA1 pyramidal neurons was insufficient to remedy the increased [Ca2+] transients associated with sustained neuronal activation [91]. Moreover, other studies indicate that the Ca2+ buffering capacity of aged CA1 pyramidal neurons remained unchanged or was even reduced [24–26]. As such, the enhanced expression and function of VGCCs likely results in enhanced intracellular [Ca2+] transients during sustained neuronal activation under normal aging conditions. Indeed, measurements of the intracellular [Ca2+] using a fluorescent indicator showed elevated [Ca2+] transients associated with neuronal activity in the hippocampal CA1 of aged rats and mice [28, 29]. Whether a similar effect results from treatment with aged blood plasma, and the type of VGCCs that are responsible for this, remains to be further investigated. Yet, our results on pre-synaptic release probability, glutamatergic transmission, and synaptic plasticity are in accordance with previous studies that report on enhanced intracellular [Ca2+] transients in the hippocampal CA1. For example, L-type VGCC blockers enhanced hippocampal LTP in aged rats, an effect that correlated with depression of the [Ca2+]-dependent AHP [33]. Moreover, a strong relationship exists between the [Ca2+] and the frequency of spontaneous postsynaptic currents [66], suggesting that small changes in presynaptic Ca2+ availability have a large effect on neurotransmitter release. An increased [Ca2+] in presynaptic terminals in the hippocampal CA1 of aged animals has been demonstrated previously [29], and increased Ca2+ buffering resulted in f-EPSP and EPSP depression, suggesting that a decreased presynaptic [Ca2+] inhibits neurotransmitter release [24, 93]. Furthermore, increased presynaptic [Ca2+] was associated with a reduced paired-pulse ratio, resulting in impaired LTP in aged mice [29]. As such, it is likely that similar alterations in Ca2+ homeostasis contribute to the results presented here. Future experiments should determine how and which peripheral agents present in aged blood plasma are responsible for this effect.
Interestingly, it was reported that exposure of young mice to aged blood plasma is associated with changes in chemokine expression, including that of CCL11 [51]. Administration of CCL11 increases the intracellular [Ca2+], which was prevented by the administration of L-type VGCC blockers [51, 94]. Moreover, chemokines are capable of affecting HVA VGCCs in the hippocampus and other brain areas [95, 96]. This implies that altered chemokine expression resulting from PLM treatment might also be implicated in the data presented here. We are currently unable to conclude whether these peripheral factors are aging specific, as we used PBS instead of age-matched plasma as a control. This was decided due to ethical considerations and allowed for a 5 times reduction (17 instead of 85) in the number of control mice needed for experiments. Also, previous research indicates that the effect of saline treatment does not significantly differ from exposure to aged-matched plasma [57]. It is, however, important to note that the use of PBS as a control complicates group comparison, e.g., by the use of pentobarbital for plasma collections.
In conclusion, the data presented in this study show that neurophysiology in the hippocampal CA1 is affected following treatment of adult mice with aged blood plasma. These changes resemble impairments present in AD and aging and include the impaired induction and susceptibility to LTP, alterations in presynaptic glutamate release, and an increase in Ca2+ current amplitude and VGCC conductance. Together, these findings demonstrate that blood-borne factors are capable of inducing impairments in adult mice at the cellular level. This further develops our understanding of how blood-borne factors impact neurophysiology and hints towards a possible therapeutic potential for blood plasma in aging and related neurodegenerative disorders.
Footnotes
ACKNOWLEDGMENTS
This work was supported by ZonMw [733050816, 2017 –EMH, JM, and CFMH] and [73305054, 2015 - JM]: The Netherlands Organisation for Health Research and Development, Dementia Research and Innovation Program “Memorabel” with additional support from Alzheimer Nederland (73305054), and by UMC Utrecht Rudolf Magnus Young Talent fellowship 2017 (JM). The authors thank Nicky van Kronenburg for practical support.