
Abstract
Background:
Amyloid-β protein precursor (AβPP) is enriched in neurons. However, the mechanism underlying AβPP regulation of neuronal activity is poorly understood. Potassium channels are critically involved in neuronal excitability. In hippocampus, A-type potassium channels are highly expressed and involved in determining neuronal spiking.
Objective:
We explored hippocampal local field potential (LFP) and spiking in the presence and absence of AβPP, and the potential involvement of an A-type potassium channel.
Methods:
We used in vivo extracellular recording and whole-cell patch-clamp recording to determine neuronal activity, current density of A-type potassium currents, and western blot to detect changes in related protein levels.
Results:
Abnormal LFP was observed in AβPP–/– mice, including reduced beta and gamma power, and increased epsilon and ripple power. The firing rate of glutamatergic neurons reduced significantly, in line with an increased action potential rheobase. Given that A-type potassium channels regulate neuronal firing, we measured the protein levels and function of two major A-type potassium channels and found that the post-transcriptional level of Kv1.4, but not Kv4.2, was significantly increased in the AβPP–/– mice. This resulted in a marked increase in the peak time of A-type transient outward potassium currents in both glutamatergic and gamma-aminobutyric acid-ergic (GABAergic) neurons. Furthermore, a mechanistic experiment using human embryonic kidney 293 (HEK293) cells revealed that the AβPP deficiency-induced increase in Kv1.4 may not involve protein-protein interaction between AβPP and Kv1.4.
Conclusion:
This study suggests that AβPP modulates neuronal firing and oscillatory activity in the hippocampus, and Kv1.4 may be involved in mediating the modulation.
Keywords
INTRODUCTION
AβPP, the protein precursor of amyloid-β (Aβ), is broadly expressed in the central nervous system, especially in axons and cell bodies [1]. As a critical protein in Alzheimer’s disease (AD) pathogenesis, symptoms including altered levels of soluble AβPP [2] and AβPP transcript mutation in the cerebral cortex [3] occur in early- and late-onset AD patients, suggesting a potential role of AβPP as a biomarker for pre-clinical diagnosis and treatment [4]. AβPP has been shown to regulate the synaptic structure, transmission, and plasticity [5, 6]. Overexpression of AβPP and presenilin-1 in mice results in impaired neocortical plasticity, as well as other symptoms and functional abnormalities similar to those of mild-to-moderate AD patients [7]. Impaired long-term potentiation (LTP) has been found in AD patients and mouse models [7, 8]. In addition, induction of LTP is associated with increased secretion of AβPP, a lack of which leads to dysfunction of the synaptic activity [9] and impaired LTP in the hippocampus [10–12]. AβPP regulates neuronal and synaptic function via transmembrane proteins, such as voltage-gated calcium channels and a potassium and chloride co-transporter (KCC2) [13–15]. However, whether and how AβPP affects ion-channel function and thereby neuronal activity remains elusive. Potassium channels are the most widely distributed type of ion channel in eukaryotic cells. There are two types of outward potassium current in mammalian hippocampal neurons: fast-inactivating A-type transient outward potassium currents (IA) and slow/non-inactivating currents [16]. The most widely distributed A-type potassium channel subtypes in the hippocampus are Kv1.4 and Kv4.2 [17]. Kv1.4 is present at high levels in the plasma membrane of axonal presynaptic regions and the axon terminal active zone; it is also found in the postsynaptic region of dendritic shafts and spines [18, 19]. In Kv1.4–/– mice, the firing rate is increased in suprachiasmatic nucleus neurons [20] and nucleus accumbens neurons [21], suggesting that loss of Kv1.4 expression leads to a higher neuronal firing rate. The use of Ba2 + pharmacology to block Kv1.4-mediated IA has revealed the contribution of Kv1.4 to the generation of the action potential (AP), the regulation of the AP rheobase, and the control of the rhythmic discharge [22]. These studies indicate the critical role of Kv1.4 in neuronal firing activity, but whether AβPP regulates neuronal function via Kv1.4 has not yet been addressed. In this study, using electrophysiological and biochemical methods, we reveal that AβPP deficiency causes abnormalities in hippocampal neuronal activity and post-transcriptional regulation of Kv1.4 expression and function, suggesting Kv.1.4 may be a target via which AβPP regulates hippocampal neuronalfunction.
MATERIALS AND METHODS
Animals and plasmids
All experimental procedures were approved by the Animal Ethics Committee at South China Normal University and were carried out in accordance with the Guidelines for Animal Care established by the National Institute of Health. AβPP–/– mice [23] and wild-type (WT) mice were obtained by crossing AβPP+/– males and females from the Model Animal Research Center of Nanjing University (Nanjing, China). Glutamic acid decarboxylase 67+/GFP (GAD67+/GFP) mice [24] (a gift from Dr. Yu-qiang Ding at Tongji University School of Medicine, Shanghai, China) were crossed with AβPP–/– and WT mice to obtain AβPP–/–-GAD+/GFP and WT-GAD+/GFP mice. Mice were raised with free access to food and water and maintained on a 12-h light-dark cycle. Both male and female mice were used in the experiments. Littermates were used for comparison. The mouse genotype was confirmed before investigations were initiated. Two-month-old mice were used for western blotting of Kv1.4, and Kv4.2 expression levels and 2-3-month-old mice were used for in vivo extracellular recording. Full-length human AβPP (hAβPP695) was obtained from the Hui Zheng laboratory at Baylor College of Medicine (Houston, TX, US). The Kv1.4 plasmid was a gift from Zhuo Fan (South China University of Technology, Guangzhou, China).
In vivo extracellular recording
The recording method is similar to the method in our previous paper [25–27]. Before recording, mice were anesthetized with urethane (Sigma, Burlington, MA, US; 2 g/kg, i.p.). A supplemental dose of 0.1–0.2 g/kg urethane was used to maintain anesthesia such that there was no withdrawal reflex on tail pinching. Mice were placed in a stereotaxic apparatus (RWD Life Science, Shenzhen, China) for surgery and extracellular recording. During experiments, mice were self-breathing, and body temperature was maintained between 36–37°C using a heating pad (Harvard Apparatus, Holliston, MA, US). The recording electrode was a glass pipette (OD, 1 mm; World Precision Instruments, Sarasota, FL, US) pulled by a mechanical micromanipulator PC-10 (Narishige, Tokyo, Japan) having 2–5 MΩ resistance when filled with 0.5 M NaCl. The electrode was presented at an angle of 45° and positioned in the hippocampal CA1 region: AP, –1.95 mm; ML, 2.75 mm; DV, 1.35 mm [28]. For each trial, the signal was amplified 1000 times by a model 3000 AC/DC differential amplifier (A-M Systems, Carlsborg, WA, US), then converted into a CED micro1401 data acquisition unit (CED Limited, Cambridge, UK). Data was collected in Spike2 software (CED Limited, Cambridge, UK).
Analysis of extracellular recording data
The point greater than two times the standard deviation of the baseline fluctuations in 1 s was the peak of a spike. Traces 2 s before and 3 s after the spike peak were extracted as the spike trace. The raw extracellular recording trace was filtered into the theta (4–7 Hz), gamma (25–80 Hz), epsilon (90–130 Hz), and ripple (140–220 Hz) frequency bands. Autocorrelation of spike firing pattern was used to analyze the rhythmicity of spike firing and to calculate the probability of spike firing rate at time t+k, where t represents a given time, and k represents a range of time lags, using self-coded MATLAB programs (MathWorks, Natick, MA, US).
Different types of neurons were classified according to the literature [29, 30]. Putative pyramidal neurons were identified by standard parameters of firing rate and spike waveform, as well as the first moment of the autocorrelation [31]. In detail, pyramidal neurons discharged with a lower firing rate (<5 Hz) and longer spike duration (0.45±0.08 ms), compared to the faster firing rate (>5 Hz) and shorter spike duration (0.40±0.02 ms) of GABAergic neurons. Furthermore, pyramidal neurons fire with complex-spike bursts composed of several spikes with 3–10 ms inter-spike intervals. This characteristic results in auto-correlograms with 3–5 ms peaks followed by a fast or slow exponential decay in pyramidal neurons. However, in GABAergic neurons, the decay phase is much slower, and the decay may even be non-exponential. The combination of these parameters allows the classification of the different types of neurons. The power spectrum, comprising various frequency bands, was analyzed using a multitaper Fourier transform (MATLAB Chronux toolbox). To determine the preferred firing phase of neurons, we decided the peaks of the local field rhythm using the threshold method and assigned the peak point the phase value of 180°. The number of spike firings in 3° phase intervals for each frequency band of the LFP were counted and plotted as a histogram to analyze the phase-locking relationship between spikes and oscillations using the MATLAB CircStats toolbox [32].
Neuronal and cell culture
Cultured hippocampal neurons were prepared essentially as described by [13]. The dissection consisted of Hank’s balanced salt solution supplemented with 25 mM HEPES, pH 7.2. Hippocampus tissues were dissected from WT and AβPP–/– littermates of postnatal day 0 to 1 pups. The tissue pieces were trypsinized and digested at room temperature (RT) for 15 min. Gently triturated in a culture medium containing 10% heat-inactivated fetal bovine serum using fire-polished pipettes, then centrifuged at 2000 rpm for 10 min. After centrifugation, cells were resuspended in a neurobasal medium containing B27 and L-glutamine (Invitrogen, Waltham, MA, US) and placed onto a 12-mm poly-L-lysine-coated coverslip at a concentration of 1–2×104 cells/ml and left at RT for 30 min to allow cells to settle and attach. One day after plating, 4 M cytosine-β-D-arabinofuranoside (Sigma, Burlington, MA, US) was added to prevent astrocyte proliferation. Cells that had spent 12–14 days in vitro (DIV) culture were used for patch-clamp recording.
Human embryonic kidney 293 (HEK293) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, containing L-glutamine, Invitrogen) supplemented with 10% (v/v) fetal calf serum (Invitrogen) in a humidified incubator with 5% CO2/95% air. Cells were passaged every three days at a ratio of 1:10. AβPP-His, hAβPP695, and Kv1.4 plasmids were transiently transfected into HEK293 cells as required. DNA-Lipofectamine 2000 reagent (Invitrogen) was used for transfection following the manufacturer’s instructions. Briefly, cells were seeded to be 70–90% confluent on transfection day. DNA (0.8μg) was diluted into 50μl Opti-MEM medium and lipofectamine 2000 (2μl) into 50μl Opti-MEM medium. After incubating for 5 min, the two solutions were mixed and incubated for 20 min, after which the DNA-lipid complex was added to the cells. The amounts of DNA and lipofectamine 2000 were adjusted according to the area of the culture wells. For electrophysiology recording, 24 h after transfection, cells were washed gently with phosphate-buffered saline (PBS) and lysed before attachment to glass coverslips for recording. Cells were collected 24–72 h after transfection for molecular and biochemistry experiments.
Quantitative real-time polymerase chain reaction (qPCR)
Fresh hippocampus tissue was fully homogenized with TRIzol reagent; total RNA was extracted using trichloromethane and isopropyl alcohol and dissolved in RNase-free water. Reverse transcription was performed on 2μg total RNA with 200 ng random primers using a cDNA synthesis kit (TaKaRa Biotechnology, Dalian, China) following the manufacturer’s protocol. To investigate the expression level of Kv1.4 genes in the hippocampus, the following gene-specific primers were used: Kv1.4-S (5’-3’), ATGAAGCCCATCACAGTCGG; Kv1.4-A (5’-3’), CCTTGACCCCTTCTTCCATCTC; β-actin-S (5’-3’), GTGACGTTGACATCCGTAAAGA; β-actin-A (5’-3’), GTAACAGTCCGCCTAGAAGCAC; 2μl of a 7.5μM solution of primers together with 2.5μl reverse transcription product were also added. The conditions for PCR amplification were 95°C for 10 min, then 40 cycles of 95°C for 15 s followed by 60°C for 60 s, and finally a 5 min extension at 60°C. Quantification of normalized gene expression was performed by the comparative cycle threshold method. Statistical analysis was performed on 2–ΔΔCt values.
Western blotting
Hippocampal tissues were dissected and homogenized using micro tissue grinders (Kimble, Boston, MA, US) in 2 ml tubes with an appropriate volume of lysis buffer for 1 to 2 min on ice. The lysis buffer contained 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA (pH 8.0), 1% SDS, and a protease inhibitor cocktail (Complete Mini; Roche, Basel, Switzerland). After homogenization, cell debris was removed by centrifugation at 14000 rpm for 10 min at 4°C. The supernatant was collected and denatured at 75°C for 20 min before further analysis. For HEK293 cell samples, 48 h after transfection, cells were rinsed twice with PBS and lysed by adding 200μl lysis buffer containing a protease inhibitor cocktail to each petri dish for 30 min on ice. Cell lysates were centrifuged at 14000 rpm for 10 min at 4°C to collect supernatant and denatured at 75°C for 20 min. Equal amounts of protein were loaded into the wells of an SDS-PAGE gel (Bio-Rad, Hercules, US). After running for 30 min at 80 V and 2.5 h at 100 V, the gel was transferred to nitrocellulose membranes for 1 h at 100 V. The membranes were blocked for 30 min using 5% non-fat milk in Tris-buffered saline (TBS) containing 0.5% Tween-20 (TBST), then probed with specific primary antibodies against AβPP (4G8, Millipore, Burlington, US; A8717, Sigma), Kv1.4 (ab78015, Abcam). Anti-γ-tubulin antibody (T6557, Sigma) was used as the loading control. After three 10 min washes with TBST, HRP-labeled secondary antibody was applied at RT for 1 h. Bands were visualized using the Immobilon Western ECL system and analyzed with Gel Pro Analysis software.
Co-immunoprecipitation
HEK293 cells were grown in DMEM (high glucose with L-glutamine) containing 10% FBS. For immunoprecipitation, HEK293 cells were plated onto 10 cm culture dishes and grown to 70% confluency. Cell transfections were performed with lipofectamine 2000 (Invitrogen), following the manufacturer’s recommended protocol. Then, 24–48 h after transfection, cells were washed once with cold PBS and homogenized in 1 ml cold lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% NP40, and protease inhibitor, pH 7.4). To remove cell debris, the lysates were centrifuged for 20 min at 14,000 rpm at 4°C. The supernatant was incubated with 2μl of each antibody at 4°C overnight. Then 30μl protein A agarose beads (Millipore) were added, and samples were incubated at 4°C for 2–3 h. To control for nonspecific binding, protein lysates were incubated with rabbit or mouse IgG, and beads were processed in parallel. Subsequently, the beads were washed three times with 500μl cold lysis buffer. Immunoprecipitants were eluted from the beads by adding 30μl SDS-PAGE sample buffer and heating for 20 min at 75°C. The eluates (15μl) were analyzed by western blotting.
In vitro whole-cell patch-clamp recording
IA was recorded from cultured hippocampal neurons (10 to 14 DIV) at 37°C using a MultiClamp 700B amplifier (Molecular Devices, San Jose, CA, US). The recording chamber was perfused with Tyrode’s solution of the following composition (in mM): 129 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 0.01 glycine, 30 glucose, 25 HEPES, 0.001 TTX, pH 7.3. The glass micropipettes had a resistance of 3–5 MΩ when filled with an internal solution containing (in mM): 110 K-gluconate, 40 HEPES, 10 EGTA, 2 Na2-ATP, 2 Mg-ATP and 0.3 GTP, pH 7.3. The prepulse protocol to obtain IA was modified from a previous publication [33]. IA was recorded as follows: firstly, the current was recorded at the holding level (–60 mV) by giving –40 to +50 mV depolarized current in 10 mV steps, with each depolarization lasting 400 ms. Secondly, keeping the same holding level, the fast-active, fast-inactive IA was inactivated by giving –20 mV, 100 ms pre-stimuli. Thirdly, IA was obtained by subtracting the two currents. To explore the input resistance of recorded neurons, 100 ms step currents were injected from –100 pA to +20 pA in 20 pA steps. To determine the rheobase for spike firing, around 200 pA current was injected and adjusted in 5 pA increments to induce the first spike in different neurons. The current needed to induce the first spike was taken as the rheobase for spike firing. Access resistance was monitored throughout the whole recording, and only recordings with stable access resistance that held current for at least 3 min were used. Access resistance was not compensated. All data were acquired and analyzed using pCLAMP software (Molecular Devices). Data were digitized at 10 kHz and filtered at 1 kHz for subsequent analyses.
Data and statistical analysis
The IA density was calculated as the peak IA induced in each current step divided by the capacitance of the recorded neurons. The time-to-peak for IA was calculated by fitting the rising phase to the single exponential curve in pCLAMP software. Mean membrane potential was divided by mean membrane current to obtain the input resistance. The AP was extracted using the threshold search method in pCLAMP software. AP threshold was defined as the first point larger than 1.2 v/s. AP amplitude was defined as the distance between the threshold and the AP peak. Half-width was the duration of the AP at half the AP amplitude. The dV/dt plot was calculated from the equation: dV(t)/dt = [V(tn)-V(tn - 1)]/(tn-tn - 1). Two-way ANOVA was used to compare peak time and IA current density between groups. Changes in the characteristics of APs and power spectra were assessed using an unpaired t-test. Western blotting and qPCR data were analyzed using a paired-sample Student’s t-test, with p < 0.05 considered statistically significant. The Kolmogorov–Smirnov test was used to compare the various groups in the autocorrelation analysis. To compare the phase-locking relationship between different groups, Watson-William’s circle test was used as described in [32], with a p < 0.05 significance level. All data are presented as mean±SEM.
RESULTS
Altered hippocampal spiking and LFP in the CA1 region of AβPP–/– mice
To explore whether AβPP deficiency affects neuronal spiking and oscillations, we performed in vivo extracellular recording in the hippocampal CA1 region of anesthetized AβPP–/– mice and littermate WT controls (Fig. 1A). LFP was filtered into different rhythms based on their frequencies, i.e., theta (4–7 Hz), gamma (25–60 Hz), epsilon (90–130 Hz), and ripple (140–220 Hz) (Fig. 1A). Analysis of the power spectrum (Fig. 1B, C) indicated that the power of theta and gamma rhythms were significantly reduced in AβPP–/– mice (theta: WT, 0.012±0.002; AβPP–/–, 0.005±6.032E-4; t = 2.831, p = 0.008; unpaired t-test) (gamma: WT, 0.002±2.34E-4; AβPP–/–, 8.292E-4±1.584E-4; t = 2.364, p = 0.024; unpaired t-test). However, the power of epsilon rhythm (Fig. 1C) (WT, 1.226E-4±1.765E-5; AβPP–/–, 2.538E-4±6.281E-5; t = –2.724, p = 0.01; unpaired t-test) and ripple rhythm was significantly enhanced (Fig. 1C) (WT, 1E-4±1.149E-5; AβPP–/–, 2.621E-4±4.874E-5; t = –4.524, p = 6.702E-5; unpaired t-test). Three types of neurons were classified based on the detected spike and autocorrelation characters. Two of these were putative glutamatergic neurons, while the third category was that of GABAergic neurons (Fig. 1D). Autocorrelation analysis showed that the spiking rate of glutamatergic neurons in AβPP–/– mice was significantly lower than that of the WT controls (Fig. 1E, z = 0.11, p = 1.11E-15; K-S test).

Impaired firing activity of hippocampal neurons as shown by extracellular recording in AβPP–/– mice. A) Representative traces of spikes and filtered LFP in the theta (4–7 Hz), gamma (25–80 Hz), epsilon (90–130 Hz), and ripple (140–220 Hz) rhythms in WT (black) and AβPP–/– (red) mice. The raw traces were obtained by extracellular recording in the CA1 region of around 2-month-old WT and AβPP–/– mice. B) Power spectral analysis of hippocampal glutamatergic neurons in CA1 from WT (n = 20) and AβPP–/– mice (n = 8). C) Statistical analysis of power spectrum results from CA1 neurons of WT and AβPP–/– mice. *p < 0.05; **p < 0.01; Student’s t-test. D) Representative autocorrelation histograms of spike firings originated from the extracellular recordings, indicating three types of firing patterns and two types of neurons. E) Autocorrelation analysis of inter-spike interval in hippocampal glutamatergic neurons from WT (n = 27) and AβPP–/– mice (n = 12).
Spike firings were counted and compared in 3° phase intervals for all four rhythms. The results showed similar spike-LFP phase-locking for theta, gamma, epsilon, and ripple rhythms. The averaged firing phase for theta rhythm in WT and AβPP–/– mice was calculated but showed no evidence of a phase bias in the theta band (Fig. 2A) (WT, 322.565°±57.923°; z = 0.489, p = 0.613; AβPP–/–, 330.242°±39.47°; z = 1.05, p = 0.35). In contrast, spike firing in both WT and AβPP–/– mice were mainly concentrated in the descending phase in the gamma range (Fig. 2B) (WT, 249.398°±11.024°, z = 13.312, p = 1.65E-06; AβPP–/–, 243.271°±14.616°, z = 7.572; p = 5.14E-04), in the ascending phase for epsilon rhythm (Fig. 2C) (WT, 139.988°±7.737°, z = 26.646, p = 2.68E-12; AβPP–/–, 125.145°±8.342°, z = 22.524, p = 1.65E-10), and in the peak phase for ripple rhythm (Fig. 2D) (WT, 171.336°±14.683°, z = 7.32, p = 6.62E-04; AβPP–/–,148.878°±21.362°, z = 3.504; p = 0.03). There was no significant difference in the lock-in phase between the mouse genotypes (Fig. 2) (theta, F = 0.273, p = 0.602; gamma, F = 0.411, p = 0.522; epsilon, F = 3.485, p = 0.062; ripple, F = 1.705, p = 0.193; Watson-William’s circle test). In conclusion, we found that glutamatergic neuronal activity was impaired in AβPP-deficientmice.
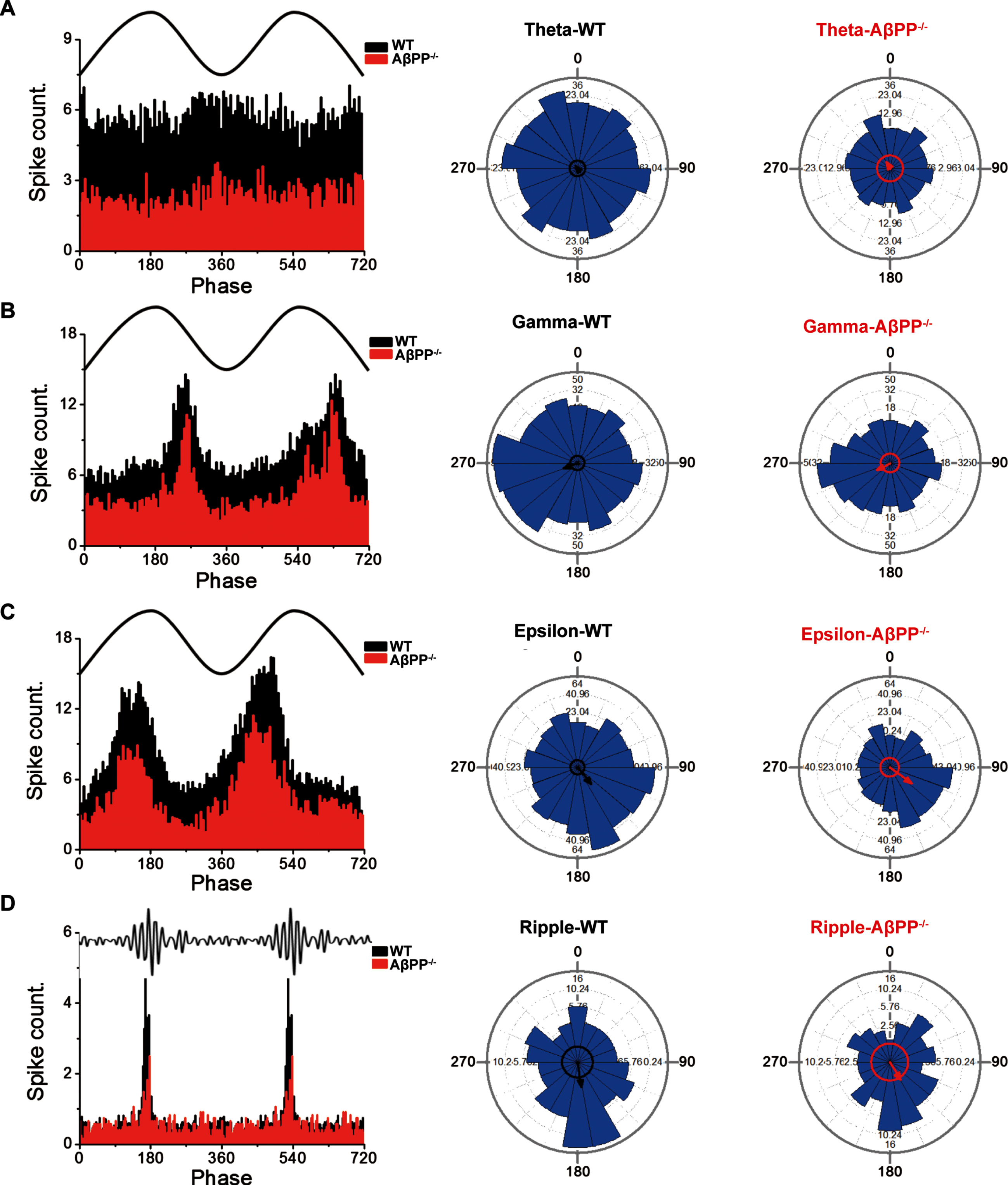
Unchanged phase-locking between spikes and oscillations of different frequencies in the hippocampus of WT and AβPP–/– mice. A) Spike count and Watson-William’s test in hippocampal neurons of WT (n = 20) and AβPP–/– mice (n = 8) with varying theta oscillation phases. The left panel shows representative traces of the theta frequency band and a histogram of the firing rate per 3° phase interval. The middle and right panels show the summation of the neuron firing rate of all neurons in 20° bins (blue bars), preferred phase vector direction (circle with an arrow), and scale vector (indicated for each 90° quartile). B) Spike count and Watson-William’s test in hippocampal neurons of WT mice and AβPP–/– mice with varying gamma oscillation phases. C) Spike count and Watson-William’s test in hippocampal neurons of WT mice and AβPP–/– mice with varying epsilon oscillation phases. D) Spike count and Watson-William’s test in hippocampal neurons of WT mice and AβPP–/– mice with varying ripple oscillation phases.
Lack of AβPP reduces excitability of hippocampal neurons
We next performed whole-cell patch-clamp recording on cultured hippocampal neurons to determine whether AβPP deficiency affected glutamatergic firing. Under the infrared differential interference contrast microscope, pyramidal neurons for recordings were identified by their typical triangular-shaped soma, which distinguishes them from the GABAergic interneurons [34]. We used 14 DIV neuronal cultures for recording. A step current was injected into glutamatergic neurons to measure the input resistance and AP rheobase (Fig. 3A). The current-voltage (I–V) curve showed that the input resistance values of WT and AβPP–/– mice were not significantly different (Fig. 3B), but the AP rheobase in AβPP–/– mice increased, suggesting that the excitability of hippocampal glutamatergic neurons in AβPP-deficient animals was markedly reduced (Fig. 3C) (WT, 147.4±15.27; AβPP–/–, 228.3±26.87; p = 0.007; unpaired t-test), albeit without changing the basic parameters of the AP, i.e., half-width, amplitude and threshold (Fig. 3D-G). Together, these data indicate that AβPP deficiency reduces excitability in glutamatergic neurons.
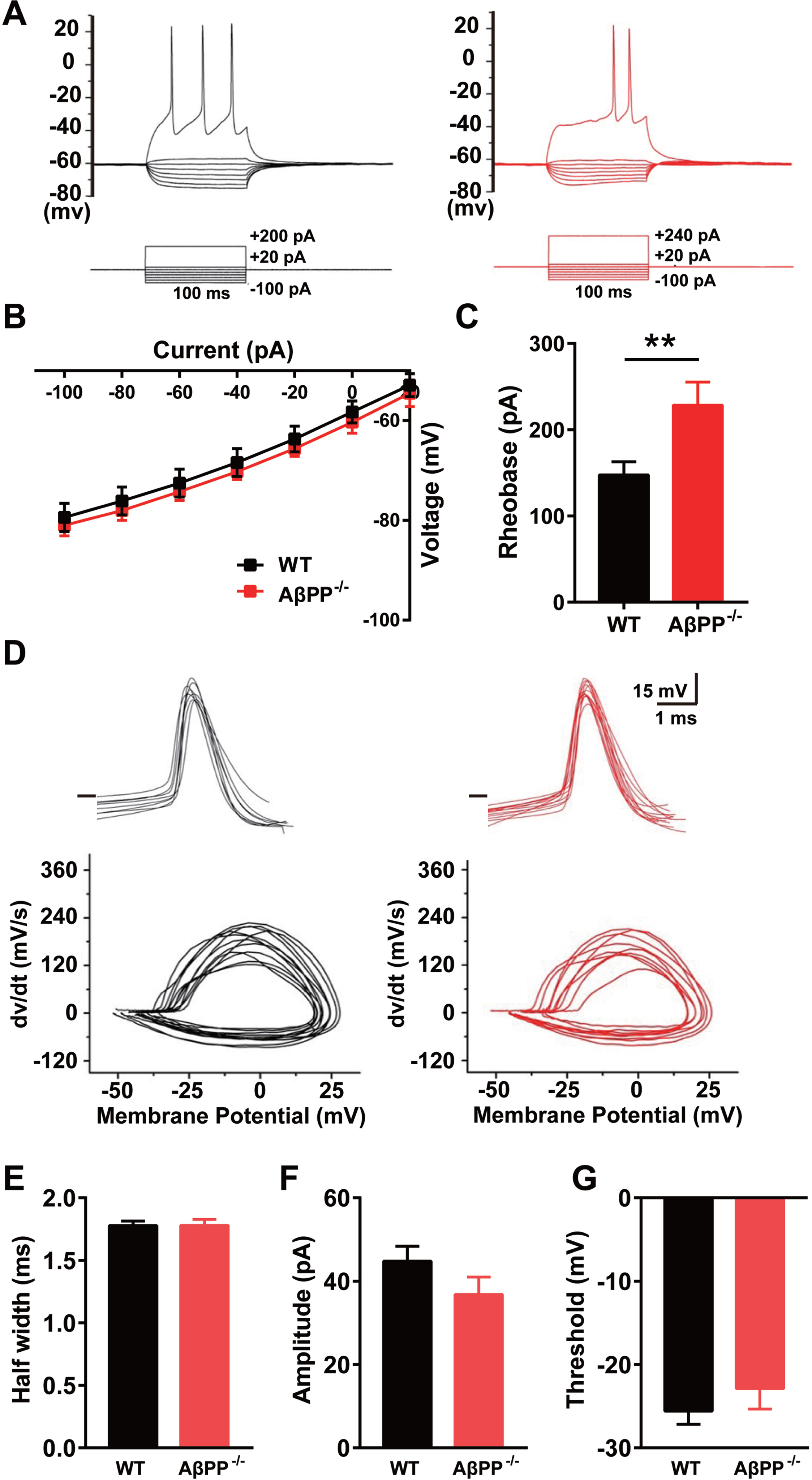
Reduced excitability of cultured glutamatergic neurons from the hippocampus of AβPP–/– mice. A) Representative traces of APs induced by step current injection in glutamatergic neurons of WT (black) and AβPP–/– mice (red). B) I–V curve allowing calculation of the input resistance of hippocampal glutamatergic neurons in WT (n = 9) and AβPP–/– mice (n = 7). C) The AP rheobase of recorded neurons in WT (n = 27) and AβPP–/– mice (n = 18). **p < 0.01; Student’s t-test. D) Sample traces of the first AP waveform induced by current injection. The phase plot of dV/dt versus V exhibits a rapid rise at spike initiation and has two components. E-G) AP parameters (half-width, amplitude, and threshold) in hippocampal GABAergic neurons in WT (n = 9) and AβPP–/– mice (n = 7).
AβPP deficiency increases levels of Kv1.4, but not via protein-protein interaction
A-type potassium channels determine somatic excitability, i.e., the electrical excitability of the somatic membrane, and have a role in AP repolarization/broadening and backpropagation under normal conditions [22, 35]. Activation of A-type potassium channels leads to a rapid depolarization of membrane potential. This property impacts repetitive firing rates and subthreshold membrane potential in many neurons, and thereby input resistance and neuron excitability [22, 36]. To test the hypothesis that loss of AβPP perturbs A-type potassium channel expression, with consequences for neuronal excitability, we first conducted immunoblots to determine the expression levels of different subtypes of A-type potassium channel. As Kv1.4 and Kv4.2 are the most abundantly expressed A-type potassium channels in the hippocampus [37], antibodies against these subtypes were used on hippocampal tissue extracts from AβPP–/– and littermate WT mice. We observed that Kv1.4 protein levels significantly increased in AβPP–/– mice (Fig. 4A, C) (WT, 1±0.09; AβPP–/–, 1.9±0.09; p = 0.04), while Kv4.2 levels remained unchanged (Fig. 4B, D) (WT, 1±0.07; AβPP–/–, 0.97±0.01; p = 0.80). To examine whether this could be reconciled with the expression of the corresponding mRNA, we measured Kv1.4 mRNA levels using qPCR (Fig. 4E). We found that Kv1.4 mRNA levels were unchanged between the two groups (WT, 1.08±0.1; AβPP–/–, 1.22±0.1; p = 0.40), suggesting that AβPP deficiency increases Kv1.4 protein levels at the post-transcriptionallevel.
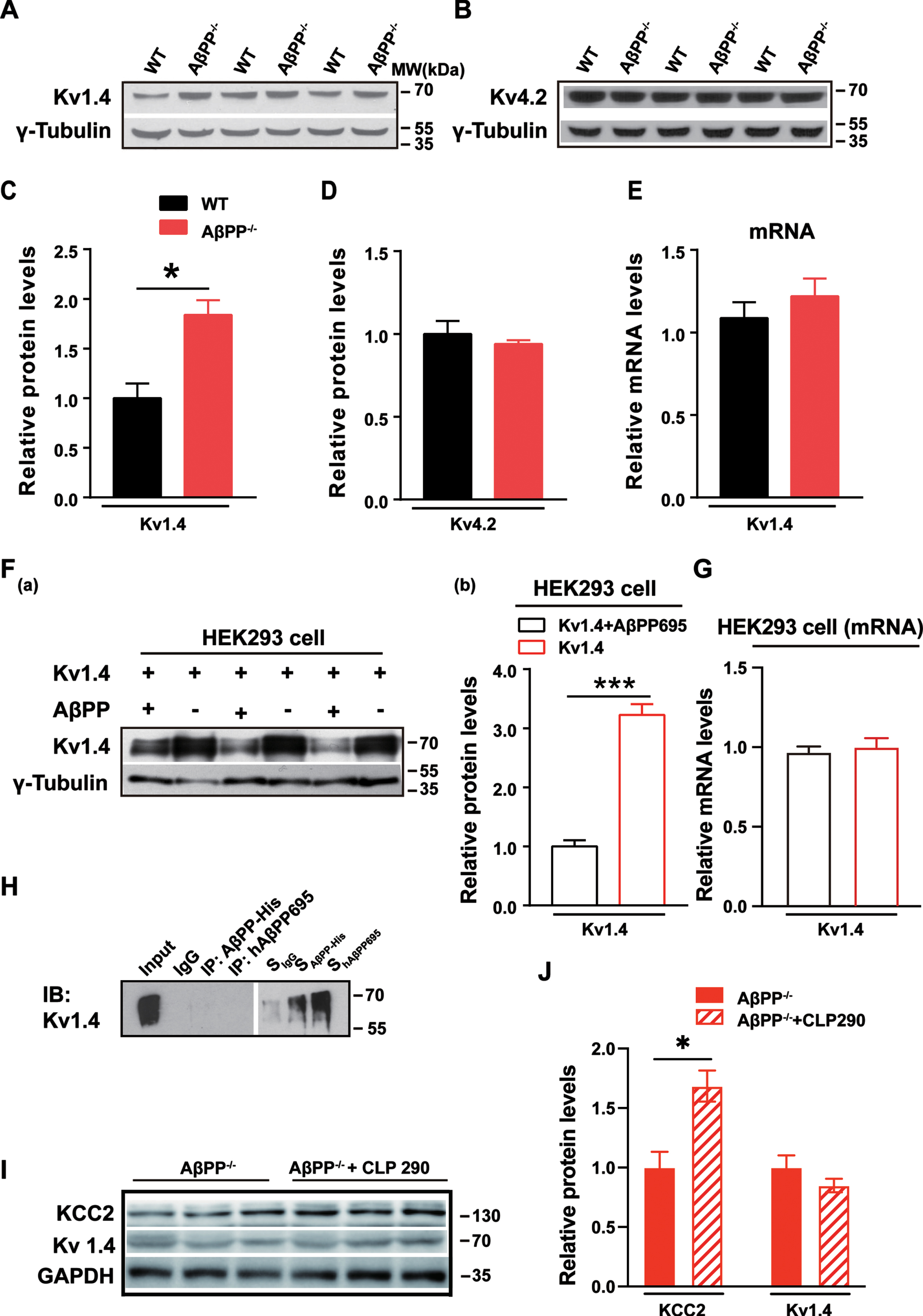
Increased post-transcriptional Kv1.4 expression in the hippocampus of AβPP-deficient mice. A, C) Immunoblots of Kv1.4 protein in the hippocampus of AβPP–/– and littermate WT mice. *p < 0.05; paired t-test. B, D) Immunoblots of Kv4.2 protein in the hippocampus of AβPP–/– and littermate WT mice. E) Kv1.4 mRNA levels in the hippocampus of AβPP–/– and littermate WT mice as assessed using qPCR. F(a), (b)) Representative immunoblots and quantitation of protein extracts of HEK293 cells transfected with Kv1.4 and AβPP. ***p < 0.005; paired t-test. G) Kv1.4 mRNA levels in the HEK 293 cells of expressing Kv1.4 and AβPP. H) Representative co-immunoprecipitation of AβPP and Kv1.4 (n = 4). I, J) Representative immunoblots of KCC2 and Kv1.4 in AβPP–/– mice after 7 days CLP290 injection compared with mice treated with vehicle. *p < 0.05; paired t-test.
To explore whether altered levels of Kv1.4 are a direct consequence of AβPP deficiency, we performed transfection experiments in the HEK293 cell line. Kv1.4 and hAβPP695 expression constructs were co-transfected as the control group, with the Kv1.4 construct and an empty pcDNA 3.0 vector being co-transfected as the experimental group.
We observed a dramatic decrease in Kv1.4 protein levels (Fig. 4F(a), (b)) in the AβPP overexpressed group compared to when AβPP was absent (Kv1.4, 3.2±0.19; Kv1.4+AβPP, 1.0±0.10; p = 0.015), suggesting that overexpressed AβPP suppressed the levels of Kv1.4 expression. The data were consistent with the results in hippocampal neurons, where Kv1.4 expression levels were significantly increased in AβPP–/– mice. Meanwhile, the mRNA levels of Kv1.4 in the HEK293 cells (Fig. 4G) were also tested, and results showed non-changed mRNA levels when overexpressed AβPP (Kv1.4, 1.0±0.03; Kv1.4+AβPP, 0.97±0.02; p = 0.40), suggesting that HEK293 cell shares the similar mechanisms of how AβPP regulates Kv1.4 with the hippocampustissue.
Next, we conducted co-immunoprecipitation experiments to determine whether there is AβPP-Kv1.4 protein interaction by transfection of hAβPP695+Kv1.4 plasmids or AβPP-His+Kv1.4 plasmids into HEK293 cells. As shown in Fig. 4H, we did not observe protein-protein interaction between AβPP and Kv1.4 (n = 4). To determine the intermediate factors that mediate AβPP regulation of Kv1.4 expression and function, we tested KCC2, a neuronal-specific transporter in the hippocampus that colocalizes with GABAAR. KCC2 levels are significantly reduced in the hippocampus of AβPP–/– mice [13]. CLP290, a novel KCC2 activator, or vehicle as a control, was injected into AβPP–/– mice intraperitoneally once daily for seven days [13]. After seven days of CLP290 injection, the KCC2 expression level was significantly increased in treated AβPP–/– mice compared with controls (Fig. 4I, J) (AβPP–/–, 1.0±0.13; AβPP–/–+CLP290, 1.68±0.13; p = 0.02). However, the expression of Kv1.4 was unchanged, suggesting that reduced levels of KCC2 in the absence of AβPP did not affect the expression of Kv1.4 (AβPP–/–, 1.0±0.1; AβPP–/–+CLP290, 0.85±0.06; p = 0.27).
AβPP deficiency alters the peak time of IA in both glutamatergic and GABAergic neurons
To evaluate whether AβPP deficiency alters Kv1.4-mediated IA, we performed whole-cell patch-clamp recording on cultured hippocampal neurons (14 DIV) from GAD67+/GFP mice, in which GABAergic neurons express GFP (Fig. 5A) [27]. IA was recorded by step current injection in the cultured hippocampal glutamatergic or GABAergic neurons. Representative traces of IA in glutamatergic and GABAergic neurons of WT and AβPP–/– mice are shown in Fig. 5B and 5C. The IA peak time in both types of neurons was significantly increased in AβPP–/– mice compared to WT (Fig. 5D, E) (WTglu versus AβPP–/–glu, p = 0.01; WTGABA versus AβPP–/–GABA, p = 0.0003; two-way ANOVA). Thus, increased levels of Kv1.4 altered IA.

AβPP deficiency increases the peak time of IA in hippocampal glutamatergic neurons. A) Image of patch-clamp recording in cultured 12–14 DIV GABAergic hippocampal neurons from GAD67 mice. Fluorescent neurons were classified as GABAergic neurons. B, C) Representative traces of IA recordings in GABAergic and glutamatergic neurons in WT and AβPP–/– mice. IA was recorded as follows: firstly, the current was recorded at the holding level (–60 mV) by giving –40 to +50 mV depolarized current in 10 mV steps, with each depolarization lasting 400 ms. Secondly, by keeping the same holding level, by giving –20 mV, 100 ms pre-stimuli, the fast-active, fast-inactive IA channels could be inactivated. Thirdly, IA was obtained by subtracting the two currents. D) Quantitative results of the IA time-to-peak in WT and AβPP–/– mice glutamatergic neurons. *p < 0.05; two-way ANOVA. E) Quantitative results of the IA time-to-peak in WT and AβPP–/– mice GABAergic neurons. ***p < 0.001; two-way ANOVA.
DISCUSSION
This study reveals that AβPP deficiency results in altered brain oscillations, comprising reduced theta and gamma power and increased epsilon and ripple power. Moreover, in line with these findings, the firing rate was reduced, caused by the increased rheobase for AP generation in hippocampal glutamatergic neurons of AβPP–/– mice (Fig. 6). Total Kv1.4 protein levels in the hippocampus were significantly increased in AβPP–/– mice, although mRNA levels remain unchanged, indicating that AβPP regulates Kv1.4 expression at the post-transcriptional level. Further mechanistic experiments on HEK293 cells revealed that AβPP and Kv1.4 did not undergo strong protein-protein interaction. Moreover, the whole-cell recording showed a significantly increased IA peak time in hippocampal neurons of AβPP–/– mice. Together, these results suggest that AβPP deficiency-induced upregulation of Kv1.4 expression and function may contribute, at least in part, to impairments in neuronal spiking and LFP in hippocampal neurons.

Graphic of the function of AβPP in hippocampal glutamatergic neurons. The indirect interaction of AβPP and Kv1.4 channels in WT mice enables regular neuronal spike firing and LFP. AβPP deficiency results in reduced theta and gamma power, increased epsilon and ripple power, and reduced neuronal excitability caused by the abnormal Kv1.4 channels.
Although a large body of research has examined the function of AβPP in neurons, less work has focused on mice at a young age in vivo. Our results showed impaired LFP and spike firing in young AβPP-deficient mice. Consistent with our findings, several other studies have shown that AβPP and its fragments affect brain networks at an older age. Zhang and colleagues recorded 3-to 4-month-old mice in which AβPP-like protein 2 (APLP2) had been knocked out. Their results showed a significantly reduced gamma-rhythm frequency in CA3 without a change the coherence of spike and field potential [38]. An increased AβPP intracellular domain (AICD) level in CA1 pyramidal cells impairs hippocampus gamma rhythm and firing rate via changes in Ca2 + and K+ conductance [39]. Other researchers used 9-month-old AβPP–/– mice and found impaired coupling of theta and gamma rhythms, which suggests that AβPP is important for this process [40]. Sharp wave-ripple (SPW-R) activity is another well-researched brain rhythm in the hippocampus; it is involved in memory consolidation and wakefulness-acquired memory. In vitro recording in the hippocampus of AβPP–/– mice indicate an unchanged SPW-R frequency [41]; however, in our in vivo experiments, SPW-R frequency increased. Since SPW-R activity is highly correlated with sleeping and waking states, the disparate results might be caused by differences in the wakefulness of the mice used in each case. In conclusion, our work shows that AβPP is essential for the modulation of oscillatory network activity, which might help to explain the impaired neuronal activity in patients with AD [42].
In the hippocampal tissue of AβPP–/– mice, the total amount of Kv1.4 protein is higher than in the wild type, but levels of Kv1.4 mRNA remain unchanged, suggesting that AβPP regulates Kv1.4 at the post-transcriptional level. AβPP might post-transcriptionally regulate Kv1.4 using small non-coding RNAs, like the microRNAs miR-17-92, miR-106a/520c, and miR-20, as executor molecules. MicroRNAs play an important role in the post-transcriptional regulation of synaptic plasticity, memory and cognition [43]. One study showed that miRNA-17-92 downregulates the expression of Kv1.4 channels and reduces IA in dorsal root ganglion neurons [44]. Furthermore, miRNA-17-92 is upregulated in AD patients, especially those with Aβ deposits, which might suggest a deficiency of AβPP to downregulate miRNA-17-92 [45]. Moreover, many microRNAs regulate the expression of AβPP, usually at the post-transcriptional level, including miR-106a/520c [46], the miR-20 family (miR-20a, miR-17-5p, miR-106b) [47], miR-101 [48], miR-16 [49], and miR-153 [50].
We found, in this study, that AβPP and Kv1.4 do not have a strong interaction. Previous work has shown that the interaction of Kv1.4 with other proteins is mediated by several domains or protein factors, including the Kv1.4 C-terminal motif [51], N-glycosylation of Kv1.4 [52], a pore region determinant [53], and postsynaptic density protein-95 [54]. Although AβPP interacts with KCC2 to affect the hippocampal neuronal activity [13], the present study shows that KCC2, which mediates GABABR signaling, does not affect Kv1.4 expression, excluding the possibility that AβPP regulates Kv1.4 via KCC2. However, multiple strands of evidence make it likely that GABABR is involved in the regulation of Kv1.4 by AβPP: we have previously shown that AβPP deficiency alters the expression of GABAR in the medial prefrontal cortex [55, 56]; in rat hippocampal neurons, transient activation of GABABR by its agonist baclofen increases IA conductance [57]; and baclofen enhances IA in rat trigeminal neurons [58]. Therefore, further research will be required to clarify this issue.
Another area with potential Kv1.4 involvement that deserves attention is spike-timing-dependent plasticity (STDP). This process regulates the strength of neuronal connections and involves, in particular, the repeated coupling of presynaptic and post-synaptic APs. An impairment of STDP mechanisms has been reported in AD patients [42].
Kv1.4 is expressed in excitatory neurons but not in somatostatin interneurons or glutamate decarboxylase 65 neurons; Kv1.4 is also not detected in inhibitory axons [59]. We thus reason that the recorded A-type current in GABAergic neurons might be induced by A-type channels other than Kv1.4, such as Kv4.2, which is highly expressed and is distributed in GABAergic neurons in the hippocampus and other brain areas [60–62]. Moreover, the Kv4.2 channel is mainly expressed in GABAergic post-synapses, which weaken the inhibition during dendritic depolarization by backpropagating APs [62]. With respect to the time-to-peak of A-type currents, the parameters were increased in both glutamatergic and GABAergic neurons, which indicates an effect of AβPP on the properties of Kv1.4 and, putatively, Kv4.2 channels. However, the hippocampus contains only about 20% GABAergic neurons, making it difficult to detect changes in the expression level of Kv4.2.
Finally, given that AβPP is the precursor of Aβ, our study may have some implications for AD pathology, e.g., potential alterations of Kv1.4-mediated function in the AD brain. Thus, the modulation of IA in glutamatergic and GABAergic neurons by AβPP might underlie the impairment of neuronal activity and connections in AD. It is worth noting that the present study was conducted using the AβPP knock-out mouse only. While this model is widely used to study the physiological function of AβPP, it has not been used to investigate AD pathogenesis. Thus, our results may not be comparable with data obtained in AD patients and AD mouse models [63, 64].
Conclusion
By combining multiple in vivo and in vitro techniques, this study uncovered a reduced firing rate and abnormal oscillatory network activity in the hippocampus of AβPP-deficient mice. AβPP deficiency induced upregulation of Kv1.4 at the post-transcriptional level and increased IA peak time, which may underlie the altered neuronal firing and oscillatory activity, providing evidence that AβPP regulates potassium ion channel activity.
Footnotes
ACKNOWLEDGMENTS
We sincerely thank Dr. Zhuo Fan and Michael M. Tamkun for the generous gift of the Kv1.4 plasmid. We also thank Xuan-xuan Zhang for the kind gift of the HEK293 cells.
FUNDING
The study was supported by grants from the National Natural Science Foundation of China (31970915, 32170950, 31871170), the Natural Science Foundation of Guangdong Province (2021A1515010804, 2023A1515010899), the Guangdong Natural Science Foundation for Major Cultivation Project (2018B030336001), and the Guangdong Grant ‘Key Technologies for Treatment of Brain Disorders’ (2018B030332001).
CONFLICT OF INTEREST
The authors declare that the research was conducted without any commercial or financial relationships construed as a potential conflict of interest.
DATA AVAILABILITY
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.