
Abstract
The aging population increases steadily because of a healthy lifestyle and medical advancements in healthcare. However, Alzheimer’s disease (AD) is becoming more common and problematic among older adults. AD-related cases show an increasing trend annually, and the younger age population may also be at risk of developing this disorder. AD constitutes a primary form of dementia, an irreversible and progressive brain disorder that steadily damages cognitive functions and the ability to perform daily tasks. Later in life, AD leads to death as a result of the degeneration of specific brain areas. Currently, the cause of AD is poorly understood, and there is no safe and effective therapeutic agent to cure or slow down its progression. The condition is entirely preventable, and no study has yet demonstrated encouraging findings in terms of treatment. Identifying this disease’s pathophysiology can help researchers develop safe and efficient therapeutic strategies to treat this ailment. This review outlines and discusses the pathophysiology that resulted in the development of AD including amyloid-β plaques, tau neurofibrillary tangles, neuroinflammation, oxidative stress, cholinergic dysfunction, glutamate excitotoxicity, and changes in neurotrophins level may sound better based on the literature search from Scopus, PubMed, ScienceDirect, and Google Scholar. Potential therapeutic strategies are discussed to provide more insights into AD mechanisms by developing some possible pharmacological agents for its treatment.
Keywords
INTRODUCTION
The World Health Organization (WHO) has stated that Alzheimer’s disease (AD) is a public health priority due to less effective prevention strategies. An estimated 139 million people worldwide will be affected by 2050. In 2019, the total global societal cost of dementia was estimated to be US$ 1.3 trillion. The WHO reported that about 50 million individuals worldwide have dementia [1], and nearly two-thirds of them battle with AD [2]. In 2019, a report from the WHO’s Global Health Estimates stated that AD and other dementias are the 7th leading cause of death worldwide, with a significant increase of 174% from 2000.
Dementia is an umbrella term used to describe a clinical syndrome of progressive cognitive decline, but its subtypes (AD, vascular dementia, Lewy body dementia and frontotemporal dementia) are classified according to the cause of dementia. AD is the most common neurodegenerative disease responsible for dementia, comprising 60% to 80% of cases. The pathological cascade of neurodegeneration has been increasingly understood, linked to the progression of AD, and characterized by the development of dementia [3]. As human longevity increases, chronic neurodegenerative disorders are having a growing impact on the health of the older adult population. Alois Alzheimer, a German neurologist, was the first to describe AD in 1906 [4] as an irreversible neurodegenerative disease that gradually destroys memory and cognitive skills, leading to the inability to carry out simple daily tasks [5]. AD is categorized as the most common type of dementia and a significant cause of death in the aging population. In 2017, the Alzheimer’s Association report stated that AD is the 6th leading cause of mortality in America, with a significant increase of 89% from the year 2000. AD is more common among older adults aged 65 years and above, but there are cases with early onset of AD in the younger population [6]. Individuals at 65 years of age have a 3% chance of developing AD. The likelihood increases to 30% in people above 85 years of age [7]. With the establishment of birth control policies in numerous nations and the advancement of medical therapies in the modern world, the population of older persons has increased. If the trend continues, more people will soon develop AD.
We searched Scopus, PubMed, ScienceDirect, and Google Scholar for relevant studies published in English. The search time included publications from 1 January 1985 until 31 May 2022. We also included highly regarded references before this time. Search terms we used were “Alzheimer’s disease”, “Alzheimer’s dementia”, “neurobiology”, and “pharmacotherapy”. In these publications, we followed up on cited references and also included relevant articles in the analysis.
The histopathology of AD includes neuronal loss, senile plaques, and neurofibrillary tangles (NFTs). The hallmark symptoms are memory loss, neurobehavioral abnormalities, and the inability to carry out daily activities. AD is a complex neurodegenerative disorder with multifaceted pathogenesis. So far, the therapeutic paradigm “one-compound-one-target” has failed. Despite the enormous efforts to elucidate the pathophysiology of this disorder, the disease is still incurable [8]. Current treatment for mild to moderate AD uses the cholinesterase inhibitors (ChEI); donepezil (Pfizer, New York, NY, USA), rivastigmine (Novartis, Basel, Switzerland), and galantamine (Janssen, Beerse, Belgium). Moderate to severe AD is treated with memantine, an N-methyl-D-aspartate receptor non-competitive as mentioned in Table 1 antagonist [9]. The summary of the currently available drugs for AD treatment is presented in Table 1. Thus, a multi-target drugs approach may be more effective in managing AD. A better understanding of the pathologies of the disease may ease the identification of possible therapeutic targets for AD. It may lead to the discovery of more effective drugs for AD patients. The current review article describes the pathophysiology and potential therapeutic targets to manage AD. Figure 1 summarizes the pathophysiology that leads to AD development.
Summary of the currently available drugs for AD treatment
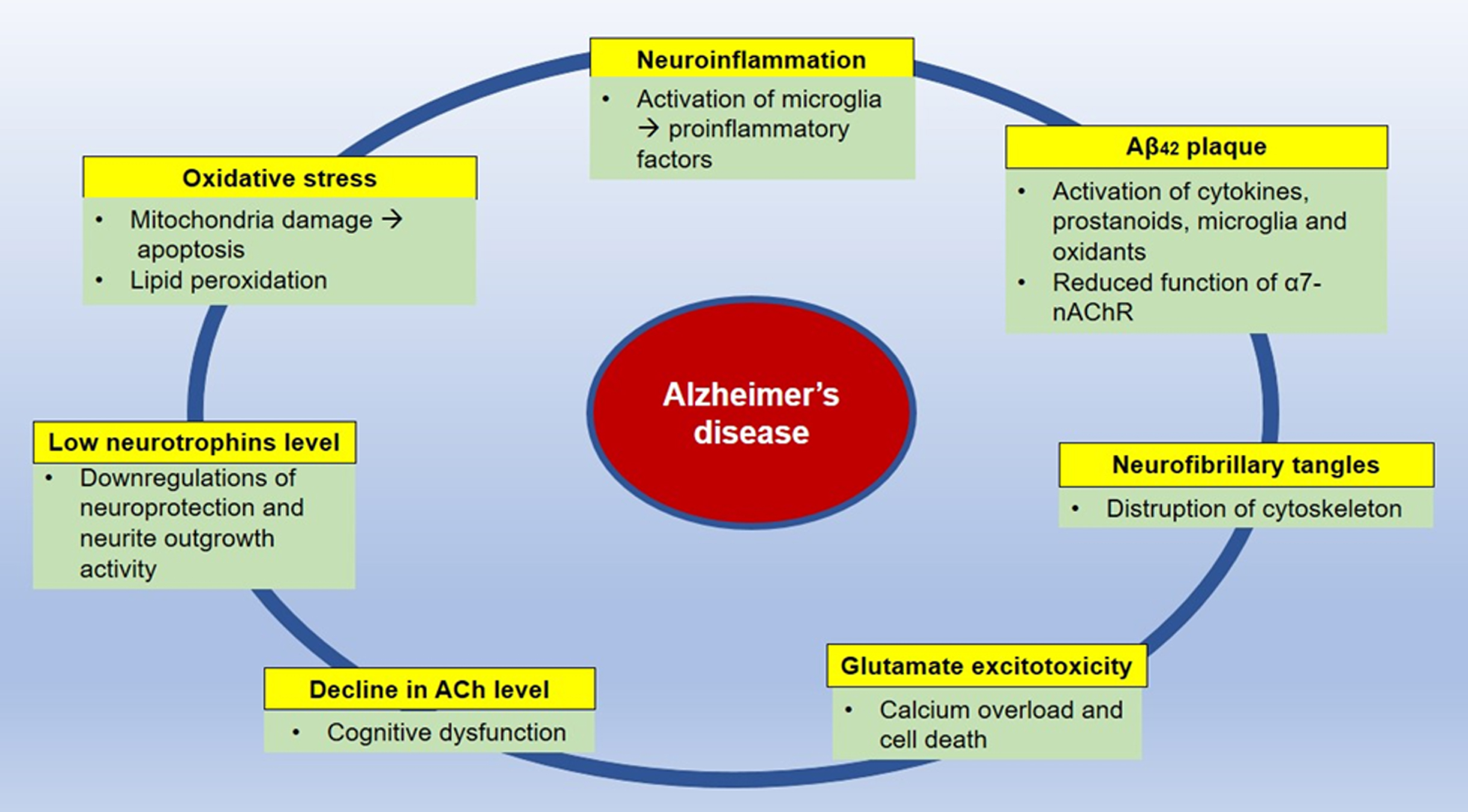
Summary of the pathophysiology of Alzheimer’s disease. ACh, acetylcholine; nAChR, nicotinic acetylcholine receptor.
PATHOPHYSIOLOGY OF ALZHEIMER’S DISEASE
AD is characterized by the abnormal accumulation of amyloid-β (Aβ) and hyperphosphorylated tau proteins, which are two hallmarks of AD. However, there are other factors such as neuroinflammation, oxidative stress, glutamate excitotoxicity, reduced neurotrophins level, and cholinergic dysfunction that contribute to the progression of the disease. This pathophysiology of AD will be further discussed in this section.
The amyloid cascade hypothesis
The amyloid hypothesis was first promulgated in 1991 by John Hardy and David Allsop [10]. Aβ plaques consist of accumulated peptides originating from the hydrolysis of amyloid-β protein precursor (AβPP) via the amyloidogenic pathway by the action of aspartyl proteases enzymes, β- and γ-secretases (Fig. 2) [11]. The enzymes β-secretases (β-site APP-cleaving enzyme 1, BACE1) and γ-secretases play an essential role in the formation of Aβ in AD patient’s brain. Both BACE1 and γ-secretases have been considered targets for inhibiting Aβ formation and counteracting AD progression. The enzyme α-secretase has not been targeted since it cleaves AβPP in the Aβ area but does not cause Aβ plaque formation [12]. α-secretase cleaves the Aβ domain of AβPP between lysine 16 and leucine 17 to form the AβPP ectodomain and α-carboxyl-terminal (CTF), C83, which is then cleaved by γ-secretase to produce the non-amyloidogenic 3 kDa fragment, p3 and APP intracellular domain (APPICD) [13]. Initially, β-secretase catalyzes the cleavage of AβPP by detaching its large ectodomain and leaving a membrane-bound C-terminal, β-carboxyl-terminal, C99 (β-CTF). Then, depending on the point of the AβPP cleavage by γ-secretase, either the 40 or 42 amino acid residues, Aβ42, and Aβ40 are produced [14]. Among these two peptides, Aβ42 is more susceptible to clumping and forming oligomers, protofibrils, insoluble fibrils, and plaques [15].
The Aβ plaques trigger the activation of cytokines, prostanoids, microglia, and the production of oxidants (most notably peroxynitrite) that function in various ways. These includes (a) killing neurons, either directly or by increasing their sensitivity to excitotoxicity; (b) boosting the production of Aβ by neurons and astrocytes – thereby closing the vicious cycle; (c) disrupting neuron structure and function by promoting excessive tau phosphorylation; (d) disrupting the protective function of astrocytes [16]; (e) oxidative stress [17]; (f) mitochondrial dysfunction [18]; and (g) endoplasmic reticulum stress [19].
Aβ is created in the healthy brain during neuronal activity and is required for synaptic plasticity and memory [20]. In pathological situations, however, aberrant Aβ buildup creates plaques that interfere with the activation of receptors essential for physiological function. Inactivation of the alpha-7 nicotinic receptor (nAChR) located on cholinergic neurons, for example, leads to synaptic dysfunction and memory loss. When Aβ42 binds to the 7-nAChR, the receptor is internalized and accumulates within the lysosomal compartment [21]. The interaction of Aβ with nAChR disrupts cholinergic signaling and causes cell death, implying that this interaction may be necessary for the etiology of AD [22, 23]. Tau phosphorylation at Serine 202, Threonine 181 and 231 via extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK-1) may also result in Aβ42 interaction with the 7-nAChR [24, 25].
Inhibition of BACE1
BACE1 has been identified as the primary therapeutic target for developing inhibitory drugs as it catalyzes the first step in cleaving AβPP to produce Aβ. Furthermore, a significant increase in BACE1 protein and activity has been observed in the AD brain compared to the normal brain [26, 27]. The treatment with a BACE1 inhibitor continuously prevented age-related cognitive impairment in transgenic AD mice. A few BACE1 inhibitors have also entered early-stage clinical trials [28]. Despite this, BACE1-deficient animal models have demonstrated a relatively benign phenotype with high survivability, implying that targeting BACE1 could be a safe treatment approach. A further study, however, revealed that severe suppression of this enzyme might result in harmful side effects such as hypomyelination and behavioral \nobreak disorders such as seizures. Aside from AβPP, BACE1 has several additional substrates relevant to myelination, such as neuregulin-1 [29].
Hence, instead of focusing on compounds that inhibit BACE 1, attention should be given to compounds that modulate BACE 1 to selectively alter Aβ production without hindering signal transduction from the Notch receptor, a single-pass transmembrane protein with extracellular, transmembrane, and intracellular domains. Another strategy to prevent Aβ production could be partial suppression of BACE 1 activity. Singer et al. revealed that in AβPP transgenic mice which were genetically modified for AβPP overexpression, a partial reduction in BACE1 using RNA interference reduced amyloid neuropathology, including Aβ deposition [30]. It also improved cognitive impairments. Furthermore, compared to homozygous BACE1-/- mice, heterozygous BACE1 (BACE1 + /-) knockout mice demonstrated normal spatial memory function, implying partial suppression of BACE1 may not affect ordinary learning and memory processes.
Classification of BACE1 inhibitors. BACE1 inhibitors were classified into two main categories: non-peptidic and peptidomimetics [12]. When developing BACE1 inhibitors, it is necessary to evaluate the drug’s capacity to cross the blood-brain barrier (BBB) and neuronal membranes in order to reach the target, due to BACE1’s position in the brain and endosome lumen [31]. BACE1 has a large catalytic pocket, which means that strong inhibitors are necessary when constructing BACE1 inhibitors. Still, at the same time, it should be small enough to exhibit suitable drug-like properties [32]. The most important characteristic of inhibitors is that they should not generate toxic cross-inhibition effects. Because the two catalytic aspartic acid residues are conserved across the class of aspartic proteases, the inhibitors should be selective over other aspartic proteases [12]. Potent peptidomimetic inhibitors tested in vitro appeared less efficient when tested in vivo, due to reduced oral availability, short half-life, and low brain permeability. The modification of peptidomimetics to make them more brain penetrant is one of the challenges in designing novel inhibitors [33].
Example of BACE1 inhibitors. JNJ-54861911, CNP520, LY3202626, Elenbecestat, Lanabecestat, and Verubecestat are examples of inhibitors that have shown high potential in vivo and have been utilized in human clinical studies to treat individuals with mild-to-moderate AD and those at risk for AD [34, 35]. Verubecestat (MK-8931) was the first BACE1 inhibitor to enter clinical phase III trials. It is in the most excellent position to validate the safety and efficacy of BACE1 inhibition [36]. However, Merck’s recent statement that one of its trials will be halted has raised concerns. In contrast, AstraZeneca’s Lanabecestat (from the aminoimidazole class) is a potent, highly permeable, orally active, BBB-penetrant with a very slow off-rate from BACE1, resulting in a lengthening of the observed effect [37]. Although designing and developing BACE1 inhibitors is difficult, it is worthwhile to find novel BACE1 inhibitors. There is compelling evidence that BACE1 inhibitors operate as disease modifiers, and their use will assist AD patients.
Inhibition of γ-secretases
Efforts have been taken to develop drugs that inhibit γ-secretase. General inhibition of γ-secretase, on the other hand, has the potential to have severe effects by interfering with different physiological and developmental processes, such as the proteolysis of non-AD components, including Notch [38]. Notch signaling regulates cell proliferation, cell fate decisions, and differentiation initiation during embryonic and postnatal development. Notch signaling controls cell renewal and binary fate decisions in the adult intestine. It differentiated and proliferated adult stem cells in the epidermis. It also retained neural progenitor identity and inhibited neuronal differentiation during early development, neurite remodeling and synaptic development plasticity in the adult brain and muscle regeneration. Notch pathway dysregulation or misexpression has been associated with cancer (e.g., T-ALL) and adult-onset illnesses (e.g., cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CASADIL) and multiple sclerosis) [39].
Familial Danish Dementia knock-in (FDDKI) mice have shown progressive synaptic and memory deficits [40] due to loss of Bri2. This protein interferes with the cleavage of AβPP by secretase through binding to AβPP, hence inhibiting the AβPP processing by secretases [41]. Depletion of Bri2 accelerates the cleavage of AβPP by secretases in FDDKI mice. Tamayev and D’Adamio [42] reported treatment with a γ-secretase inhibitor (GSI) results in memory decline in FDDKI mice. β-cleavage of AβPP yields β-CTF and the amino-terminal-soluble (βAβPPs) fragments. γ-secretase processing of β-CTF generates Aβ, the leading cause of AD. A high level of β/α-CTFs AβPP fragments was observed in synaptic fractions isolated from hippocampi of FDDKI mice treated with GSI, which links to deleterious effect on memory. Hence, it is suggested that βAβPPs/β-CTF, but not Aβ are the toxic species that cause memory loss. However, more investigation is required to confirm this finding.
Enzymatic/glial degradation, transcytotic delivery, and perivascular drainage
Aβ40 and Aβ42 are known as neurotoxic agents. Their clearance rate is estimated to be 8% per hour in the normal human cerebrospinal fluid. However, it can plummet to more than 30% in patients with AD. Aβ stable isotope labeling kinetic or SILK studies found that production and clearance of soluble Aβ isoforms are similar for Aβ38 and Aβ40. Aβ42 turnover is altered with increasing age [43]. Enzymatic/glial degradation, transcytotic delivery, and perivascular drainage are the primary mechanisms by which Aβ is removed. Firstly, activation of Aβ-degrading enzymes such as neprilysin (NEP), angiotensin-converting enzyme, and insulin-degrading enzyme facilitate the breakdown of Aβ. The introduction of NEP into the brain of AβPP transgenic mice using viral vectors has improved cognitive function by decreasing Aβ pathology [44]. NEP is a 97 kD cell surface-associated enzyme that degrades short peptides in the central and peripheral nervous systems [45]. Like all identified Aβ-degrading proteases, NEP degrades monomeric Aβ. However, the Aβ sequence from mutated AβPP makes Aβ monomers more resistant to NEP-catalyzed cleavage. In vitro, NEP was reported to degrade oligomeric forms of synthetic Aβ. Nonetheless, it was unable to destroy naturally secreted Aβ oligomers isolated from cultured cells, showing that modifications to the production of Aβ oligomers may be required [46]. The capability of NEP in the degradation of Aβ requires further investigation. Secondly, suppression of the receptor for advanced glycation end products (RAGE), which facilitates transcytosis influx of circulating Aβ into the brain, can enhance transcytotic delivery. The successful usage of the RAGE inhibitor, TTP488, in Phase II testing has resulted in a Phase III clinical trial for AD patients. Thirdly, the perivascular drainage system appears to be propelled by the motive force created by cerebral artery pulsations, implying that vasoactive medications can help with Aβ clearance. Cilostazol, a specific inhibitor of type 3 phosphodiesterase, is one of the medications stimulating this mechanism [47]. According to Puzzo et al. most clinical treatments designed to lower Aβ levels, have failed. Researchers argue that the difficulties of making an early diagnosis to begin early treatment is the main reason for failed “anti-amyloid” therapy [48].
Antibody-based immunotherapeutic approach
The latest current treatment of AD using an antibody-based immunotherapeutic approach has been developed using human B-cell clones prompted by neo-epitopes exhibited in pathological Aβ aggregates [49]. When a group of human memory B cells was screened for reactivity against aggregated Aβ, the expression of Aducanumab was determined. Aducanumab is a human monoclonal antibody that is not only selective for aggregated Aβ, but also soluble oligomers and insoluble fibrils [50]. Aducanumab revealed the ability to pass the BBB in preclinical tests, resulting in decreased brain amyloid deposition [50]. Aducanumab, marketed under the brand name Aduhelm, is the first AD drug approved by the United States Food and Drug Administration (FDA) since 2003 and the first targeted therapy for the illness. This is the first licensed medication that seeks to address the underlying causes of AD rather than just the symptoms. The FDA approved the medicine in June 2021 through its accelerated approval pathway, which expedites the approval of treatments that are believed to benefit individuals with AD. Aducanumab is a monoclonal antibody directed against Aβ, where it binds to the Aβ aggregate. It is designed to specifically target an epitope that is often inaccessible in the Aβ monomer. Aducanumab works by inhibiting the formation of amyloid plaques in the brain via the interaction above, potentially decreasing neurodegeneration and disease development [50]. Numerous patients, their families, and advocacy groups have supported the approval, stressing that while Aducanumab is not a cure, it may serve as a starting point for more enhanced treatments and encourage more significant innovation in AD treatment discoveries [51]. The Aβ hypothesis is widely accepted. The development of drugs to attenuate the effects of Aβ in AD brains is still under investigation. The discovery of effective drugs against Aβ, either to inhibit overproduction or clearance of Aβ plaques still holds promise to benefit ADpatients.
Tau hypothesis
The tau protein is a microtubule-associated protein primarily present in adult neurons. It plays an essential role in microtubule assembly and neuronal microtubule stabilization [52, 53]. Tau protein buildup was discovered from plaques in the brains of AD patients in 1988, implying for the first time that tau protein could be a probable cause of dementia as observed in AD [54]. An abnormal rise in the number of hyperphosphorylated tau proteins in the cytosol caused the protein to clump together into paired helical filaments and straight filaments, which are often called NFTs. These result in disruption of the cytoskeleton’s structural and regulatory activities [55]. This disruption impacts severe biological processes in the nervous system, such as maintaining proper shape, axonal transport, synaptic dysfunction, and neurodegeneration [56]. Pharmacological interactions such as phosphorylating kinase inhibition and tau phosphatase activation have been proposed as intervention options to combat taudysfunction.
Inhibition of phosphorylating kinases
Kinase controls biological activity by activating protein by phosphorylation of specific amino acids utilizing ATP as a phosphate source. Protein phosphorylation plays a role in both normal and pathological processes. In AD, for example, phosphorylation of AβPP may affect the ability of β-secretase to cleave AβPP to generate Aβ1–42 [57]. Furthermore, both β-secretase and γ-secretase, which are responsible for the production of Aβ, are regulated by phosphorylation [58, 59].
Activating three tau protein kinases throughout aging, namely glycogen synthase kinase 3 (GSK3), Erk1/2, and p38, results in tau hyperphosphorylation and synaptic protein loss, long-term potentiation, and memory degradation in rats [60]. Kinases such as CDK5 or PKA make tau a more attractive substrate for GSK3 [61, 62], but other kinases such as PKB, PKC, or PKN suppress GSK3 activity [63-65]. GSK3β, p38 active form, JNK, CK1, PKA, PKN, Fyn, and c-Abl colocalize with NFT in AD brains [66–69]. CK1 and DYRK1A mRNA levels were raised in these diseased brains [70, 71], as were the expression levels of p38, Erk1/2, and JNK1/2/3 [72, 73]. These findings suggest that a dysregulation of protein kinases may contribute to tau hyperphosphorylation. However, disagreement exists on which of these kinases is the most critical for tau hyperphosphorylation in human illness. Efforts must be made to minimize the adverse effects of blocking these kinases, as kinases are required for the regular function of many signaling pathways [74].
Activation of tau phosphatases
Protein phosphatases (PPases) are classified according to their capacity to dephosphorylate serine, threonine, or tyrosine residues. PP1, PP2A, PP2B (also known as calcineurin), PP4, PP5, and PP6 are members of the phosphoprotein phosphatase family, whereas the metal-dependent protein phosphatase (PPM) family contains several forms of PP2 C and mitochondrial PPases. The brain has a high concentration of serine/threonine PPases, with PP1, PP2A, PP2B, and PP5 being particularly abundant and implicated in AD. In recent investigations, protein-tyrosine phosphatases (PTPases), particularly striatal-enriched tyrosine phosphatase, have also been involved with AD [75].
PPases could inhibit protein kinases involved in tau hyperphosphorylation. The role of phosphatase is to remove phosphate groups from molecules, hence lowering hyperpolarization. In normal human brains, measurement of phosphatase activities reveals a predominance of PP2A activity (71%) over other phosphatases such as PP2B (7%), PP5 (11%), or other phosphatases (11%) [76, 77]. Total phosphatase activity is reduced by half in AD brains [77], indicating that specific tau phosphatases are required for AD [78]. A considerable decrease in protein phosphatase activity is the primary cause of tau hyperphosphorylation in the AD brain.
Protein phosphatase 2A activator. Previous biochemical experiments were used to dephosphorylate hyperphosphorylated phosphatase tau using several PPase preparations immunoprecipitated from the human brain. They have demonstrated that PP2A is the primary phospho-tau phosphatase [77–80]. PP2A is extensively expressed in all tissues of vertebrates and is involved in the dephosphorylation of thousands of distinct phosphoproteins. PP2A is abundantly expressed in the brain, where it interacts with various substrates [81]. A decreased activity of PP2A was observed in AD due to reduced levels, increased inhibition, and alterations in its specificity and subcellular localization. Reduced mRNA expression of the PP2A catalytic subunit may cause decreased levels of this protein [82]. A protein called SET or inhibitor 2 of PP2A was highly expressed in AD brains. This protein binds to and inhibits PP2A [83].
Additionally, activating GSK3 increased the inhibitory phosphorylation of PP2A at Y307. Inhibition of GSK3 decreased the inhibitory impact both in vitro and in vivo. These findings suggest a correlation between PP2A and GSK3. Alterations in PP2A activity resulted in an integrity breakdown of the cytoskeleton. It induces apoptosis by phosphorylating pro-apoptotic molecules at S112 and S136, and Akt at T308 and S473 [85]. Several studies have been published on this subject [86–89]. As a result, decreased PP2A activity appears to be a substantial factor for increased tau phosphorylation. Only the ABαC isoform of PP2A has been shown to bind to and dephosphorylate tau efficiently. While other types of PP2A may be involved in additional disease-related pathways, the data indicate that the predominant PP2A deficit in AD pathogenesis is methylation ABαC deficiency. PP2A appears to dephosphorylate AβPP at Thr668, where it is phosphorylated by Cdk514 and c-jun N-terminal kinase, inhibiting the formation of neurotoxic peptides [90].
PP2A’s dephosphorylation of kinases involved in neuroinflammatory pathways decreases kinase activity, hence decreasing inflammation [91]. As a result, lower PP2A levels and activity in the AD brain may increase inflammation. When the PP2A inhibitor, okadaic acid was stereotactically injected into rats brain, several typical signs of AD such as Aβ deposition, tau hyperphosphorylation, and neurodegeneration were observed [92, 93]. However, because okadaic acid actions are non-selective, other protein phosphatases, such as PPI or PP4– 6, may also be inhibited at the concentrations used in these in vivo investigations. Cantharidin, microcystin-LR, and nodularin are also PP2A inhibitors but are less used in research [94, 95].
The decreased activity of PP2A is related to the presence of two endogenous protein inhibitors, I1PP2A and I2PP2A. In AD brains, nuclear I1PP2A and cytoplasmic I2PP2A activities were elevated by 20% [83, 96]. Inhibiting I2PP2A cleavage appears to be a promising therapeutic strategy to block tau pathology [97, 98]. Thus, addressing these inhibitors may be another strategy for restoring PP2A in AD.
Several findings indicate that decreasing PP2A activity promotes AD progression. Accordingly, increasing PP2A activity should benefit AD management. Hence, direct PP2A activators or endogenous biochemical regulatory mechanisms are generated to achieve this goal. Several drugs are being explored to increase PP2A activity. (a) sodium selenate has been tested in an AD model. This anionic PP2A activator has been shown to decrease tau hyperphosphorylation and to improve cognitive performance [99, 100], (b) memantine has been shown to inhibit I2 activity towards PP2A [101], (c) metformin, which increases PP2A levels by inhibiting its degradation, has been shown to be beneficial in AD models [102], (d) the tau fibrillization inhibitor, phenothiazine methylene blue, has been identified, (e) davunetide, an eight amino acid peptide derived from the activity-dependent neuroprotective protein activity-dependent neuroprotector homeobox protein, reduced tau phosphorylation in tau transgenic mice and 3x transgenic AD mice [15], (f) eiconsanoyl-5-hydroxytryptamide, an inhibitor of PP2A demethylation, promoted PP2A activity, decreased protein phosphorylation, and improved cognitive function in rodent models of neurodegeneration [103], and g) SIG-1012 inhibited protein phosphatase methylesterase 1and efficiently increased PP2A methylation and activity in hippocampal neurons. SIG-1012 administration has been demonstrated to decrease tau phosphorylation in rats [104].
The neuroinflammatory hypothesis
Researcher’s strategies on AD have shifted significantly during the last few decades. There are currently few studies elucidating the neuroinflammation pathology in the brain of AD patients [105]. In AD, Aβ plaques and NFT activate the inflammatory responses to eliminate the foreign substances. Microglia act as neuroprotection, defending the brain against infectious agents. When harmful chemicals threaten neuronal cells, microglia are driven to generate new receptors and signaling molecules to support the damaged regions [106]. Prolonging the immunological cell response in the presence of Aβ plaques, on the other hand, creates instability and an abnormal balance of inflammatory components. Microglia generate proinflammatory cytokines such as IL-18, TNF-α, IL-6, PGE2, INFγ, and IL1, resulting in neurodegeneration and aggravating neurological diseases [106–109]. Chronic immunological responses in the brain eventually result in neuronal degeneration and cognitive impairments. Simultaneously, extensive research suggests that chronic inflammation within the brain may exacerbate the symptoms of the disorders by accelerating the generation of Aβ and NFT [110–113].
The usage of non-steroid anti-inflammatory drugs (NSAIDs)
Researchers discovered a prolonged immune response in neuroimaging studies and in postmortem tissues of AD patients and in rat models [114–117]. To bolster the theory, epidemiological research revealed that people with rheumatoid arthritis who received NSAIDs were consistently free of AD [118, 119]. This finding may provide support for a connection between the etiology of AD and neuroinflammation.
Since introduction of the neuroinflammation theory, therapeutic agents have been created and extensively explored to suppress or slow down the aberrant immune response associated with AD. NSAIDs are currently one of the most promising therapies for AD. These drugs primarily work through four distinct mechanisms to exert their anti-inflammatory effects. This involves inhibition of cyclooxygenase (COX)-1 and -2 expression, decreasing PGE2 levels, decreasing Aβ42 levels, and activating peroxisome proliferator-activated receptor-gamma (PPARγ) [120].
COX is a unique protein found in the central nervous system (CNS) that catalyzes the production of prostaglandin G2 from arachidonic acid and its reduction to prostaglandin H2 and other prostaglandins. COX-2 expression was increased during inflammation in both neurons and reactive microglia in various studies [121–124]. COX-1 and COX-2 inhibitors also demonstrated promising results in attenuating the neurotoxicity of chronic neuroinflammation and apoptosis in animal models and in cell culture [125, 126]. Aspirin [127], indomethacin [128], etoricoxib [122], etodolac [129], and diclofenac [130] are all examples of NSAIDs that target COX. However, not all COX inhibitors have credible efficacy in treating AD. Selective COX-2 inhibitors, such as celecoxib and rofecoxib, had no effect on cognitive impairments in AD [131].
The second goal of NSAIDs is to decrease prostaglandin E2 level in AD patients to avoid the development of the Aβ plague. Prostaglandin E2, a COX product, is observed to be higher in the cerebrospinal fluid of patients with probable AD, resulting in an increase in the expression of AβPP and the production of the proinflammatory cytokine, IL-6 [132, 133]. The NSAID dexamethasone is reported to limit the release of prostaglandin E2 in astrocytes and astroglioma cell lines (U251 and U373 MG) by inhibiting the production of microsomal prostaglandin E synthase-1, an enzyme involved in prostaglandin E2 synthesis [134, 135]. The advantage of lowering prostaglandin E2 rather than suppressing COX activity is that it can help minimize drug-related adverse events such as gastrointestinal damage and renal discomfort [135].
Specific NSAIDs decrease Aβ42 peptide level directly and thus alleviate AD’s severity. These NSAIDs include diclofenac, diflunisal, fenoprofen, flurbiprofen, meclofen, piroxicam, R-flurbiprofen, S-flurbiprofen, ibuprofen, indomethacin, and sulindac sulphide, but not naproxen, aspirin, celecoxib, meloxicam or SC-560 [136–139]. The ineffectiveness of certain NSAIDs indicates that the decrease in Aβ42 levels may be independent of COX activity, the principal mode of action of NSAIDs. Whether other NSAID-responsive pathways mediate this activity, such as by modulating lipoxygenase activity or transcription controlled by PPARs, warrants further investigations. In vivo investigations indicated that novel flurbiprofen compounds with high bioavailability and a long half-life decreased the Aβ42 level in transgenic mice [139]. Additionally, this process is considered to be independent of COX activity, as COX-deficient cells did not exhibit significant changes in the level of Aβ42 peptide [136].
Another way NSAIDs work is by activating the PPARγ, a member of the nuclear receptor family of transcription factors that regulate many proinflammatory genes. NSAIDs such as indomethacin, fenoprofen, and ibuprofen have been shown to activate the receptors and inhibit the production of pro-inflammatory cytokines, NF-B, STAT, and AP-1, in the brain [140]. Additionally, studies have discovered that other PPARγ agonists, such as rosiglitazone and pioglitazone, reduce the neural damage caused by inflammation [141, 142]. While NSAIDs may be a potential therapy for AD, not all NSAIDs are beneficial. Certain substances exacerbate or facilitate the generation of Aβ in the brain [143]. Celecoxib, a COX-2 inhibitor, similarly did not benefit AD patients aged 70 and above [144]. Thus, extensive research is required to identify the most effective NSAIDs for treating neuroinflammation in AD with the fewest possible side effects.
Probiotics therapy
The brain-gut-microbiota axis is disrupted in AD [145]. Disturbances in the brain-gut-microbiota axis, including the CNS and enteric nervous system (ENS), contribute to AD pathogenesis. It is well established that the gut microbiota increases local and systemic inflammation as a result of lipopolysaccharides (LPS) produced by pathogenic bacteria and the production of proinflammatory cytokines [146]. LPS can cause prion amyloids to adopt a more pathogenic β-pleated sheet shape [147]. Changes in the nature of the gut microbiota may result in increased permeability of the intestinal barrier and BBB, hence promoting inflammation at the gut, systemic, and CNS levels [148, 149]. Additionally, the gut bacteria secreted a significant amount of amyloid. Aβ was also formed in the ENS and CNS. The gut microbiota’s role in the pathophysiology of AD is extensively demonstrated in animal models of AD [145]. A deeper understanding of the gut microbiota’s role in developing of AD and the strong relationship between gut dysbiosis increased intestinal permeability, and neurological dysfunction opens the door to new therapeutic approaches [150]. Modifications in the gut microbiota can alter brain activity, leading to the possibility of therapeutic treatment of the microbiome in AD and other neurological illnesses [151]. Numerous studies demonstrate that probiotics have a positive effect by increasing intestinal epithelial integrity, protecting against barrier disruption, lowering pro-inflammatory responses, and suppressing the beginning or propagation of neuroinflammation and neurodegeneration [152]. There are preclinical and clinical studies that demonstrate the therapeutic potential of Lactobacilli and Bifidobacteri probiotics in reducing neuroinflammation [153], memory deficit [154], antibiotic treatment [151], fecal microbiota transplantation [155, 156], and dietary intervention [157].
Gene therapy strategies
With the advancement of DNA recombinant technology over the last few decades, scientists have begun to search for an AD cure using a more targeted approach known as gene therapy. The following section discusses some potential gene therapy strategies that have been investigated so far. IL-4 and IL-10 are anti-inflammatory cytokines that appear to be critical regulators of brain neuronal activity. A deficiency of these cytokines has resulted in hippocampus long-term potentiation (LTP) deficits associated with aging [158, 159]. One study indicated that cerebral injections of IL-4 through an adeno-associated viral vector could induce persistent cytokine production in APP/PS1 transgenic mice. This boosted the M2a phenotype of microglia in the frontal brain and hippocampus and lowered Aβ plaque levels [160]. Additionally, it was discovered that long-term production of IL-10 reduces astro/microgliosis, promotes neuron development, and improves spatial learning [161]. One of the receptors identified on microglia is the protein Triggering Receptor Expressed on Myeloid Cells 2 (TREM2), which also contributes to AD development [162–165]. Rare mutations in the TREM2 gene result in protein variations that have negative consequences. For instance, both the R47 H and R62 H genotypes have been substantially related to the development of Aβ plaques and hence, an increased chance of developing AD [166, 167]. In mice, complete deletion of the TREM2 gene ameliorated the illness by decreasing the amount of Aβ plaque-associated macrophages and particular inflammatory cytokines [162–164]. The study by Jay and colleagues [163] further implied that TREM2 absence reduces amyloid pathology in the early stages of the disease but increases it at later stages. Additionally, scientists have found and examined additional microglial genetic risk factors associated with neuroinflammation such as CD33, CR1, ABCA7, and SHIP1 [165, 168].
Another high-risk gene for AD associated with neuroinflammation is Apolipoprotein E (APOE), a regulator of lipoprotein metabolism required for cholesterol transport, neuroplasticity, and inflammation in the CNS [169]. Three common alleles encode it: ɛ2, ɛ3, and ɛ4. APOE ɛ2 relates to a lower chance of developing AD later in life, but APOE ɛ4 increases the risk of developing AD [167, 170]. Pro-inflammatory cytokines (IL-6 and TNF-α) were enhanced in the brains of animals expressing APOE ɛ4 compared to their APOE ɛ3 counterparts, implying isoform-specific effects of the APOE [171]. As a result, medication directed against APOE ɛ4 may be a promising therapeutic strategy for slowing the progression of the disease.
Signal blocking molecules
Signal-blocking compounds may be utilized therapeutically to prevent the cytokine cascade from chronic hyperactivity, resulting in chronic neuroinflammation. For example, XPro1595 is used to decrease TNF-α, a critical cytokine involved in the immunological response in AD patients [172–174]. Other signaling molecules, such as those blocking Rho-kinase 1 and NADPH-oxidase 2, show promise in treating AD [175, 176]. Another treatment option for AD is resveratrol. The activator of silent information regulator 2, homolog 1, which is abundant in grape wine, is useful in ameliorating the cognitive problems associated with AD [177–180].
Stem cell therapy
Most therapeutic strategies targeting AD pathology mainly focus on preventing the development of AD or delaying the onset of the disease. However, many of them cannot restore the deleterious neuron loss with neuroinflammation in AD. The result of stem cell therapy may pave the way for a revolutionary treatment of AD by regenerating neurons or delivering therapeutic molecules to the brain. Both preclinical and clinical experiments utilizing stem cell therapy demonstrated promising results in treating the inflammatory pathology associated with AD. For example, transplantation of neural stem cells/neural progenitor cells into APP/PS1 transgenic mice decreased the level of pro-inflammatory cytokines IL-1, IL-6, TNF-α, and PGE2 by reducing the glial and TLR4-mediated inflammatory pathway, resulting in improved cognitive functioning [165]. Numerous studies have also reported that transplanting mesenchymal stem cells into AD transgenic mice model resulted in a variety of beneficial effects, including decreased microglial activation and Aβ deposits, inhibition of inflammatory cytokine release, inhibition of neuron cell death, and reversal of cognitive deterioration [181, 182]. This evidence strengthens the case for stem cell therapy as a potentially effective treatment for AD.
Multitarget-directed ligands (MTDLs) drugs
Instead of just taking two or more distinct medications, the creation of MTDLs composed of a mixture of molecules containing tacrine and an antioxidant moiety could effectively address multiple oxidative stress components. Examples include the combination of tacrine and melatonin, tacrine and coumarin, tacrine and gallamine, tacrine and resveratrol, tacrine and scutellarin, tacrine and anisidine, and tacrine and trolox [183–186]. Wenzel and Klegeris [187] discussed the efficacy and mechanism of inhibiting cathepsin B, dual specificity phosphatase 2, and mono glycerol lipase. These models have demonstrated promising preclinical outcomes in vivo and in vitro [187]. MTDLs have antioxidant and anti-inflammatory properties, suggesting that they may be used to inhibit AD-associated neuroinflammation.
The Oxidative Stress Hypothesis
A fundamental causal factor for AD is commonly believed to be oxidative stress. When a person ages, reactive oxygen species (ROS) (e.g., O2•–, H2O2, HO•) and reactive nitrogen species (RNS) (e.g., NO, ONOO-) gradually accumulate in the body, resulting in a functional loss in essential organs and the brain. They are responsible for irreversible memory impairment [188]. Increased cholesterol levels in the brain, particularly in neurons, make them more susceptible to oxidative damage [189]. Increased amounts of oxidation products from lipids, proteins, DNA, and RNA, have been shown to enhance the growth of Aβ40 and Aβ42 in memory-related brain regions of AD patients, such as the hippocampus and cortex, but not in other locations [190, 191].
Mitochondria, which serve as an oxygen metabolism site, are the principal source of ROS production when their regular function is disrupted [192–194]. The intermediates produced by the electron transport cycle in the mitochondria, including superoxide anion (O2) and hydrogen peroxide (H2O2), are highly reactive and are the primary cause of oxidative stress in AD [35]. ROS cause damage to the mitochondria, particularly at the DNA site, and may finally result in cell death via apoptosis [195]. Additionally, activation of NADPH oxidase generates superoxide and other ROS [196].
Another symptom of oxidative stress in AD is the dysregulation of metal ions in the brain, such as iron (Fe) and zinc (Zn), via metalloproteins [35, 198]. Free loosely bound Fe and copper (Cu) are believed to be involved in the generation of ROS, where they are reduced to Cu (I) and Fe (II), respectively, to create superoxide and hydroxyl radicals [196]. They can, however, act as antioxidants when bound to antioxidant enzymes such as superoxide dismutase 1 (SOD1) and catalase. Thereby, they neutralize dangerous ROS. Lipid peroxidation occurs when ROS attack and oxidize the membrane’s lipids and proteins. The result of this reaction, 4-hydroxynonenal (4-HNE), has been discovered in the hippocampus of AD patients at elevated levels [199]. 4-HNE is toxic because it tends to change macromolecules within the cell. This exacerbates the disease by boosting the generation of ROS [77]. Without a doubt, oxidative stress contributed to or exacerbated the pathogenesis of AD. Thus, identifying ROS production key indicators and the mechanism behind is critical for developing effective pharmacotherapies that can lower the chance of developing AD.
Applications of antioxidant compounds
Based on the oxidative stress markers, reduced ROS levels in the brain appear to be an approachable strategy for treating oxidative stress-induced AD. This can also be accomplished using endogenous and exogenous antioxidant compounds. Antioxidants found in foods are vitamin C, vitamin E, omega-3 fatty acids, selenium, and polyphenols. They have been shown to have antioxidant and neuroprotective characteristics, suggesting that they may be used as a potential treatment for oxidative stress [193, 201]. Phytochemical compounds from herbal medicines with antioxidant andanti-inflammatory activities have the potential to treat AD such as flavonoids, phenolic acid, tannins, terpenoids, anthocyanins, diterpene, triterpene, cannabinoids, and ursolic acid [202].
Glutathione (GSH) is a significant endogenous antioxidant in the brain, capable of reacting with ROS to form oxidized glutathione (GSSG) and then returning to GSH via GSH reductase [203]. The level of GSH is dramatically lowered in AD pathogenesis driven by oxidative stress [204]. However, research indicates that the level of this antioxidant can be restored or enhanced using N-acetylcysteine and -glutamylcysteine ethyl ester as another possible therapy option for AD [203].
Inhibition of monoamine oxidase B (MAO-B) enzymes
Monoamine oxidases, flavin-containing enzymes located in the outer mitochondrial membrane, catalyze the oxidative deamination of neurotransmitters like noradrenaline, dopamine, and serotonin. The formation of H2O2 causes oxidative stress due to its enzymatic activity [205]. Patients with AD have elevated MAO-B enzyme activity, which increases with age. By suppressing MAO-B, the degradation of neurons in the brain of AD patients can be slowed down [206]. Several established mechanisms of MAO-B inhibitors in preventing neurodegeneration include: (a) inhibitors that reduce MAO-B-generated free radicals and prevent DNA and membrane destruction, (b) inhibitors that protect neurons from apoptosis in cell culture, and (c) inhibitors that reduce the secretion of neurotoxic products [207].
Glutamate excitotoxicity
Glutamate is the primary excitatory neurotransmitter in the brain. It is stimulating numerous postsynaptic receptors, including the N-methyl-D-aspartate (NMDA) receptor, which has been linked to memory and dementia [208]. Excessive glutamatergic stimulation may result in excitotoxicity. This is associated with neuronal calcium overload and neurodegenerative diseases [209]. An exaggerated calcium influx activates cellular enzymes such as proteases, lipases, and DNAses [210]. When these enzymes and their metabolic products are activated, they destroy the neuronal cell membrane, genetic material, and structural proteins, ultimately resulting in cell death. It was, therefore, advocated that the glutamatergic pathway constitutes a therapeutic target for treating AD [211].
Memantine was approved as a treatment for AD patients by the European Medicines Agency (EMEA) in 2002 and by the FDA a year later. In multiple large-scale, controlled clinical trials, memantine showed significant symptomatic effectiveness in moderate-to-severe AD [212]. Memantine is well tolerated, and with dose reduction, adverse symptoms such as disorientation, hallucinations, somnolence, dizziness, and nausea are resolved [213].
Memantine is a non-competitive NMDA-receptor antagonist that can attenuate excitotoxicity. Memantine has a low affinity for binding and is voltage-dependent. In contrast to the Mg2+ ion, which has a weaker binding affinity, memantine continues to block the NMDA receptor channel during sustained stimulation at intermediate levels under pathological conditions. When transiently high glutamate concentrations are present, memantine dissociates from the receptor, and normal neurotransmission continues [214].
Memantine has been shown to prevent dendritic drebrin. Drebrin is an actin-binding protein that forms stable F-actin and is highly concentrated within dendritic spines. It is lost because of synaptic degeneration caused by soluble Aβ-derived oligomers (ADDLs) [215], as well as to guard against ROS generated by ADDLs via interaction with activated NMDA receptors [216]. In animal models, the combination of acetylcholinesterase (AChE) inhibitors with memantine improved memory performance test more than either therapy alone [214]. This finding lends credence to the multi-target drug hypothesis, which states that simultaneously targeting distinct AD pathophysiology pathways may yield a more efficient treatment for AD.
Downregulation of neurotrophins level
Neurotrophins are secretory proteins involved in the nervous system’s growth, survival, development, function, and plasticity [217, 218]. The nerve growth factor (NGF) plays a vital protective role in specific brain cells, most notably basal forebrain-derived neurons [219], by inhibiting tau hyperphosphorylation in vitro and in vivo [220]. Thereby, NGF promotes neurite development [221]. The malfunction of NGF and its receptors results in the selective degeneration of cholinergic neurons of the basal forebrain during the late stages of AD. Chronic NGF deficiency has been implicated in promoting AD-like neurodegenerative illness, including tau hyperphosphorylation and plaque accumulation [222, 223]. Additionally, AD was associated with an increase in precursor (proNGF) levels and a decrease in mature (mNGF) levels (derived from proNGF), which resulted in the loss of cortical connections and atrophy of cholinergic neurons in the basal forebrain [224].
Apart from NGF, brain-derived neurotrophic factor (BDNF) is also critical for neuronal survival, differentiation, and morphological regulation [225]. Interacting with the tyrosine kinase B (TrkB) receptor, BDNF promotes dendritic arborization and axonal branching [225, 226]. BDNF is a key regulator of the hippocampus’s acquisition and consolidation of spatial memory. It redistributes synaptic protein on the presynaptic membrane, thereby strengthening high-frequency transmission in the excitatory synapse of the CA1 region. Hence, in order to sustain LTP for 8 hours to days, BDNF and other synaptic plasticity-related proteins must be upregulated [227]. A decrease in the brain’s BDNF levels is one of the earliest signs of AD development [228, 229]. Additionally, the level of BDNF in the brain of AD patients was considerably decreased as a result of Aβ42 modulation. This reduced phosphorylated cAMP response element-binding protein (p-CREB) and primary BDNF transcripts in neuroblastoma cells [230, 231]. Thus, neurotrophin levels may be an appropriate target for therapeutic strategies preventing and treating AD.
Clinicians and investigators have been motivated by the potential benefits of using NGF to treat neurological illnesses. NGF supports nerve regeneration and has considerable potential for use in various other applications [232]. Numerous studies have been undertaken recently to provide substantial insight into the biochemical and molecular features of NGF and BDNF-induced neurite outgrowth, with the potential for novel treatment techniques. Additionally, investigations on natural products used in traditional medicine have been conducted to determine their efficacy as AD healers. In an AD model, a new alkaloid, HupA, isolated from Huperzia serrata, reduced cognitive impairments and increased NGF expression [233]. Apart from that, 6-shogaol, a ginger bioactive, increased NGF, PSD-95, and synaptophysin expression in hippocampus tissue [234]. Embelin, benzoquinone compound (C17H26O4) from the fruits of Embelia ribes, alleviated the decreased expression of BDNF, CREB1, APP, Mapt, SOD1, and NFκB mRNA levels caused by chronic cerebral hypoperfusion rats [235]. An anticholinesterase agent can promote neurite outgrowth, possibly by increasing NGF and BDNF levels in the brain [236, 237]. Ganstigmine and donepezil can be administered intraperitoneally or orally to correct cholinergic and behavioral impairments in AD11 mice [238].
Kalirin, a multidomain guanine nucleotide exchange factor, was previously shown to activate the ERK, Akt, and CREB pathways in vivo [239]. Shih and colleagues [240] examined the influence of the adapter protein SH2B1 on BDNF-generated signaling and morphogenesis of cortical neurons and PC12-TrkB cells. The results indicate that SH2B1 promotes BDNF-induced neurite outgrowth primarily via the MEK-ERK1/2 pathway. In agreement with this finding, the baseline level of pAKT(S473) was much higher in PC12-SH2B1 + TrkB cells than in control cells, although no spontaneous neurite outgrowth was observed. According to these studies, neurite regeneration improves cognitive functioning by increasing NGF and BDNF levels in the brain.
Cholinergic hypothesis
Acetylcholine (ACh) is a neurotransmitter that plays a critical function in memory and attention. ACh is essential in modulating memory acquisition, encoding, consolidation, reconsolidation, extinction, and retrieval [241]. The activity of the choline acetyltransferase enzyme, which is responsible for ACh biosynthesis, was significantly decreased in the neocortex, amygdala, and hippocampus of AD brains, resulting in a significant decrease in the level of ACh in the synaptic cleft [241]. There was also a loss of cholinergic neurons observed in AD [242]. The increasing degeneration of cholinergic neurons and the resulting decrease in ACh levels in the brain, notably in the temporal and parietal neocortex and hippocampus, results in cognitive impairment in people with AD. In vitro activation of muscarinic phospholipase C (PLC) associated receptors promote the physiologic, non-amyloidogenic processing of AβPP [243], and muscarinic agonists reduce the neurotoxicity of Aβ [244]. GSK-3 is responsible for the phosphorylation of tau in AD. The cholinergic-stimulated PLC pathway is used to inhibit this kinase. A cholinergic system impairment in AD may increase GSK-3 activity via decreased PLC activation and, consequently, hyperphosphorylation of tau proteins [245, 246].
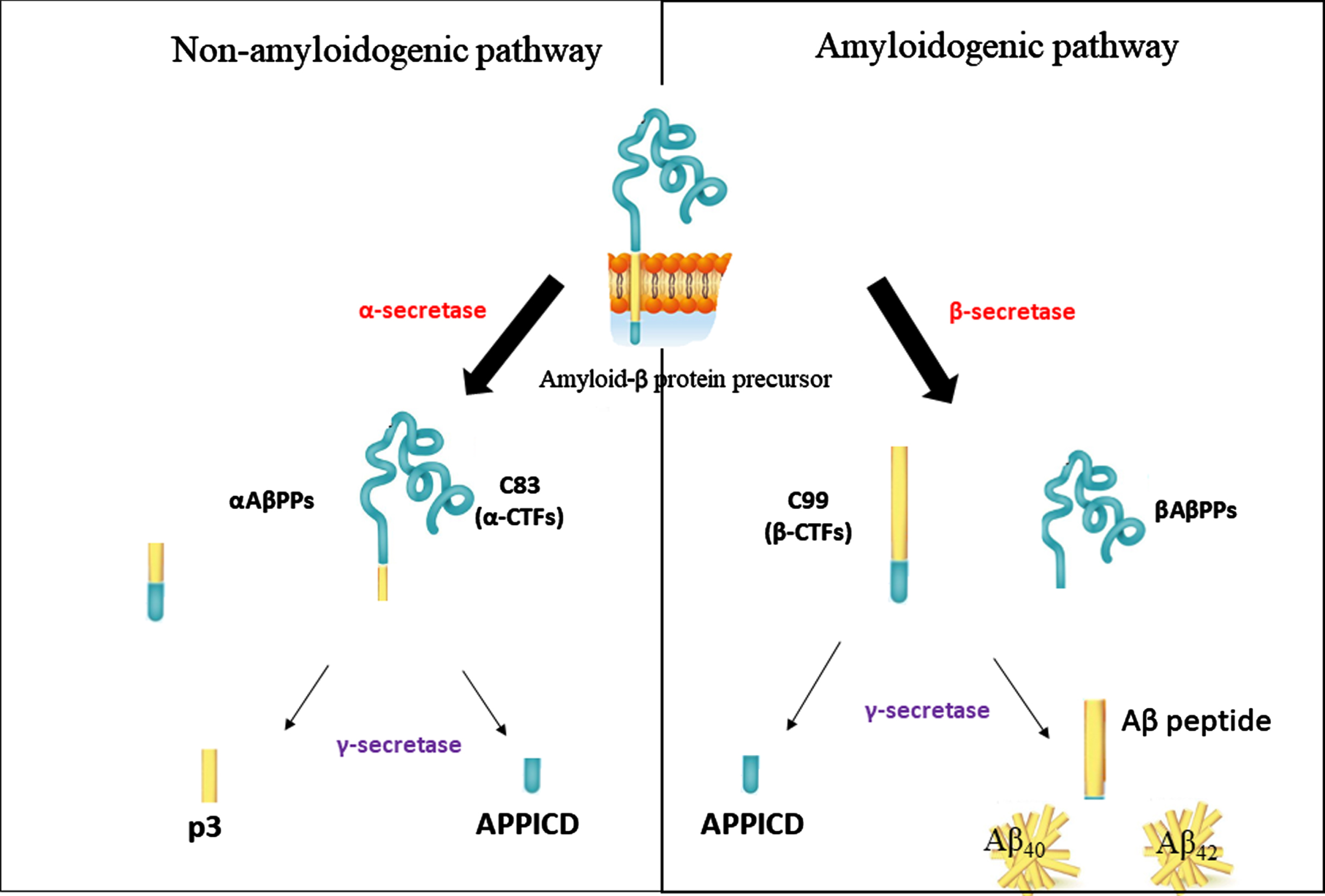
Production of Aβ from amyloid-β protein precursor processing (AβPP) in amyloidogenic and non-amyloidogenic pathways by β- and α-secretases. In the non-amyloidogenic pathway, an AβPP breakdown into a soluble AβPP, αAβPPs, and α-carboxyl-terminal, C83 (α-CTFs) by enzyme α-secretase. In the amyloidogenic pathway by β-secretase, AβPP releases soluble amyloid precursor protein, βAβPPs and β-carboxyl-terminal, C99, β-CTFs. Both pathways release AβPP into the intracellular domain (APPICD). The final products of γ-secretases are the amyloid-β peptide, Aβ, and p3 fragments.
Inhibition of cholinesterase enzymes
AChE and butyrylcholinesterase (BuChE) are the two primary enzymes responsible for the hydrolysis of ACh in the brain, resulting in a reduction in cholinergic function. Thus, inhibition of these enzymes enhances cholinergic function, and clinical improvement is reported in patients with AD [247]. ChEIs are currently accessible therapeutics that have been endorsed for the treatment of mild to moderateAD.
The use of an AChE inhibitor alters the cholinergic anti-inflammatory system, a physiological process that reduces cytokine production and tissue destruction during inflammation [248]. ACh released from the vagus nerve causes an influx of Ca2+ into macrophages by interacting with α7 nAChR expressed on the surface of macrophages. This led to the activation of the nuclear factor κappa B (NFκB), resulting in the suppression of inflammatory cytokine production, including TNFα, high mobility group box of proteins and IL-6 [249]. Given the critical role of inflammation in the etiology of AD, AChE inhibitors may benefit AD patients by lowering brain inflammation while maintaining or improving the ACh level.
Although AChE inhibitors are currently licensed for use in mild-to-moderate AD, it may be helpful to expand their application to the very early and late stages of the disease, even though stabilization of functional and cognitive symptoms with inhibitors is considered a significant clinical outcome. However, rather than enduring an illness without therapy, alleviating symptoms with these inhibitors may lessen the burden on patients and cares. Additional research using randomized control trials with long-term follow-up is needed to demonstrate the treatment’s cost-effectiveness and influence on illness progression, quality of life, and time until institutionalization. Figure 3 summarizes the therapeutic intervention together with the pathological condition of AD.

Summary of the therapeutic intervention for the treatment of AD. Ca2+, calcium ions; Na2+, sodium ions; NMDAR, N-methyl-D-aspartate receptor; AMPAR, α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)-type glutamate receptors; BACE 1, β-site APP-cleaving enzyme 1; NEP, neprilysin; ACE, angiotensin-converting enzyme; IDE, insulin-degrading enzyme; RAGE, advanced glycation end products; Aβ, amyloid-β; ACh, acetylcholine; nAChR, alpha-7 nicotinic receptor; AChE, acetylcholinesterase; BuChE, butyrylcholinesterase; NFT, neurofibrillary tangles; PP2A, protein phosphatases 2A; NSAIDs, non-steroidal anti-inflammatory drugs; APOE ɛ4, Apolipoprotein E epsilon 4; PPARγ, peroxisome proliferator-activated receptor-gamma; MTDLs, multitarget-directed ligands; GSH, glutathione; MAO B, monoamine oxidase B; ROS, reactive oxygen species; NGF, nerve growth factor; ERK, extracellular signal-regulated kinase; TrkB, tyrosine kinase B; BDNF, brain-derived neurotrophic factor; CREB, cAMP response element-binding protein.
Example of cholinesterase inhibitors
Physostigmine, an alkaloid derived from the seeds of Physostigma venenosum, was the first ChEI to be explored for use in AD treatment. It can cause various adverse reactions, including nausea, vomiting, headaches, diarrhea, and dizziness [250]. Tacrine was the first medication with good inhibitory activity against AChE and BuChE to be licensed for treating AD patients in 1993. However, tacrine was withdrawn from the market due to its adverse effects and hepatotoxicity [251].
Currently, the ChEIs donepezil, rivastigmine, and galantamine are available for AD treatment. Donepezil is a synthetic piperidine derivative that was approved in 1996/1997 as a competitive and non-competitive AChE inhibitor. Rivastigmine, a carbamyl derivative, is a non-competitive AChE inhibitor that was approved in 1997. Galantamine is a reversible, competitive AChE inhibitor approved in 2000 [252]. Rivastigmine’s capsular form has adverse consequences. Nonetheless, reformulated rivastigmine in the transdermal patch produces fewer gastrointestinal side effects. Examples of potential AChE inhibitors that are under investigation are physostigmine derivatives, phenserine tolserine. esolerine, tesofensine, naturally derived (huperzine A., huperzine B. (i) beta-cyclogeraniol diglycoside, (ii) cycloartenol, (iii) p-hydroxybenzoic acid, (iv) vanilloloside, (v) 5-O-methyladenosine from Nelumbo nucifera, galangin from Rhizoma alpiniae Officinarum, cardanol derivatives and synthetic analogs (Phenyl-5,6-dimethoxy-1-oxo-2,3-dihydro-1H-2-indenylmethanone analogs, N-Alkyl-7- methoxytacrine hydrochlorides and ladostigil) [253]. Fifty-seven extracts from 177 potential Malaysian plant extracts were found to exhibit potent cholinesterase inhibition ranging from 50% to 100% against both AChE and BuChE enzymes [254]. Other medicinal plants which have claimed to possess nootropic and anticholinesterase activities were roots of Clitorea ternatea [255, 256]. This plant was found to reduce the level of AChE in the frontal cortex and hippocampus. Hence, it restored the memory impairment induced by chronic cerebral hypoperfusion.
Donepezil. In comparison to patients treated with various inhibitors, patients treated with donepezil showed the most beneficial pharmacological action in terms of cognitive improvement, responder’s rate (40– 58%), dropout cases (5– 13%), and side effects (6– 13%) [257]. Hippocampus degeneration was reported to occur more slowly in AD patients treated with donepezil compared to untreated individuals [258].
Donepezil decreased brain and blood AChE activity by 66% and 32%, respectively. It increased cerebral ACh by 35% in a two-week in vivo study without affecting choline acetyltransferase, vesicular ACh transporter, choline transporter, or muscarinic receptor levels [259]. Donepezil increased the expression of nicotinic receptors in rat cortical neurons, hence promoting neuroprotection [260]. It stimulated non-amyloidogenic pathways of AβPP processing via PKC [261]. It inhibited AChE-induced Aβ aggregation [262]. Parnetti et al. observed that donepezil medication decreases AChE activity in cerebrospinal fluid patients, but does not affect BuChE activity, Aβ42, tau, or phosphorylated tau proteins[263].
Donepezil has been shown in clinical investigations to enhance cognitive performance in patients with severe AD symptoms [257]. Additionally, donepezil retards the deposition of Aβ plaques [264]. Unlike donepezil, which inhibits only AChE, rivastigmine inhibits both BuChE and AChE. Apart from reducing cholinesterase activity, galantamine improves nicotinic receptor activity by engaging with binding sites not occupied by ACh or nicotinic agonists [265]. However, these drugs may have gastrointestinal adverse effects such as nausea, diarrhea, anorexia, or stomach discomfort [264].
CONCLUSION
Management of AD without any side effects is still a challenge in medicine and drug discovery. Currently, the available drugs only provide symptomatic relief but do not have disease-modifying properties to cure AD. AD is a multifactorial disease state which is not caused by a single factor. Consequently, it requires a multi-targeted drug approach to counteract all or at least the major pathogenesis pathways of AD. However, polypharmacy, which employs multiple drugs for a single ailment, may produce more off-target effects than a single drug. Suppose, however, that the therapeutic value of a multi-targeted medicine outweighs the potential adverse effects and does not eventually impair the patient’s ability to regain health. In that situation, the additional risk is almost certainly worth it. This review has identified the most crucial therapeutic targets for preventing and treating AD such as the formation of Aβ plaques, tau hyperphosphorylation, neuroinflammation, oxidative stress, glutamate excitotoxicity, downregulation of neurotrophins level, and dysfunction of the cholinergic system through a literature search. AD can be described as a future global dilemma when the global population grows older. Therefore, a deeper understanding of the factors that lead to its progression and pathogenesis is necessary to search for new and more effective treatments.
Footnotes
ACKNOWLEDGMENTS
We would like to thank the Alexander von Humboldt Foundation for the Georg Foster Research Award in collaboration with the Department of Psychiatry and Psychotherapy, University Clinic, Friedrich-Alexander-University Erlangen-Nuremberg, Schwabachanlage 6, 91054 Erlangen, Germany and Universiti Sains Malaysia for the RUI grant [1001.CDADAH.8012302].