
Abstract
Background:
Cerebrospinal fluid (CSF) biomarkers in patients with cerebral amyloid angiopathy-related inflammation (CAA-ri) have demonstrated inconsistent results.
Objective:
We investigated the relationship between CSF amyloid-β protein (Aβ) and vascular pathological findings to elucidate the mechanisms of Aβ elimination from the brain in CAA-ri.
Methods:
We examined Aβ40 and Aβ42 levels in CSF samples in 15 patients with CAA-ri and 15 patients with Alzheimer’s disease and cerebral amyloid angiopathy (AD-CAA) using ELISA as a cross-sectional study. Furthermore, we pathologically examined Aβ40 and Aβ42 depositions on the leptomeningeal blood vessels (arteries, arterioles, and veins) using brain biopsy samples from six patients with acute CAA-ri and brain tissues of two autopsied patients with CAA-ri.
Results:
The median Aβ40 and Aβ42 levels in the CSF showed no significant difference between pre-treatment CAA-ri (Aβ40, 6837 pg/ml; Aβ42, 324 pg/ml) and AD-CAA (Aβ40, 7669 pg/ml, p = 0.345; Aβ42, 355 pg/ml, p = 0.760). Aβ40 and Aβ42 levels in patients with post-treatment CAA-ri (Aβ40, 1770 pg/ml, p = 0.056; Aβ42, 167 pg/ml, p = 0.006) were lower than those in patients with pre-treatment CAA-ri. Regarding Aβ40 and Aβ42 positive arteries, acute CAA-ri cases showed a higher frequency of partially Aβ-deposited blood vessels than postmortem CAA-ri cases (Aβ40, 20.8% versus 3.9%, p = 0.0714; Aβ42, 27.4% versus 2.0%, p = 0.0714, respectively).
Conclusion:
Lower levels of CSF Aβ40 and Aβ42 could be biomarkers for the cessation of inflammation in CAA-ri reflecting the recovery of the intramural periarterial drainage pathway and vascular function.
Keywords
INTRODUCTION
Cerebral amyloid angiopathy (CAA) is characterized by amyloid deposition in blood vessel walls of the brain and leptomeninges. Sporadic amyloid-β protein (Aβ)-type CAA is the most common form of CAA among elderly people [1]. Elimination failure of interstitial fluid (ISF) from the brain parenchyma, such as by dysfunction of the intramural periarterial drainage (IPAD) and glymphatic system [2, 3], could be associated with CAA development [4]. CAA-related inflammation (CAA-ri) is an extremely rare CAA phenotype caused by an exaggerated immune reaction against deposited Aβ in blood vessel walls showing elevated levels of cerebrospinal fluid (CSF) anti-Aβ antibody and microglial activation [5–10]. Neuropathological studies on patients with CAA-ri demonstrated inflammation and vasculitis with an absence of Aβ in blood vessels [9]. Macrophages and multinucleated giant cells containing phagocytosed Aβ in the cytoplasm were observed. However, there were some blood vessels with missing Aβ on the vessel walls, showing no apparent presence of giant cells [9].
Regarding CSF biomarkers, lower levels of Aβ40 have been reported in patients with CAA [11]. Anti-Aβ antibody levels in the CSF have been a useful biomarker for the diagnosis and treatment of CAA-ri [6, 10]. However, examination of anti-Aβ antibodies is not straightforward. Another biomarker is necessary to diagnose CAA-ri as soon as possible, to provide appropriate therapies before irreversible brain damage occurs. Several previous studies investigating CSF biomarkers for Alzheimer’s disease (AD), including Aβ40, Aβ42, and phosphorylated tau protein (p-tau), in patients with CAA-ri, have demonstrated inconsistent results. In comparison with CAA cases or age-matched control cases, several reports have found lower concentrations of CSF Aβ40 and Aβ42 were revealed in CAA-ri cases, while others showed no apparent difference [6, 12–15]. Some reports have demonstrated lower biomarker levels in post-treatment CAA-ri [6, 15]; however, another study showed that biomarkers did not change between pre- and post-treatment samples [12]. The major limitation of these previous studies was the low probability of CAA-ri diagnosis due to the fact that few cases were pathologically confirmed.
In this study, we examined Aβ40, Aβ42, and tau proteins in CSF samples from patients with CAA-ri before and after treatment, and AD with CAA (AD-CAA). We also determined the vascular pathological deposition of Aβ40 and Aβ42 using biopsy brain samples from patients with acute-phase CAA-ri and autopsy brain samples from patients with CAA-ri in the chronic phase after treatments to elucidate the elimination mechanisms of Aβ from blood vessel walls in CAA-ri. We thereby assessed whether CSF Aβ40, Aβ42, and tau proteins could be used as biomarkers for the diagnosis and treatment of CAA-ri.
METHODS
Patients with CAA-ri
We retrospectively collected patient information, CSF, and biopsy samples from patients with CAA-ri from hospitals in Japan between 2013 and 2020. To identify and include appropriate patients, we used the clinical diagnostic criteria previously proposed by Chung et al. [16] for definite CAA-ri and designated as Aβ-related angiitis (ABRA). For probable and possible CAA-ri, we used the clinical diagnostic criteria proposed by Auriel et al. [17].
In total, 15 patients with CAA-ri were enrolled (Tables 1 and 2). Samples from several patients (patient 1, 2, 6, 7, 12–15) were used in our previous study [8]. CSF samples before immunosuppressive treatments (pre-treatment CAA-ri group) were obtained from 11 patients (Table 1). The median interval between clinical presentation and CSF sample collection before treatment was three (range 0.5–12) months. We also obtained samples after immunosuppressive treatments in Patients 1 and 6. Patient 1 was treated with two courses of intravenous methylprednisolone (1 g/day for three consecutive days), followed by oral corticosteroid administration (60 mg/day). Patient 6 was treated with one course of intravenous methylprednisolone (1 g/day for three consecutive days), followed by oral corticosteroid administration (60 mg/day).
Patient characteristics of the pre-treatment CAA-ri group
Patient characteristics of the pre-treatment CAA-ri group
Aβ, amyloid-β protein; CAA-ri, cerebral amyloid angiopathy-related inflammation; N.A., not available; p-tau, phosphorylated tau protein; t-tau, total tau protein.
Patient characteristics of the post-treatment CAA-ri group
Aβ, amyloid-β protein; CAA-ri, cerebral amyloid angiopathy-related inflammation; N.A., not available; p-tau, phosphorylated tau protein; t-tau, total tau protein.
CSF samples after treatment only were obtained from four patients (post-treatment CAA-ri group) (Table 2). The data on time from treatment to CSF sample collection was unavailable. Six brain biopsy samples from six patients were enrolled before initiation of any immunomodulatory therapies. In one patient, we obtained brain samples before treatment; however, only CSF samples obtained after treatment were available.
For autopsy samples of patients with CAA-ri, we used formalin-fixed paraffin-embedded brain tissue (n = 2). Details of the clinical course and neuropathological findings of these patients have been described previously [18, 19]. Patient 1 was a 75-year-old Japanese man with a 3.5-year history of ABRA at autopsy. A brain biopsy performed one month after disease onset from the right parietal lobe showed granulomatous vasculitis with Aβ deposition on the vessel walls. Shortly after the brain biopsy, corticosteroids were administered for nearly 10 months, with no obvious improvements. The patient died from pneumonia, and an autopsy was performed five hours after the death. Neuropathological examination revealed widespread and severe Aβ-type CAA showing vasculopathic changes with a few active cerebral vasculitic lesions [18]. Patient 2 was a 78-year-old Japanese man with a 2-year history of ABRA. A brain biopsy was performed 18 days after onset from the left temporal lobe, showing granulomatous vasculitis with Aβ deposition on the vessel walls. The patient received corticosteroids, cyclophosphamide, plasma exchange, and intravenous immunoglobulin therapy, with partial improvement in the clinical manifestations [19]. The patient died of interstitial pneumonitis, and an autopsy was performed five and a half hours after death. Neuropathological examination revealed a widespread occurrence of Aβ-type CAA with infarcts.
In summary, the samples examined in this cross-sectional case-control study were as follows: 11 CSF samples from pre-treatment CAA-ri, including five cases where brain biopsy was performed and four CSF samples from post-treatment CAA-ri including one case where brain biopsy was performed before any treatment. As a retrospective study, we used CSF samples from two patients with CAA-ri (Patients 1 and 6) before and after treatment. Two brain autopsy samples were examined.
Patients with AD-CAA
For the group of patients with AD-CAA (n = 15), we used previously published data [11]. We enrolled patients who visited our academic memory clinic between April 2001 and September 2015. Patients satisfied the criteria for probable AD according to the National Institute of Neurological and Communicative Disorders Association criteria [20]. In addition, the patients presented with microbleeds only in the cerebral cortex/subcortex and satisfied the Boston criteria for CAA-related hemorrhage [21].
CSF biomarker examination
We obtained CSF samples by lumbar puncture at the time of evaluation from all patients with CAA-ri and AD-CAA who underwent CSF examination as part of their diagnostic and follow-up procedures. Samples were centrifuged at 1500 rpm at 4°C for 5 min, and the resulting supernatants were stored at –80°C until further analysis. Sandwich enzyme-linked immunosorbent assay (ELISA) was used to determine levels of CSF-Aβ40 (Human Amyloid β (1–40) Assay Kit; IBL, Gunma, Japan), CSF-Aβ42 (Innotest β-amyloid (1–42); Fujirebio, Belgium), CSF-tau for total tau protein (t-tau) (Innotest hTAU-Ag; Fujirebio), and CSF-ptau for p-tau (Innotest Phospho-tau (181p); Fujirebio), according to the manufacturer’s instructions [11].
Neuropathological examination
Formalin-fixed paraffin-embedded brain tissue samples were obtained wherever possible. Six patients with pre-treatment CAA-ri underwent brain biopsy (acute CAA-ri group). Brain biopsy was performed on lesions delineated using magnetic resonance imaging. The biopsied brain areas were as follows: frontal cortex (two patients) and occipital cortex, parietal cortex, parieto-occipital cortex, and temporoparietal cortex (one patient each). Brain tissues, including the arachnoid, pia mater, and cerebral gray and white matter, were resected. Regarding autopsy brain tissues (CAA-ri autopsy group), we examined the frontal cortex. Brain tissues were stained with hematoxylin and eosin and Klüver-Barrera stain. Immunolabeling was performed using an EnVision System (Dako, Glöstrup, Denmark). The following primary antibodies were used: Aβ (monoclonal, 4G8; Covance, Indianapolis, IN, USA; 1 : 5000), Aβ35 - 40 (monoclonal, 1A10; Immuno-Biological Laboratory, Fujioka, Japan; 1 : 1000), Aβ1 - 42 (rabbit polyclonal; Immuno-Biological Laboratory; 1 : 100), and, p-tau (monoclonal, AT8; Innogenetics, Ghent, Belgium; 1 : 2000). Diaminobenzidine was used as the chromogen. According to the manufacture’s datasheet, antibody against Aβ35–40 (1A10) is human Aβ40 specific (https://www.ibl-japan.co.jp/en/search/product/detail/id=3520).
To assess the affected blood vessels in the leptomeninges, we counted the number of Aβ-positive leptomeningeal arteries, arterioles, and veins on the biopsy specimens using a 40×objective lens. Leptomeningeal arteries and arterioles were identified by vessel size and presence of media. The blood vessels without apparent media were identified as veins. In autopsy cases, we counted the number of Aβ-positive arteries, arterioles, and veins from the first longitudinal frontal sulcus to the second longitudinal frontal sulcus covering the middle frontal gyrus.
Statistical analysis
Values are presented as the medians and ranges. We used the Kruskal-Wallis test to compare the study groups, followed by the Mann-Whitney U test with Bonferroni correction to compare each group. Statistical significance was defined as p < 0.05, as determined by the Kruskal-Wallis test, and as p < 0.017 in the Mann-Whitney U test with Bonferroni correction. Statistical analyses were performed using Prism 9 software (GraphPad, San Diego, CA, USA).
Ethical approvals
This study was approved by the Medical Ethics Committee of Kanazawa University. Informed consent was obtained from all patients.
RESULTS
Clinical profiles
We enrolled 15 patients with CAA-ri and 15 with AD-CAA (Tables 1–3). Patients in the pre-treatment CAA-ri group (median [range], 77 [71–84] years, p < 0.001) and post-treatment CAA-ri group (75.5 [74–80] years, p = 0.016) were significantly older than patients with AD-CAA (71 [59–82] years).
Due to the insufficient volume of the CSF samples, Aβ40, Aβ42, p-tau, and t-tau could not be measured in one, one, one, and eight patients, respectively.
The median concentrations of Aβ42 and p-tau were significantly lower in the post-treatment CAA-ri group (Aβ42, 167 pg/ml; and p-tau, 21 pg/ml) than in the pre-treatment CAA-ri (Aβ42, 324 pg/ml, p < 0.01; and p-tau, 44 pg/ml, p = 0.011) and AD-CAA (Aβ42, 355 pg/ml, p < 0.01; and p-tau, 61 pg/ml, p < 0.01) groups (Fig. 1A–C, Table 3). The median Aβ40 level in the post-treatment CAA-ri group (1770 pg/ml) tended to be lower than those in the pre-treatment CAA-ri (6837 pg/ml, p = 0.056) and AD-CAA (7669 pg/ml, p = 0.046) groups. One case of post-treatment CAA-ri showed elevated levels of t-tau in the CSF; however, t-tau levels were not significantly different between pre-treatment CAA-ri and AD-CAA cases (Fig. 1D).
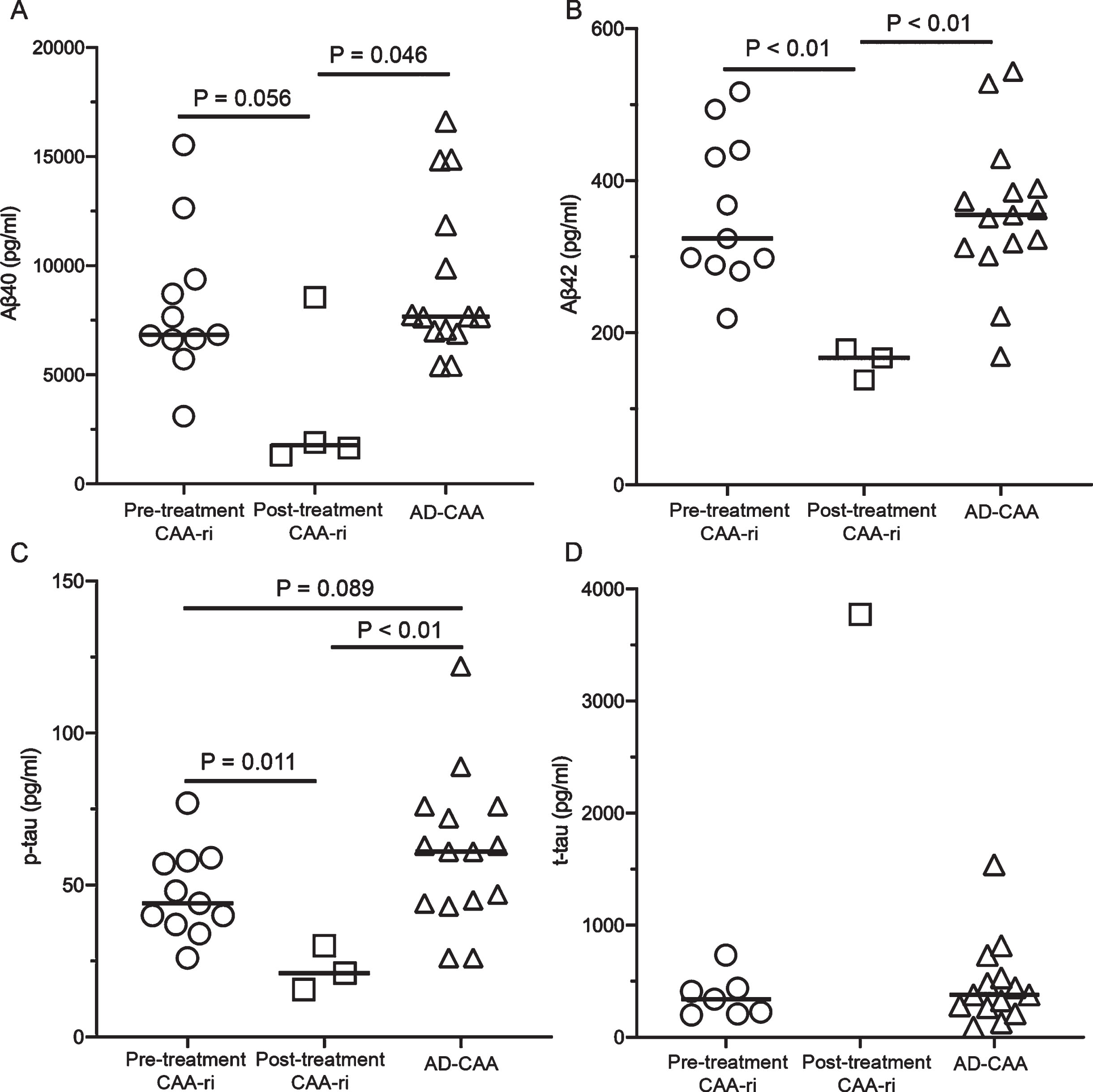
Cerebrospinal fluid amyloid-β protein (Aβ) (A, B) and tau protein (C, D) levels in patients with cerebral amyloid angiopathy-related inflammation (CAA-ri) and Alzheimer’s disease with CAA (AD-CAA). The CAA-ri group is divided into samples taken before treatment (pre-treatment CAA-ri group) and after treatment (post-treatment CAA-ri group). Aβ40, Aβ1 - 40; Aβ42, Aβ1 - 42; p-tau, phosphorylated tau protein; t-tau, total tau protein.
Profile of the cases and results of cerebrospinal fluid analysis
Aβ, amyloid-β protein; AD, Alzheimer’s disease; CAA, cerebral amyloid angiopathy; CAA-ri, cerebral amyloid angiopathy-related inflammation; n, number; N.A., not available; p-tau, phosphorylated tau protein; t-tau, total tau protein. *p value among the three groups was calculated using Kruskal-Wallis test. ¶Statistically significant in comparison to the AD-CAA group. ‡Proposed clinical criteria for CAA-ri [16, 17] and CAA [21] were applied to CAA-ri and AD-CAA cases, respectively. §The value was significantly lower than that in both the pre-treatment CAA-ri and AD-CAA groups.
Serial examination of CSF levels of Aβ40 performed in two patients demonstrated decreased levels of Aβ40 in one case; however, the other case showed no apparent difference before and after treatment (Fig. 2).
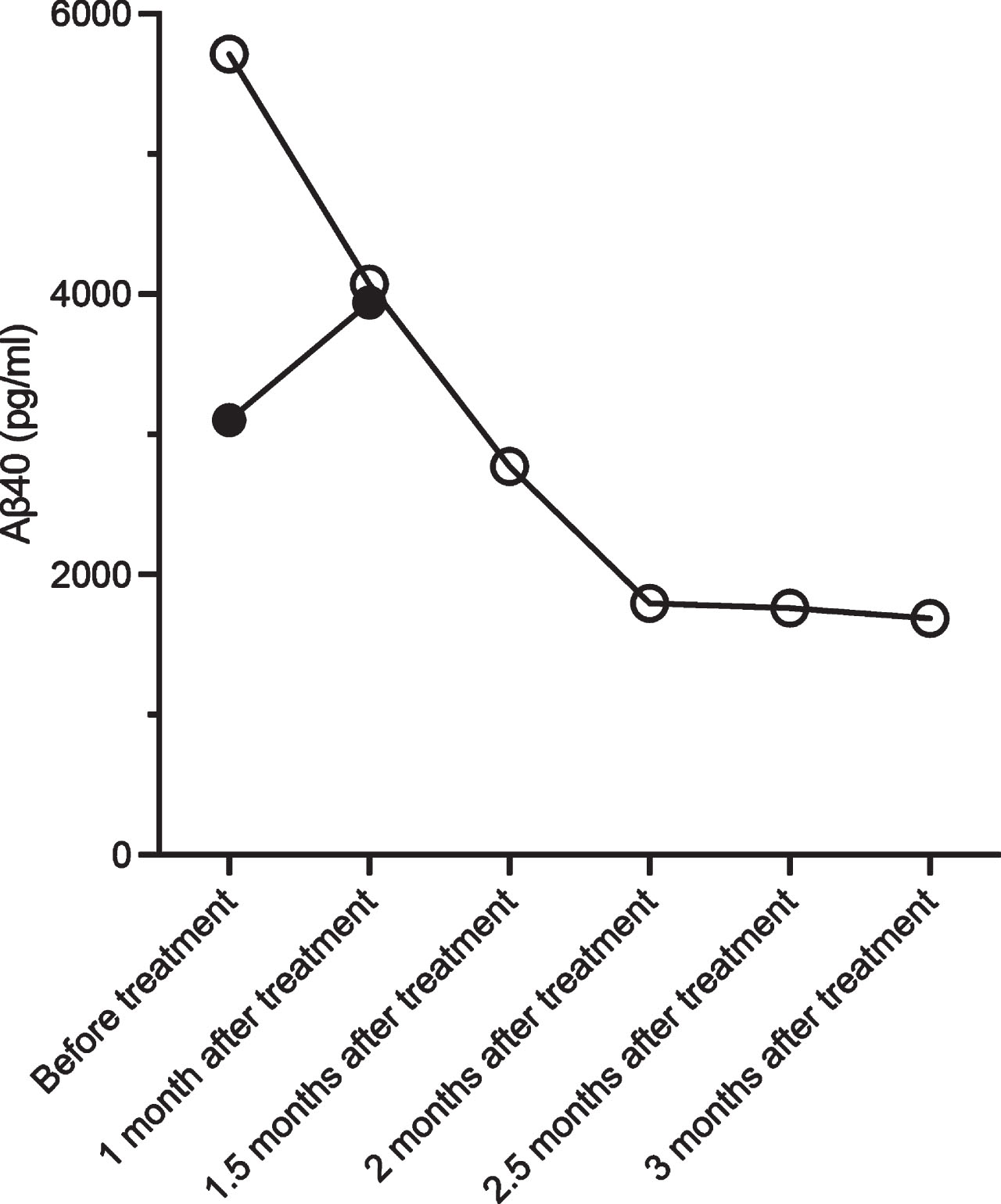
Cerebrospinal fluid analysis in two patients (patients 1 and 6 in Table 1) with cerebral amyloid angiopathy-related inflammation before and after treatment. Patient 1 (open circles) shows a decrease in Aβ40 levels after treatment, whereas patient 6 (black dots) shows an increase in Aβ40 levels after treatment.
Pathological examination
We used eight brain samples, including six from the pre-treatment CAA-ri group (acute CAA-ri) and two from the CAA-ri autopsy cases after immunomodulatory treatments (CAA-ri autopsy) (Table 4). Regarding leptomeningeal blood vessels, small-sized arteries, arterioles, and veins showed severe CAA with apparent vasculopathic changes, such as concentric splitting as well as Aβ40 and Aβ42 deposition in vessel walls in all cases (Fig. 3A–I). Pre-treatment CAA-ri cases showed granulomatous transmural vasculitis with multinucleated giant cells and lymphocytes in the subarachnoid spaces (Fig. 3A, D). Multinucleated giant cells and macrophages containing Aβ in the cytoplasm were present (Fig. 3B, C, E, F). Most arteries demonstrated circumferential Aβ40 and Aβ42 deposition; however, several arteries also showed Aβ40 and Aβ42 deposition in parts of arterial walls (Fig. 3B, C, E, F). Most leptomeningeal blood vessels in the CAA-ri autopsy group demonstrated circumferential Aβ40 (Fig. 3H) and Aβ42 (Fig. 3I) deposition.
Summary of pathological findings
Aβ, amyloid-β protein; CAA-ri, cerebral amyloid angiopathy-related inflammation.

Neuropathological findings of cerebral amyloid angiopathy-related inflammation (CAA-ri) cases. Severe granulomatous vasculitic lesions with amyloid-β protein (Aβ) deposition on the leptomeningeal blood vessels are observed in two patients with pre-treatment CAA-ri (A–F). (A–C) and (D–F) show serial sections from the different cases. Serial sections of the autopsied CAA-ri case (Patient 1) shows apparent Aβ-type CAA without inflammation (G–I). Some Aβ on the leptomeningeal small arteries and arterioles is missing (arrows) with the presence of Aβ-containing multinucleated giant cells and macrophages (B, C, E, F), while circumferential Aβ deposition on the leptomeningeal arterioles is observed in the autopsied case of CAA-ri (H, I). A few veins show some Aβ deposition in vessel walls in the autopsied CAA-ri case (H, I; arrowheads). Hematoxylin and eosin (A, D, G), and immunostaining for Aβ35 - 40 (B, E, H) and Aβ1 - 42 (C, F, I). Scale bars = 100μm (A–I).
Table 4 summarized the quantification of the affected blood vessels in the leptomeninges of the acute CAA-ri and CAA-ri autopsy groups. The percentage of arteries showing partial Aβ40 and Aβ42 positivity (median rate of partially Aβ-positive arteries among all Aβ-positive arteries) were higher in the acute CAA-ri group than in the CAA-ri autopsy group (Aβ40, acute CAA-ri group 20.8% versus CAA-ri autopsy group 3.9%, p = 0.071; Aβ42, acute CAA-ri group 27.4% versus CAA-ri autopsy group 2.0%, p = 0.071). Meanwhile, the median ratio of Aβ40-positive vein and Aβ40-positive arteries was not significantly different between the acute CAA-ri (7.5%) and CAA-ri autopsy (10%, p = 0.857) groups. The results of the examination of Aβ42-positive veins and arteries were also similar (acute CAA-ri group 7.4% versus CAA-ri autopsy group 7.5%; p > 0.999).
DISCUSSION
We demonstrated that CSF levels of Aβ42 and p-tau in patients with post-treatment CAA-ri were significantly lower than those in patients with pre-treatment CAA-ri and AD-CAA. In addition, Aβ40 levels in CSF samples from patients with post-treatment CAA-ri tended to be lower than those in patients with pre-treatment CAA-ri and AD-CAA levels. Pathological examination demonstrated a higher frequency of partially deposited Aβ in arteries and arterioles in patients with acute CAA-ri, while Aβ deposition in veins showed no apparent difference between acute and autopsied CAA-ri cases.
Our results showed that CSF levels of Aβ40, Aβ42, p-tau, and t-tau would not be useful CSF biomarkers for the diagnosis of the acute phase of CAA-ri in comparison to patients with AD-CAA, although previous studies showed that CSF levels of Aβ in patients with CAA-ri were lower than those in age-matched controls and AD and CAA patients [13–15]. In this study, the AD-CAA group was significantly younger than the pre-treatment and post-treatment CAA-ri groups. In a previous study, reduced clearance from old-aged wild-type mouse brains was observed in comparison to clearance from middle-aged mouse brains [22]. Since ISF clearance could be facilitated in the young human brain, further age-matched studies are necessary to confirm these results.
CSF levels of Aβ40, Aβ42, and p-tau could be markers for effective treatments in patients with CAA-ri. Similar results have been reported in the previous studies; however, our case series included a higher frequency of definite CAA-ri cases (5 of 11 cases, Table 3) than previous reports [6]. Since the sequential examination of Aβ40 in this study was inconsistent between cases (Fig. 2), further studies using a large number of definite cases of CAA-ri are essential to confirm our preliminary results. Although CAA-ri is an extremely rare disorder [7], appropriate treatment can improve patient prognosis [23]. The largest longitudinal cohort study in patients with CAA-ri demonstrated transient and potentially relapsing inflammatory characteristics of the clinical and radiological manifestations of CAA-ri [24]. Anti-Aβ antibodies and inflammatory cytokines, such as matrix metalloproteinases, and platelet-derived growth factor-BB, could be biomarkers of this disorder [6, 10]; however, more conventional CSF markers are necessary to initiate effective therapies as soon as possible.
Pathological examination of the pre-treatment CAA-ri cases in this study demonstrated obvious granulomatous transmural vasculitis, while a higher frequency of blood vessels showed Aβ deposition in parts of the walls. Meanwhile, the ratio of arteries and veins showing deposition of Aβ40 and Aβ42 demonstrated no significant difference between pre-treatment CAA-ri cases and autopsied CAA-ri cases that had received anti-inflammatory treatment (Table 4). Our results suggest that Aβ deposited in the arterial walls could be eliminated by phagocytic mechanisms and the IPAD pathway during inflammation; however, elimination of ISF through the glymphatic system could not be changed in patients with CAA-ri because the number of Aβ-positive veins did not increase after treatment (Fig. 3H, I). Moreover, damaged blood vessels during acute inflammation may be a source of Aβ in the CSF. These mechanisms, however, have less influence on Aβ levels in the CSF because of similar Aβ levels in patients with pre-treatment CAA-ri and AD-CAA (Fig. 1A, B).
Disturbance of elimination through the IPAD pathway due to inflammation and the lack of an increase in the compensatory elimination of ISF from the glymphatic system can cause severe brain edema, which is observed in patients with CAA-ri [9]. Increased circumferential Aβ-deposited vessels in cases of autopsied CAA-ri and decreased CSF levels of Aβ40 and Aβ42 in patients with post-treatment CAA-ri could be caused by recovery of the IPAD pathway regarding ISF elimination and a relative decrease in elimination through glymphatic systems to the CSF after treatment with CAA-ri. Recovery of normal vascular function after cessation of inflammation normalizes Aβ clearance across the blood-brain barrier. An experimental study revealed that 25% of the overall Aβ efflux in humans was attributable to direct transmural clearance across the blood-brain barrier [25]. In addition, severe brain edema may decrease Aβ production in the neurons. Since amyloid-related imaging abnormality-edema/effusion (ARIA-E) stems from vascular wall disruption caused by an increase in CAA after immunization and interaction between Aβ on the vessel walls and injected anti-Aβ antibodies [26], knowledge regarding Aβ elimination in patients with CAA-ri could provide a clue to identify the pathomechanisms of ARIA-E.
There has been much debate regarding the existence of glymphatic systems in human and animal brains [22, 28]. However, efflux of substances injected into the brain towards the CSF has been demonstrated in studies using animal models [29]. The perivascular space around venules and veins collects fluid drained from the brain parenchyma to dispose of substances in the CSF [28]. Furthermore, there is some seepage of fluid across the ependyma into the ventricles [28]. These findings suggest that the efflux of ISF could move into the CSF directly from the brain parenchyma. A schematic overview of the proposed pathomechanisms for the elimination of Aβ deposited on the vessel walls in CAA-ri is presented in Fig. 4.

Schematic overview of amyloid-β protein (Aβ) elimination in cerebral amyloid angiopathy-related inflammation (CAA-ri). Interstitial fluid (ISF) containing Aβ is cleared from the intramural periarterial drainage pathway (IPAD) and glymphatic system (A). In the CAA-ri state before treatment, inflammation disturbs the elimination of ISF, leading to brain edema (B). Aβ deposited in vessel walls is cleared by phagocytic mechanisms and clearance towards the cervical lymph nodes via the IPAD pathway, whereas clearance towards the glymphatic system shows no change (B). After treatment for CAA-ri, recovery of clearance from the IPAD pathway causes a relative decrease in ISF elimination towards the glymphatic systems, leading to a lower concentration of Alzheimer’s disease biomarkers in the cerebrospinal fluid (C). SMC, smooth muscle cell.
Regarding the lower CSF levels of p-tau in patients with post-treatment CAA-ri, animal studies have shown that the IPAD and glymphatic systems clear extracellular tau protein from the brain [30, 31]. There have been no reports of human studies showing that tau proteins are eliminated from the IPAD pathway and glymphatic system; however, the presence of tau oligomers in the arterial walls of human brains has been demonstrated [32]. It is uncommon for tau aggregation to be observed in the human cerebral and leptomeningeal blood vessels. The CSF biomarker results in this study reflect the disturbance of tau elimination from the brain through the IPAD pathway, which is similar to the pathomechanism of Aβ elimination. Further studies regarding the elimination of tau protein from the human brain are essential to elucidate the pathomechanisms underlying the lower concentrations of p-tau in patients with post-treatment CAA-ri.
This study has several limitations that should be recognized. Firstly, the number of patients included in the study was small; subsequent studies are therefore essential to confirm our results as the analysis in this study was only exploratory. Further, not all case of CAA-ri were confirmed by pathological examination. Moreover, patients with AD-CAA were diagnosed using only clinical information. Hence, the possibility of another diagnosis cannot be excluded in patients with probable CAA-ri and AD-CAA. We obtained CSF data from AD-CAA cases reported in a previous study. The ELISA methods used for cases of CAA-ri in this study were the same; however, different measurement times could provide different results. In this study, we did not examine the CSF levels of anti-Aβ antibody, which is a more reliable biomarker in patients with CAA-ri. This study was a cross-sectional analysis that used samples from different subjects. To confirm our results, prospective or retrospective studies using samples from the same patients are necessary.
In conclusion, lower levels of CSF Aβ40, Aβ42, and p-tau could be biomarkers for the cessation of inflammation in CAA-ri, reflecting recovery of the IPAD pathway and vascular function. Aβ deposited on the vessel walls can be cleared by phagocytic mechanisms and the IPAD pathway during acute inflammation. No increase in compensatory elimination of ISF through the glymphatic system would be observed in patients with CAA-ri.
Footnotes
ACKNOWLEDGMENTS
We would like to acknowledge the assistance of the doctors across various institutions throughout Japan who provided us with the cerebrospinal fluid samples and brain biopsy specimens used in this study.
We would also like to thank Yumiko Kakuda, Ristuko Goto, Miyuki Honda, and Hisako Fujimura for their excellent technical assistance.
FUNDING
This study was partially supported by JSPS KAKENHI [Grant Number JP26860234 (K.S.), and 19K07958 (K.S.)]; Takeda Science Foundation (K.S.); a grant from the Amyloidosis Research Committee, Intractable Disease Division of the Japanese Ministry of Health, Labour, and Welfare [Grant Number 20FC1022 (K.S., K.O., M.Y.)]; and a grant from the Research Committee on Collection of Biosamples from Patients with CAA on Intractable Diseases from the Japanese Ministry of Health, Labour, and Welfare (H21-Nanchi-Ippan-072) (M.Y.).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article.