
Abstract
Background:
Mitochondrial (MT) dysfunction is a hallmark of Alzheimer’s disease (AD). Amyloid-β protein precursor and amyloid-β peptides localize to MT and lead to MT dysfunction in familial forms of AD. This dysfunction may trigger subsequent types of pathology.
Objective:
To identify the MT phenotypes that occur early in order to help understand the cascade of AD pathophysiology.
Methods:
The 5xFAD mouse model was used to explore the time course of MT pathologies in both sexes. Protein biomarkers for MT dynamics were measured biochemically and MT function was measured using oxygen consumption and ATP assays.
Results:
We discovered progressive alterations in mitochondrial dynamics (biogenesis, fission, fusion, and mitophagy) and function (O2 consumption, ATP generation, and Ca2+ import) in the hippocampus of 5xFAD mice in both sexes as early as 2 months of age. Thus, mitochondrial dynamics and function become altered at young ages, consistent with an early role for mitochondria in the AD pathological cascade.
Conclusion:
Our study offers the baseline information required to understand the hierarchical relationship between the multiple pathologies that develop in this mouse model and provides early biomarkers for MT dysfunction. This will aid in dissecting the temporal cascade of pathologies, understanding sex-specific differences, and in testing the efficacy of putative mitochondrial therapeutics.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) causes the failure of many different tissue, cellular, and molecular systems [1–3]. These include: 1) the generation of toxic cleavage products of amyloid-β protein precursor (AβPP) [4], 2) impairments in the autophagy-lysosomal pathway leading to protein aggregation [5], 3) chronic inflammation initiated by microglial activation [3, 6], 4) astrocyte dysfunction leading to disrupted neurotransmitter uptake and the release of pro-inflammatory cytokines [7, 8], 5) neuronal hyperactivity followed by hypoactivity with disease progression [9], 6) synapse failure/loss and reduced dendritic complexity [10], 7) glutamate excitotoxicity [11], 8) increased oxidative stress [12, 13], 9) blood-brain barrier breakdown [14], 10) deficits in brain glucose consumption [15], and 11) impaired mitochondrial (MT) dynamics and function [16, 17].
The causal relationship between these pathological hallmarks remains speculative. Yet, substantial evidence indicates that broad MT dysfunction may be an upstream failure in sporadic AD. For instance, MT regulate the set point for neural activity, potentially accounting for neuronal activity changes [18]. Toxic peptides of AβPP generated as the primary effect in genetic forms of AD cause MT damage [19], explaining how genetic forms of AD lead to MT pathology. MT DNA is a recently recognized agonist of innate immune responses [20–22]; so the release of MT DNA upon MT damage can account for inflammasome activation and chronic inflammation [23]. MT localize near synapses to meet their energy demands and calcium buffering [24, 25], offering the view that synapse loss and its consequences are downstream effects of an impaired MT system [26]. Furthermore, proteins that mediate MT fission are required for normal synapse formation [27]. To highlight a few studies using mouse and cell culture models, a study using 7-month-old hAbKI AD mice showed increased expression of MT fission genes and reduced gene expression of MT fusion, MT biogenesis, and mitophagy associated genes [28]. Another study reported upregulation of MT energy metabolism and apoptosis related genes in Tg2576 mice suggesting impaired MT metabolism in this mouse model [29]. MT targeted antioxidants rescued primary neurons and N2A cells from Aβ-induced MT toxicity highlighting the importance of MT based therapeutics in AD [30]. Thus, MT dysfunction ties together many of the major CNS failures in AD. This viewpoint emphasizes the need to understand the nature and developmental time course for MT dysfunction and to relate these to other aspects of AD pathology [15].
We have utilized the transgenic 5xFAD model of AD to take a major step towards this goal. The 5xFAD mice overexpress APP 695 with the Swedish (K670N, M671L), Florida (I716V), and London (V717I) familial Alzheimer’s disease (fAD) mutations along with human presenilin 1 (PS1) with two fAD mutations, M146L and L286V [31]. This mouse model has become increasingly dominant in studies of AD due to the fast and aggressive accumulation of plaques, neuroinflammation, and neuronal loss [31, 32]. However, a detailed understanding of the onset and progression of MT dysfunction in this mouse model remains lacking. In addition, few studies of mouse models have addressed the role of sex in the developmental time course of AD pathology. Our study fills these gaps. We conducted biochemical and functional assays on MT isolated from the hippocampi of female and male 5xFAD mice at 2, 4, 6, and 8 months of age. Importantly, our results indicate that significant MT dysfunction is evident as early as 2 months of age in the 5xFAD mice and offer MT biomarkers that are altered at early times in this model. The identification of early MT pathology is important for understanding its relationship with other forms of pathology and to assay the effects of potential therapeutics early in the onset of the disease.
MATERIALS AND METHODS
Mouse model
5xFAD transgenic mice were used in this study (MMRRC Stock No: 34840-JAX). Experiments were performed during the light part of the diurnal cycle (12:12h). A 5xFAD homozygous colony was established for the study. Age-matched B6/SJL mice were used as controls. Mice were grouped by sex and age (n = 4-5/group). Housing, animal care, and experimental procedures were consistent with the Guide for the Care and Use of Laboratory Animals and approved by the institutional IACUC.
MT isolation from hippocampus
Differential centrifugation was used to isolate functional MT as described earlier with slight modifications [33]. Briefly, mice were euthanized by cervical dislocation and brains were quickly excised on ice. Hippocampi were collected in ice-cold isolation buffer: mannitol (210 mM), sucrose (70 mM), HEPES (5 mM), and EGTA (1 mM), pH 7.5 supplemented with 0.5% BSA prior to use. Hippocampi were homogenized in 1 ml of isolation buffer and the lysate was centrifuged at 800× g for 10 min at 4°C. The supernatant was collected and centrifuged at 8000× g for 15 min at 4°C to obtain the MT pellet. The pellet was washed twice with 1 ml of isolation buffer followed by centrifugation at 8000× g for 15 min at 4°C. Protein quantification was performed using the BCA protein estimation kit (Thermo Fisher Scientific).
Western blotting
The MT pellet was lysed in 1X RIPA buffer supplemented with protease and phosphatase inhibitors for 30 min on ice. Lysates were centrifuged at 14,000× g for 15 min and the supernatant used to quantify protein content. Protein samples were denatured by boiling with SDS-Sample Buffer at 95°C for 6 min. A total of 30 mg protein was loaded in each lane of the SDS-PAGE followed by transfer on to PVDF membrane. Resulting blots were incubated in Ponceau S staining solution (Tocris #5225) for 5 min to quantify total protein. The membranes were blocked in 5% non-fat dry milk for 1 h at room temperature followed by overnight incubation with primary antibodies: anti-TFAM (1:3000, Abcam; ab131607), anti-Sirt3 (1:1000, Cell Signaling Technology; CST #5490), anti-MFN2 (1:1000, Cell Signaling Technology; CST #9482), anti-FIS1 (1:2000, Proteintech; 10956-1-AP), anti-MTP18 (1:1000, Abcam; ab198217), anti-DRP1 (1:1000, Abcam; ab184247), rabbit anti-LC3B (1:3000, Abcam; ab51520), rabbit anti-p62 (1:10,000, Abcam; ab109012), rabbit anti-OPTN1(1:1000, Cell Signaling Technology; CST #58981S), rabbit anti-PINK1 (1:500, Novus Biologicals; BC100-494), rabbit anti-Parkin (1:1000, Cell Signaling Technology; CST #32833S), anti-BECN1 (1:1000, Cell Signaling Technology; CST #3495S), anti-pMFF (1:1000, Cell Signaling Technology; CST #49281S), anti-MFF (1:1000, Cell Signaling Technology; CST #84580S), anti-FUNDC1 (1:1000, Cell Signaling Technology; CST #49240S), anti-MICU1 (1:1000, Cell Signaling Technology; CST #12524), anti-MCU (1:1000, Cell Signaling Technology; CST #14997) diluted in blocking buffer at 4°C with gentle mixing. The membranes were washed 3 times in 1X TBST (Tris-buffered saline with 0.1% Tween) buffer and then incubated with either goat anti-rabbit IgG HRP (1:10000, Jackson ImmunoResearch #111-035-144) or goat anti-mouse IgG HRP (1:5000, Abcam) conjugated secondary antibodies for 1 h at 23°C. Membranes were washed 3 times in 1X TBST buffer and proteins were visualized using the ECL reagent (Advansta, K-12042-D20) on a Chemidoc imager. ImageJ software was used to analyze the scanned blots and to quantify signalintensities.
MT respiration
The oxygen consumption rate (OCR) of isolated brain MT (from 2, 4, 6, and 8 months of age male and female mice) was measured by the XFe96 Extracellular Flux analyzer (Seahorse Bioscience). Isolated brain MT were diluted to 200μg/ml in cold MAS-BSA buffer (220 mM mannitol, 70 mM sucrose, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA) + substrate (10 mM pyruvate/malate) and plated into seahorse XF96 V3 PS cell culture microplates. Wells without MT were used for background correction. The plate was centrifuged at 2000× g for 20 min at 4°C to help attach the MT to the plate. After centrifugation, 130μL of MAS-BSA buffer + pyruvate/malate substrate was added to the wells and the plates were incubated at 37°C for 10 min. Oxygen consumption was triggered by the addition of ADP. Two baseline measurements were obtained before any injection, and two response measurements were obtained after each injection followed by additional 30 s of mixing. The final concentrations of compounds after injections were: 1 mM ADP, 2μM oligomycin, 4μM FCCP, and 20μM rotenone/antimycin A. OCR was calculated by the Seahorse XF96 softwarepackage.
ATP kinetic assay
Isolated MT from hippocampi were diluted to 0.01 mg/mL protein in ATP assay buffer: sucrose (225 mM), KH2PO4 (44 mM), Mg (CH3COO)2, 4H2O (12.5 mM), and EDTA (6 mM), pH 7.5. Standard reaction buffer (SRB) was prepared as per the manufacturer’s instructions (ATP determination kit, Invitrogen, A220066). A diluted MT suspension was added to the SRB followed by the substrate mix containing pyruvate (1 mM), palmitoyl-L-carnitine (0.05 mM), alpha-ketoglutarate (10 mM), and malate (1 mM) to energize the MT. The mix was transferred to a 384 black well plate and the reaction was initiated with the addition of 10 mM ADP. Fluorescence emission intensity was recorded in CLARIOstar (BMG Labtech) plate reader.
Statistical analyses
Data are represented as mean±SEM. SDS-PAGE analysis using MT from mice hippocampi was performed using 4 different mice for the statistical analyses. Data were plotted and analyzed with GraphPad Prism version 9 (GraphPad Software, La Jolla, CA, USA). Unpaired student’s t-test was used to determine significant differences between the two groups; p < 0.05 was the cutoff for assessing statistical significance.
RESULTS
MT biogenesis is altered in 5xFAD mice
The MT transcription factor A (TFAM) is a nuclear protein that plays a key role in MT DNA replication. TFAM upregulation is linked to increased MT DNA, suggesting that the protein helps coordinate the nuclear and MT genomes for MT biogenesis [34]. Sirt3 is a MT deacetylase that promotes MT biogenesis by deacetylating TFAM and enhancing its activity [35, 36]. To measure MT biogenesis in 5xFAD mice, we quantified TFAM and Sirt3 expression in hippocampal MT using western blot analysis. Male 5xFAD mice at 2 months of age had reduced TFAM levels and Sirt3 was trending towards significance, but female mice showed no detectable difference compared to the controls (Fig. 1). At 4 months of age, TFAM expression was increased in females and decreased in males. Sirt3 levels were reduced in females with a trend towards reduction in males. At 6 months, TFAM and Sirt3 levels were significantly decreased in 5xFAD females, with males also exhibiting a significant reduction in Sirt3. Surprisingly, these changes were not observed in 8-month-old male mice, although Sirt3 remained reduced in 5xFAD females. These results suggest an early defect in MT biogenesis, with the appearance of that defect first in male mice.

MT biogenesis markers. Western blots of B6/SJL and 5xFAD hippocampal MT probed for TFAM and Sirt3 abundance. Bar plot data represent the mean±SEM. Statistical differences were determined by unpaired Student’s t-test with significance set at p < 0.05.
MT fission:fusion balance is disturbed in 5xFAD mice
We searched for changes in fission:fusion proteins using hippocampal MT using the same procedures. The MT fusion protein, MFN2, was dramatically reduced in females at 2 months (Fig. 2), becoming ∼5.5 fold lower than in controls by 8 months. Males failed to show a significant decrease until 6 months of age. The fission protein, MFF, is the primary receptor for DRP1 [37]. Phosphorylation of MFF by AMPK leads to DRP1 recruitment to mitochondria resulting in mitochondrial fission [38]. MFF levels remained relatively unaffected in both females and males except for the ratio between the phosphorylated and dephosphorylated form that increased at 6 and 8 months (Supplementary Figure 1). FIS1 showed decreased expression in both sexes at 4 month of age (Fig. 2). The expression of MTP18, a novel regulator of MT fission, was significantly depressed in both 5xFAD sexes across all ages (Fig. 2). Another fission marker, DRP1, showed decreased expression in females at 2 months. A significant reduction in DRP1 levels in males was observed by 4 months. The most striking result is that MTP18 is reduced in both sexes at all ages.

MT fission:fusion markers. Western blots of B6/SJL and 5xFAD hippocampal MT probed for MFN2, MTF18, FIS1, and DRP1 abundance. Bar plot data represent the mean±SEM. Statistical differences were determined by unpaired Student’s t-test with significance set at p < 0.05.
Blockade in 5xFAD mitophagy
Mitophagy is critical in regulating MT quality control and compromised mitophagy has been implicated in the pathogenesis of AD [39]. To probe the induction of mitophagy in 5xFAD mice, we monitored the conversion of soluble microtubule-associated protein 1A/1B-light chain 3B (LC3BI) to LC3BII, a lipid bound form found predominantly in autophagosomal membranes. Specifically, we quantified the levels of LC3B-II and the LC3BII/I ratio in the MT fraction isolated from the hippocampus of 2-, 4-, 6-, and 8-month-old B6/SJL and 5xFAD mice. A significant increase in LC3BII levels was observed starting at 4 months of age in 5xFAD females and at 6 months in males (Fig. 3). Intriguingly, both 5xFAD females and males showed a significant increase in LC3BII/I ratio at 2 months of age, and this ratio remained elevated at 4, 6, and 8 months of age (Fig. 3). Our observations are consistent with the previous studies showing LC3BII recruitment to MT in the 5xFAD and 3xTg models of AD [40, 41]. Together, elevated LC3BII levels and enhanced LC3BI to II conversion indicate increased mitophagy induction in 5xFADmice.
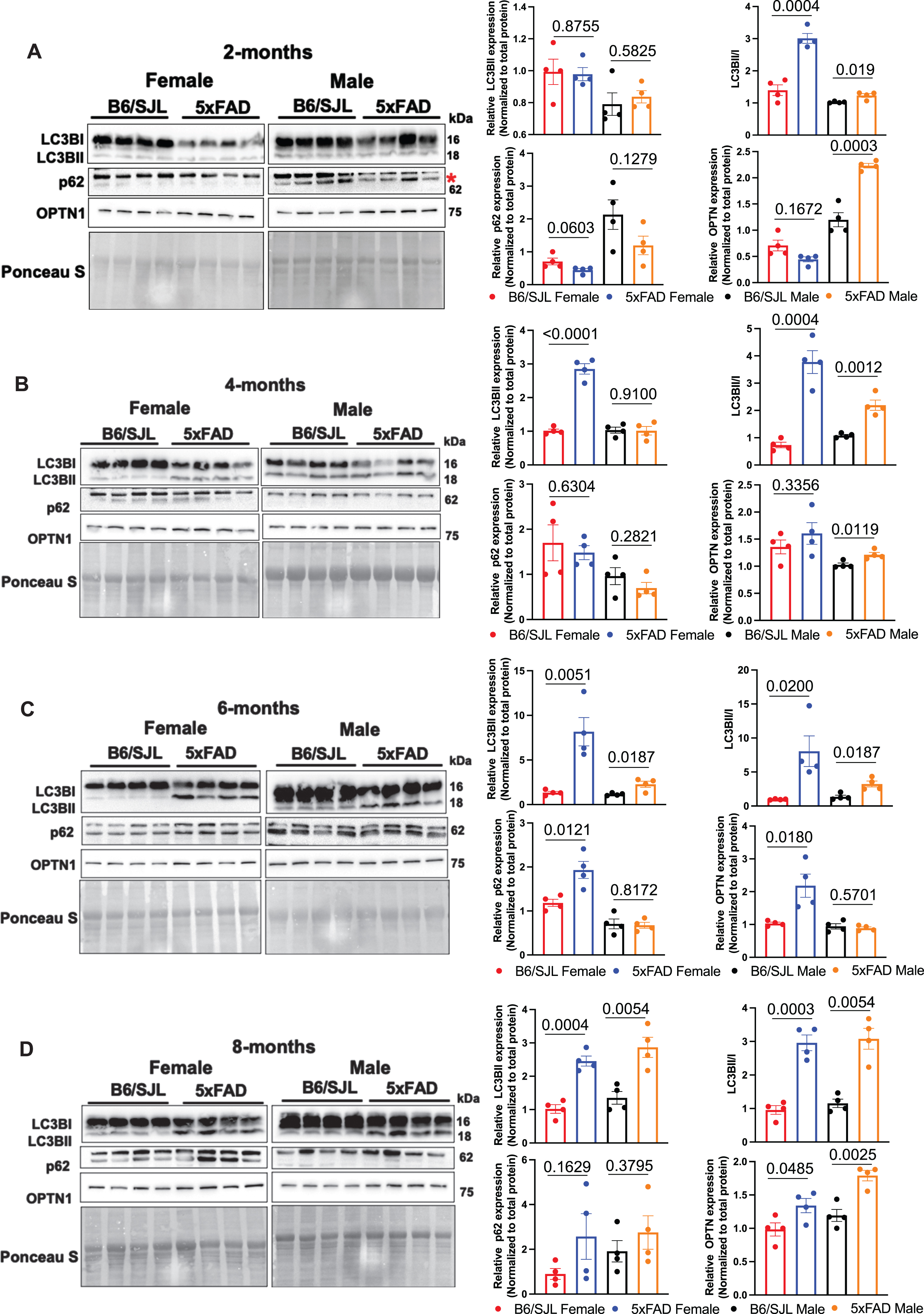
Blockade in 5xFAD mitophagy. Western blots of B6/SJL and 5xFAD hippocampal MT probed for LC3BI, LC3BII, p62, and OPTN abundance. Bar plot data represent the mean±SEM. Statistical differences were determined by unpaired Student’s t-test with significance set at p < 0.05.
To measure the progression of mitophagy, we quantified the mitophagy adaptor SQSTM1/p62 which is degraded after mitophagy is induced. Remarkably, the levels of SQSTM1/p62 in the MT fraction remained unchanged in both the sexes at all ages except for 6-month-old females, which showed a significant increase (Fig. 3). These results suggest that the progression of normal mitophagy is blocked in the 5xFAD mice. To test this possibility, we measured the levels of OPTN, another mitophagy receptor critically involved in the elimination of damaged MT [42]. The levels of OPTN remained unchanged in the females at 2 and 4 months of age but increased significantly at 6 and 8 months of age (Fig. 3). Males exhibited a completely different pattern, being increased as early as 2 months and remaining significantly elevated. Thus, the enhanced LC3B-II accumulation and unaltered/increased p62 and OPTN levels indicate that even though there is increased mitophagy initiation in the 5xFAD mice, the subsequent steps in the mitophagy pathway are blocked.
We also measured the levels of FUNDC1, an outer MT membrane (OMM) residing protein that acts as a major mitophagy receptor and promotes hypoxia induced MT clearance independent of the PINK1/Parkin pathway [41, 44], which is discussed below. Strikingly, our results showed a significant decrease in the levels of FUNDC1 in the MT fraction starting at 2 months of age in both females and males (Supplementary Figure 1). This suggests a defect in the initiation of receptor-mediated mitophagy early in both sexes. Our results offer the conclusion that in 5xFAD mice, the PINK1/Parkin-independent mitophagy pathways are compromised very early in both sexes.
Defective PINK1/Parkin-dependent mitophagy
The PINK1 (PTEN-induced kinase1)/Parkin cascade has recently been identified as the major pathway for quality control mitophagy in neuronal cells [45, 46]. Thus, we tested PINK1 expression levels in the hippocampal MT fraction. PINK1 levels began declining at 2 months in females and 4 months in males (Fig. 4). These results were consistent with decreased PINK1 levels seen in APP transgenic mice and in immortalized mouse primary hippocampal (HT22) neurons expressing mutant APP [47, 48]. Reduced PINK1 should cause decreased recruitment of Parkin to the OMM. We found that Parkin levels also declined as early as 2 months of age (Fig. 4). This observation was consistent in both the sexes.

Defects in PINK/Parkin dependent pathways. Western blots of B6/SJL and 5xFAD hippocampal MT probed for PINK1, Parkin, and BECN1 abundance. Bar plot data represent the mean±SEM. Statistical differences were determined by unpaired Student’s t-test with significance set at p < 0.05.
Parkin levels on the MT could be reduced from defective translocation. Therefore, we examined the levels of Beclin (BECN1), a component of the PI3 kinase complex that recruits Parkin to damaged/depolarized MT [49]. Interestingly, BECN1 levels start declining at 4 months in females and remain low (Fig. 4). In males, BECN1 levels were reduced at 2 and 4, but not at 6 and 8 months of age, suggesting that, compensatory mechanisms come into play to restore the BECN1 levels in older males, and that reduced Parkin on MT may not be completely dependent on BECN1 levels.
MT calcium uptake is defective
Calcium signaling is perturbed in AD, which affects key enzymes and several organelles including MT as reported earlier in studies of mouse AD models and human postmortem brains [50, 51]. To address the relationship between calcium dysregulation and MT dysfunction, we measured protein levels for key regulators of MT calcium import, MICU1 and MCU.
MCU is a Ca2+ activated MT calcium uniporter present on the inner membrane of MT. MT calcium uptake 1, MICU1, is a component of the regulatory complex along with MICU2 and EMRE, which regulates the activity of MCU and serves as the gatekeeper to control Ca2+ entry into the mitochondria [52, 53]. MICU1 is crucial in stabilizing cristae junctions and maintaining structural mitochondrial membrane framework [54]. Our results indicated a significant reduction in MICU1 and MCU protein expression at 2 months of age in 5xFAD males but not in females (Fig. 5). However, this depressed expression level did not extend to 4-month-old males. Reduced MCU expression was evident in females beginning from 4 months of age, which continued until 8 months, MICU1 was depressed in 5xFAD females at 6 and 8 months of age. Both proteins were reduced in 5xFAD males at 6 months of age, but this reduction was not detected at 8 months. Overall, the deficit in MICU1 and MCU protein abundance was consistent across all ages in 5xFAD females but not in 5xFAD males.

Calcium uptake machinery is altered in 5xFAD mice. Western blots of B6/SJL and 5xFAD hippocampal MT probed for MICU1 and MCU abundance. Bar plot data represent the mean±SEM. Statistical differences were determined by unpaired Student’s t-test with significance set at p < 0.05.
Early defects in MT bioenergetics
Impaired MT biogenesis, MT dynamics, and mitophagy would be expected to result in decreased oxidative phosphorylation and impaired MT respiratory efficiency. To test this, the MT OCR was measured in freshly isolated MT from mice of both genotypes using the Seahorse XFe96 extracellular flux analyzer. Purified MT were energized by pyruvate and malate as respiratory substrates and MT respiratory parameters including basal respiration, ATP production, maximal respiratory capacity, and spare respiratory capacity (SRC) were quantified. We detected no significant differences in basal respiration using MT isolated from any age or sex (Fig. 6). No significant differences in ATP-coupled OCR were found across age and sex (Fig. 6). In contrast, maximal respiratory capacity appeared to be compromised in both males and females at all ages (Fig. 6). SRC is a measure of the ability of the MT to meet the demands for extra energy, beyond the basal level, in response to stressed conditions. SRC was depressed at all ages in female 5xFAD mice. Males also showed depressed SRC at most ages. These results indicate that abnormal MT bioenergetics emerge early in 5xFAD mice and that the pronounced bioenergetic parameters include maximal respiration and SRC.
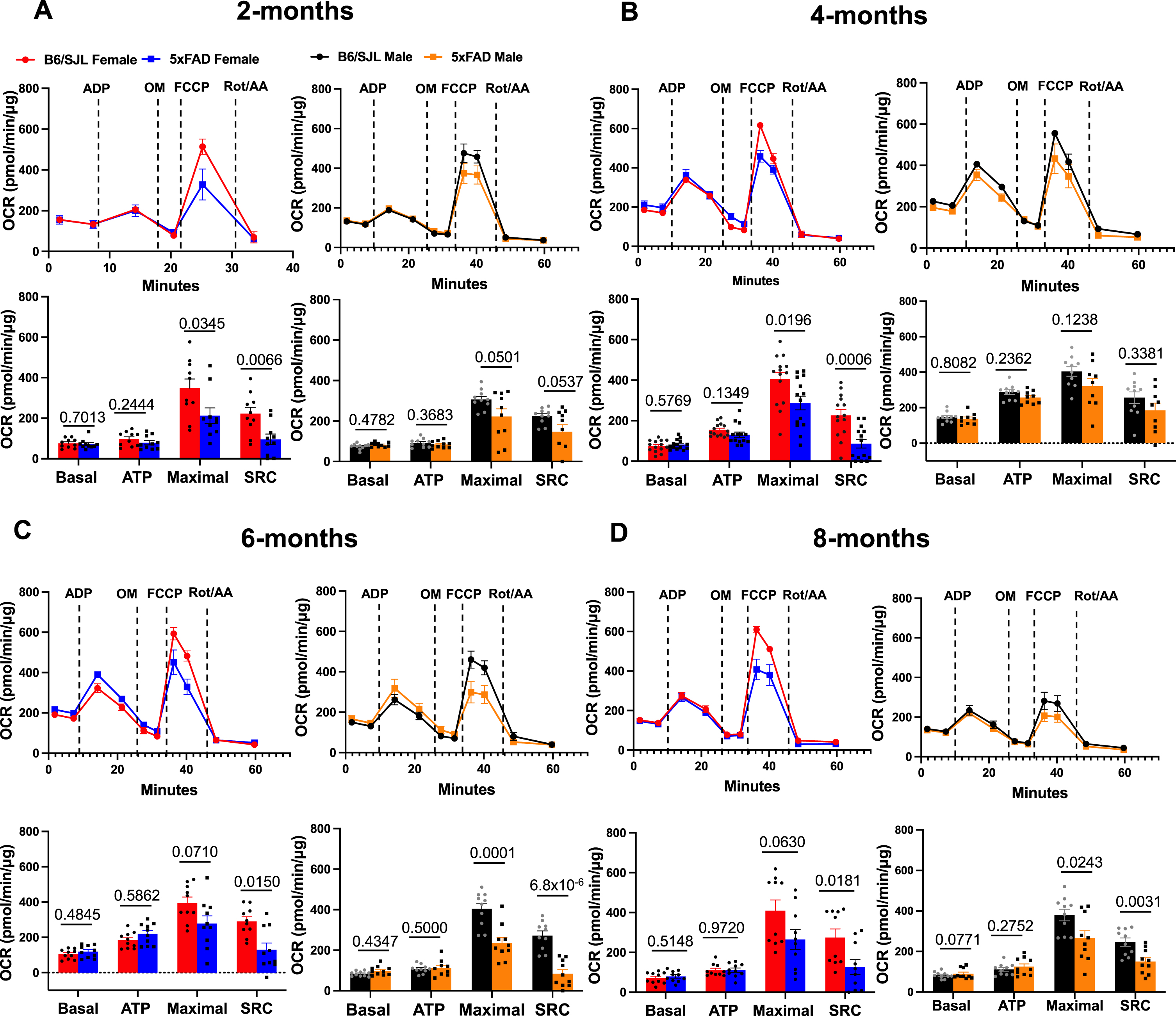
Defective maximal respiration and SRC in 5xFAD mice. Extracellular flux analyzer profiles and quantification of oxygen consumption rate in 5xFAD and B6/SJL mice at A) 2, B) 4, C) 6, and D) 8 months of age. Statistical differences were determined by unpaired Student’s t-test with significance set at p < 0.05.
ATP production is not compromised
Prior studies have reported abnormalities in the MT ETC complexes due to AD pathology [55], and the alpha chain of ATP synthase— a subunit of complex V— has been detected in the neurofibrillary tangles that form in degenerating neurons of AD patients [56]. In addition, reduced ATP levels were previously reported in AD postmortem brains and 5xFAD mice [57, 58], which is consistent with the expected impact of MT degeneration and loss of ATP synthase activity. Considering these reports, our surprising results showing normal ATP-coupled OCR in the 5xFAD model (Fig. 6) prompted us to measure ATP generation directly, rather than using the proxy of OCR.
We utilized a luminescence based real-time ATP assay. MT isolated from the brains of these mice revealed no change in ATP production in 2-month-old 5xFAD females, whereas male mice showed a slight but significant increase in ATP (Fig. 7). Both 4- and 6-month-old female and male 5xFAD mice exhibited significantly increased ATP compared to control mice (Fig. 7). This increase persisted in 8-month-old males but was lost in females of the same age (Fig. 7). These results suggest a modest increase in MT ATP production in the 5xFAD mice, perhaps from compensatory pathways, but no decrease in MT ATP generation at least until 8 monthsof age.
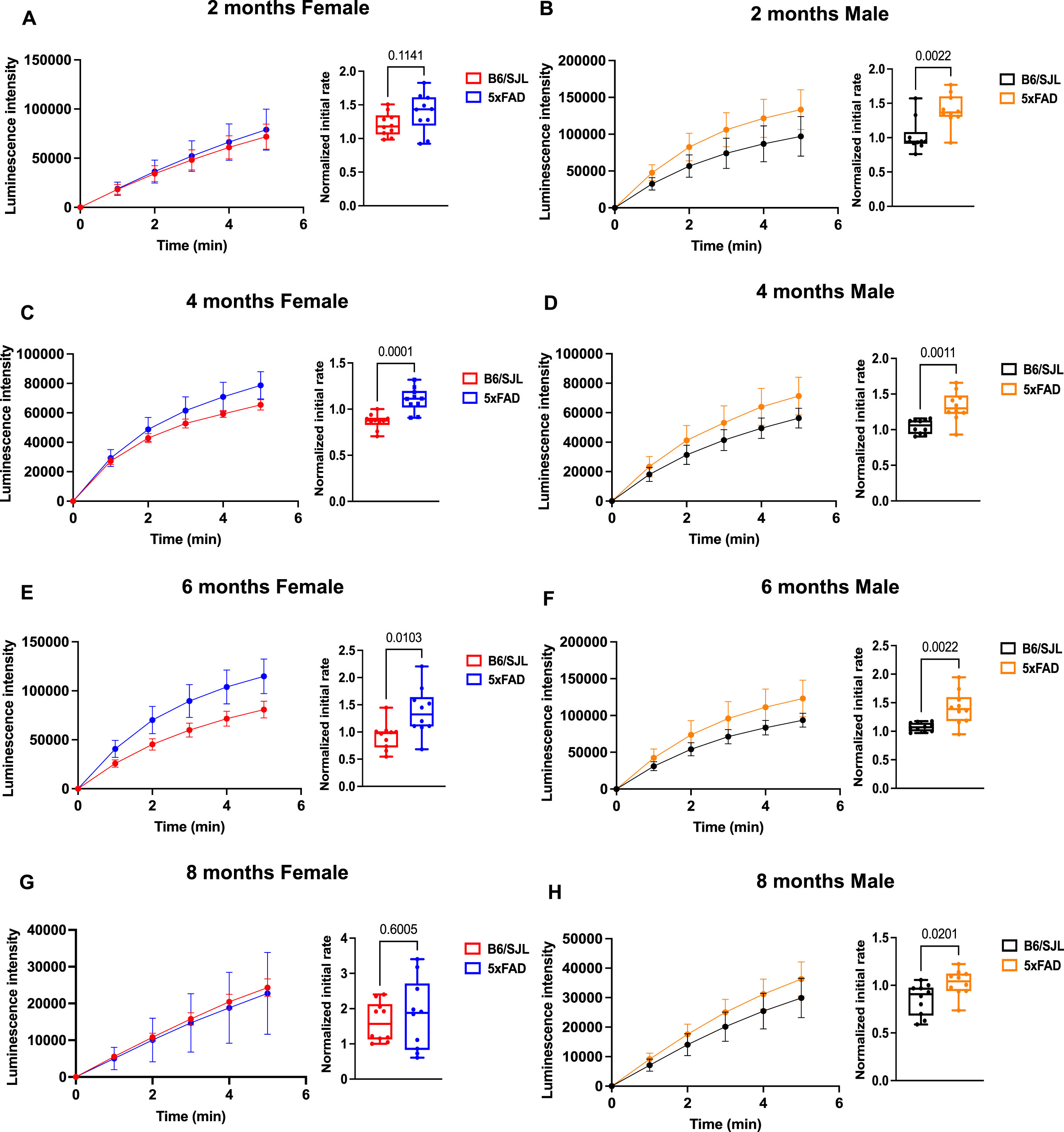
ATP production is elevated in 5xFAD mice. Luminescence intensity graphs and bar plots of the initial rate plot suggest increased MT ATP generation in 5xFAD mice at A) 2, B) 4, C) 6, and D) 8 months of age. Statistical differences were determined by unpaired Student’s t-test with significance set at p < 0.05.
DISCUSSION
Understanding the cascade of molecular, cellular, and circuit failures that occur in AD requires the identification of the earliest precipitating event(s). For genetic forms of AD, and the mouse models that have been designed from understanding these genetic forms, the precipitating factor is abnormal AβPP processing. Abnormal AβPP processing may also cause sporadic AD, but this possibility remains debated, with several other candidate initiating events remaining as contenders. Despite this unknown, and the likelihood that sporadic AD is a class of related diseases with different etiologies, mouse models designed from familial forms of AD remain the best in vivo system to dissect the cascade of pathologies that ensue from abnormal AβPP processing. Among the many mouse models studied to date, the 5xFAD model has emerged in recent years as the one most frequently employed.
We employed this model to probe MT pathology across age and sex. MT dysfunction remains as an attractive contender for causing at least some forms of sporadic AD, as encompassed in the “Mitochondrial Cascade Hypothesis for Sporadic Alzheimer’s Disease.” [59]. And it is possible that MT dysfunction occurs immediately downstream of abnormal AβPP processing in familial AD. Despite this possibility, a systematic study of MT abnormalities across age and sex using the 5xFAD model was lacking. Thus, we documented the MT abnormalities and their trajectory across age and sex as a critical baseline for understanding the hierarchical relationship between the multiple pathologies that develop in the model, identifying early biomarkers of MT dysfunction for developing protective therapies, and understanding the effects of sex on AD-related molecular and cellular pathologies.
We identified several important biomarkers for MT dynamics dysfunction that occur by 2 months of age in 5xFAD mice (Table 1). We also detected early bioenergetic alterations that include deficits in maximal OCR and SRC in both sexes. These data indicate that very early MT pathology is broad across multiple aspects of MT biology and function, rather than being specific to one function. Previous studies have reported that intraneuronal accumulation of Aβ42 starts at 1.5 months preceding amyloid deposition and gliosis at 2 months [17], positioning broad MT dysfunction perhaps coincident with the cytoplasmic accumulation of Aβ42.
Summary of the age and sex dependent changes in MT biomarkers and MT bioenergetics in the 5xFAD mouse model
# indicates trending p values (0.05 < p < 0.1).
The developmental trajectories observed for some biomarkers are complex. For instance, TFAM exhibits reduced expression in female 5xFAD at 4 and 6 months, but this reduction recovers by 8 months. MCU abundance in males is initially depressed but appears to reach normal levels by 8 months. There are at least two general explanations for these observations. The most likely is that compensatory processes emerge to rectify unbalanced homeostasis and physiology. The alternative is simply assay variability. However, we employed 4 mice from each genotype for western blotting data, but the general conclusions made were obtained using a second cohort (n = 4). This generates confidence in the conclusions, especially for changes that reach a highly significant level (p < 0.01). But it is possible that some observations made are due to the inherent variability in quantifying the western blotting data. Our data also offer interesting differences in protein expression between the sexes. It is difficult to know what underlies these differences between the sexes, although we expect that some are due to changing hormonal levels that characterize the estrous cycle in the female mice [60, 61]. Further study is required to elucidate the underlying mechanisms.
In summary, our study offers the most comprehensive data on MT pathology that occurs in a mouse model for AD, specifically the 5xFAD model. It provides a valuable resource to further study the relationship of MT pathology to other types of pathology that occur in this model, to test putative therapeutics designed to protect MT from the disease, and to further probe how sex can differentially affect some of the MT markers/phenotypes that we observed. Nevertheless, mitochondrial dynamics and function, including biogenesis and mitophagy, for instance, are extraordinarily complex biological processes. Additional work remains to extract additional details of mitochondrial defects that occur in the 5xFAD model and to test the conclusions made.
Footnotes
ACKNOWLEDGMENTS
We thank Dr. Deepthi Yedlapudi for her work in initiating this study.
FUNDING
This study was supported by philanthropic funds from the Community Foundation for Palm Beach and Martin Counties and other anonymous philanthropic sources.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and its supplementary material.