
Abstract
Frontotemporal dementia (FTD) can manifest as diverse clinical phenotypes and is frequently caused by mutations in different genes, complicating differential diagnosis. This underlines the urgent need for valid biomarkers. Altered lysosomal and immune functions proposedly contribute to FTD pathogenesis. Cathepsins, including cathepsin S, are enzymes preferentially expressed in brain in microglia, which influence lysosomal and immune function. Here, we examined whether alterations in serum cathepsin S levels associate with specific clinical, genetic, or neuropathological FTD subgroups, but no such alterations were observed. However, further research on other lysosomal proteins may reveal new biologically relevant biomarkers in FTD.
INTRODUCTION
Frontotemporal dementia (FTD) is an umbrella term for a group of clinical syndromes characterized by atrophy in frontal and temporal regions. These syndromes include behavioral variant frontotemporal dementia (bvFTD), primary progressive aphasias, the FTD-plus phenotypes, namely corticobasal syndrome and progressive supranuclear palsy (PSP), and FTD with concomitant motoneuron disease (FTD-MND) [1, 2]. The definite diagnosis of FTD remains a challenge, especially in the early phases of disease, due to the overlapping cognitive, motor, and neuropsychiatric features with other neurodegenerative diseases and primary psychiatric disorders as well as due to the lack of disease-specific biomarkers. Early accurate diagnosis of FTD is a crucial step toward developing disease-modifying therapies and effective disease management strategies. Two major neuropathological subtypes, the TAR DNA-binding protein 43 (TDP-43) proteinopathies and tauopathies, account for approximately 90 % of FTD cases [2]. The C9orf72 hexanucleotide repeat expansion (C9-HRE) and GRN mutations associate with TDP-43 proteinopathy in FTD patients, whereas FTD patients carrying mutations in the microtubule-associated protein tau gene (MAPT) are characterized by tau-positive inclusions. Interestingly, specific mutations in GRN, gene encoding progranulin protein, have also been shown to link with lysosomal storage disease in addition to FTD spectrum diseases [3].
Lysosomes are cellular organelles involved in the degradation and recycling of intracellular and extracellular material, including lipids, proteins, nucleic acids, and carbohydrates [4]. Autophagy, on the other hand, is a process responsible for the sequestration and degradation of long-lived proteins, dysfunctional organelles, and invading pathogens. This degradation process takes place in autolysosomes, which form upon the fusion of autophagosomes with lysosomes [5]. Recent studies have provided evidence that lysosomal dysfunction associates with the onset and progression of FTD and amyotrophic lateral sclerosis (ALS) [6]. Mutations in several genes encoding proteins that are mechanistically related to lysosomal and autophagosomal function, including C9-HRE, sequestome 1 (SQSTM1/p62), ubiquilin 2 (UBQLN2), dynactin subunit 1 (DCTN1), TANK-binding kinase 1 (TBK1), optineurin (OPTN), and valosin containing protein (VCP) have been linked to both diseases [7]. Interestingly, many of these genes are highly expressed in microglia, which are key immune cells of the brain. These proteins localize to lysosomes or modulate their function, trafficking, lysosomal acidification, and autophagy.
The most abundant lysosomal proteases are the family of cathepsins that play a vital role in protein degradation, energy metabolism, and immune responses [8]. There are eleven cathepsins encoded in the human genome (B, C, F, H, K, L, O, S, V, W, and X) [9]. Cathepsin B is the most studied cathepsin expressed in the central nervous system (CNS), and its elevated levels in serum correlate with cognitive dysfunction in Alzheimer’s disease (AD) patients [10]. Cathepsins have also been shown to cleave progranulin [11], which regulates inflammatory responses in the CNS and is linked to FTD pathophysiology [12]. Cysteine cathepsins, such as cathepsin S, have been reported to contribute to neurodegeneration and deposition of protein aggregates in neuronal cells [13] and associate with AD progression [14]. Cathepsin S is expressed throughout in the brain and body, but is preferentially expressed in the microglia in the CNS, suggesting that it impacts their function and interaction with neurons [15]. Cathepsin S is unique among the cysteine cathepsin family due to its restricted tissue expression, association with antigen-presenting cells localized in lymph nodes and spleen as well as other immune cells, such as macrophages and microglia, and its ability to remain active in environments with neutral pH, such as extracellular space [16]. Therefore, cathepsins, and especially cathepsin S, might be interesting biomarker candidates in FTD, but has remained to be studied further.
Here, we have used the ultrasensitive single molecule array (Simoa) method to measure the levels of serum cathepsin S in a large multicenter cohort of FTD patients and healthy control subjects. We compared the serum cathepsin S levels between the FTD patient groups harboring the different major genetic mutations, clinical phenotypes, or different predicted prevailing neuropathologies. Furthermore, we also compared the cathepsin S levels between FTD spectrum patients and healthy controls.
MATERIALS AND METHODS
Patients and controls
This study was performed in compliance with the Declaration of Helsinki, and it was approved by the local Institutional Ethical Review Boards in Kuopio, Finland (ethical permit no. 10.5.2013/16/2013), and Brescia, Italy (ethical permit no. NP5231). The study subjects have given a written informed consent before participating in the study. A total of 226 serum samples were analyzed (Table 1). The FTD group included 144 FTD patients. Of these, 58 patients were recruited at the Department of Neurology, Kuopio University Hospital (Finland) and 86 at the Centre for Neurodegenerative Disorders, University of Brescia (Italy). The cohort consisted of 92 bvFTD, 24 PSP, 23 nonfluent variant primary progressive aphasia (nfvPPA), 3 FTD-MND, and 2 semantic variant primary progressive aphasia (svPPA) patients (Table 1). Patients were diagnosed by a neurologist specialized in neurodegenerative disorders in the tertiary level outpatient clinics. Patients were diagnosed according to the current diagnostic criteria for each FTD subtype [17–19].
Serum cathepsin S levels according to cohort details
bvFTD, behavioral variant frontotemporal dementia; C9-HRE, Chromosome 9 open reading frame 72 hexanucleotide repeat expansion; FTD, frontotemporal dementia; FTD-MND, frontotemporal dementia with motor neuron disease; GRN, gene encoding progranulin; MAPT, microtubule associated protein tau; nfvPPA, non-fluent variant primary progressive aphasia; PSP, progressive supranuclear palsy; svPPA, semantic variant primary progressive aphasia; TDP-43, TAR DNA-binding protein 43; Tau, tau protein (tauopathy); y, years.
Genotyping data were available from 55 patients; 21 were C9 HRE carriers and 31 GRN mutation carriers, and 3 patients carried MAPT mutations (Table 1). The predicted neuropathologies of 79 patients were deduced from available data. Based on their genetic status patients were divided into FTD-TDP and FTD-Tau groups so that C9orf72 HRE and GRN mutation carriers were allocated to FTD-TDP group and MAPT-mutation carriers were allocated to FTD-Tau group. Clinical phenotype was also used to deduce predicted neuropathology: FTD-ALS patients were allocated to FTD-TDP group and PSP patients were allocated to FTD-Tau group. All in all, 53 were predicted to have TDP-43 pathology and 26 tauopathy. The rationale for these deductions has been described in previous literature [20].
The healthy control (HC) group consisted of 82 participants (Table 1). They had undergone clinical examination by a neurologist, cognitive testing and neuropsychological assessment, and were subsequently confirmed not to have any neurodegenerative disorder. Of the HC group participants, 19 were recruited from Oulu University Hospital (Finland) and 63 from Kuopio University Hospital (Finland).
Simoa analysis of cathepsin S levels in serum samples
Cathepsin S levels were quantified from a total of 226 serum samples (FTD patients and HC) according to manufacturer’s instructions using the digital immunoassay technology on a Single Molecule Array (Simoa) HD-1 Analyzer, software version 1.5.1809.12001 and human Cathepsin S Discovery Kit (REF# 102064) [Quanterix, Billerica, MA, USA] [21]. Before measurements, the samples were thawed at room temperature, gently mixed, and centrifuged (10,000× g, 5 min, +20°C). Samples were randomized on plates and measured in duplicates. The samples were diluted 1:100 before measurement. A maximum coefficient of variation (CV) of sample replicates of 20% was accepted. The concentrations of all samples were above the lower limit of quantification (1.95 pg/mL) and within the dynamic range (0–200 ng/mL) of the assay.
Statistical analyses
Serum cathepsin S levels were compared between major FTD clinical phenotypes or causative mutation carriers and HC. The statistical analyses were performed using IBM SPSS Statistic Version 27 and GraphPad Prism version 9.3.1. Due to not normally distributed cathepsin S data according to visual inspection, natural logarithmic transformation was performed. Differences between cathepsin S levels were evaluated with a multivariate linear regression model adjusted for age at sampling and sex. A p-value of ≤0.05 was considered as statistically significant. HC were assumed not to carry GRN, MAPT, or C9 HRE mutations.
RESULTS
Serum cathepsin S levels were first compared between the total group of FTD patients and HC. There was no statistical difference detected (p = 0.171, B = –0.058) between the groups (Fig. 1A). Sex or age did not have a statistically significant effect on cathepsin S levels in the total study cohort (p = 0.218, B = –0.050 and p = 0.088, B = –0.004, respectively). When examined separately, cathepsin S levels did not correlate with age (p = 0.075, r = –0.197) or sex (p = 0.584, r = –0.061) in healthy controls nor in the FTD patient group; age (p = 0.386, r = –0.073) or sex (p = 0.354, r = –0.078). When the FTD patients were subgrouped according to different clinical phenotypes, comparison of serum cathepsin S levels in bvFTD patients (n = 92) to those in HC did not reveal statistically significant differences (p = 0.453, B = –0.031) either. The same was true also when comparing serum cathepsin S levels in HC to the levels in PSP (n = 22) and nfvPPA patients (n = 22) (p = 0.177, B = –0.103 and p = 0.166, B = –0.090, respectively) (Fig. 1A). There were only 3 FTD-MND patients and 2 svPPA patients and, therefore, the data were insufficient for drawing any conclusive comparisons between HC and these clinical subgroups (Fig. 1A). Data on disease severity (Clinical Dementia Rating with NACC FTLD) was available for 127 of the 144 patients. However, cathepsin S levels did not correlate with the FTLD-CDR score in the FTD patient group (p = 0.237, r = 0.106).
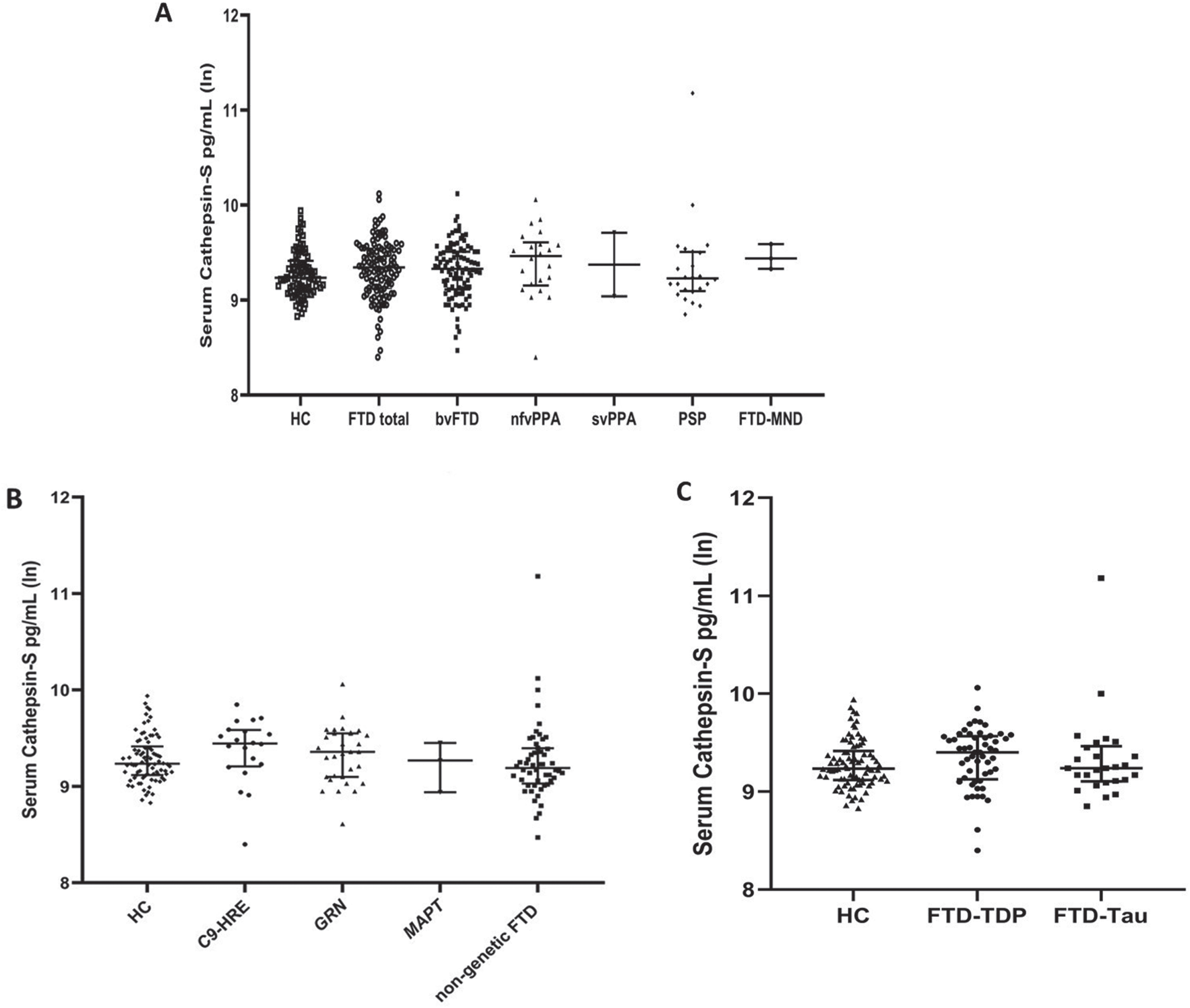
Serum cathepsin S levels in FTD patients of different clinical phenotypes (A), carriership of different genetic mutations (B), and predicted underlying neuropathologies (C) and healthy controls. The Y-axes are presented using logarithmic scale.
Next, serum cathepsin S levels in GRN mutation-carrying FTD patients (n = 31) were compared to those in HC or GRN mutation non-carrying FTD patients. These analyses did not show a difference between the groups (p = 0.580, B = –0.030 and p = 0.909, B = –0.007, respectively) (Fig. 1B). Comparing C9 HRE-carrying FTD patients (n = 21) to HC or to C9 HRE-non-carrying FTD patients did not reveal any statistically significant differences in the serum cathepsin S levels either (p = 0.220, B = –0.081 and p = 0.290, B = 0.075) (Fig. 1B).
Finally, we compared the serum cathepsin S levels in patients subgrouped according to their predicted neuropathologies. Neither the FTD patients with predicted TDP-43 neuropathology (FTD-TDP; n = 53) nor those with predicted tau neuropathology (FTD-Tau; n = 26) showed differences in serum cathepsin S levels when compared to each other (p = 0.920, B = –0.009) or HC (p = 0.256, B = –0.055 and p = 0.275, B = –0.076, respectively) (Fig. 1C). Comparison of the serum cathepsin S levels in patients with predicted tau neuropathology to those with predicted TDP-43 neuropathology did not indicate any correlation even when adjusted for FTLD-CDR score (p = 0.872, B = 0.018).
DISCUSSION
In this study, our objective was to assess whether alterations in serum cathepsin S levels associate with different clinical phenotypes of FTD. In addition, we assessed if serum cathepsin S levels were changed in patients carrying the main FTD causative mutations or having predicted TDP-43 or tau neuropathologies. These analyses were done in order to evaluate if serum cathepsin S levels could be utilized to differentiate the different FTD patient subgroups from each other or from HC. However, our analyses did not reveal any statistical differences between the different groups, suggesting that altered serum cathepsin S levels do not specifically correlate with any clinical FTD subtypes, predicted TDP-43 or tau neuropathologies, or the major known causative genetic mutations in C9orf72, GRN, or MAPT.
Cathepsin S is preferentially expressed in microglia, which are key mediators of immune responses in the CNS. Microglial activation may at least partially be triggered by the abnormal accumulation of the tau or TDP-43 proteins, which are fundamental neuropathological changes in FTD patient brain and associate with neurodegeneration [22]. Activated microglia, in turn, can induce the expression and secretion of lysosomal cathepsins, especially cysteine cathepsins, including cathepsin S, during early stage of neuroinflammation [23]. Moreover, cathepsin S is released by microglia and macrophages after stimulation with inflammatory cytokines or pro-inflammatory lipopolysaccharide, which can lead to the degradation of extracellular matrix components, thereby supporting the migration of microglia to the inflammation site [23]. Cathepsins are also important regulators of the function of lysosomes, a cellular compartment whose dysfunction is associated with neurodegenerative diseases, and they also cleave the FTD-linked progranulin protein [24]. Proteolytic processing of progranulin to granulin peptides is a key regulator of progranulin function in mediating anti- and proinflammatory effects. Thus, from a biological point of view, changes in cathepsins might possess biomarker potential in FTD. However, the present results in this multi-centered international FTD cohort suggest that serum cathepsin S measurements do not provide a practical diagnostic tool for FTD or its differential diagnostics. However, future studies may reveal whether assessment of other cathepsin family members or lysosomal proteins in biofluids could be utilized as biomarkers in FTD diagnostics. For example, cathepsins B, D, and L have been implicated in different neurodegenerative disorders, including FTD, AD, and Parkinson’s disease, and therefore might represent new possible biomarker candidates worth further examination in FTD patients [25]. The strengths of this study are a reasonable sample size, multicenter data, and ultrasensitive analysis methodology. The main weakness of the study was that confounding factors affecting serum cathepsin S levels could not be assessed. Cathepsin S is also expressed in peripheral tissues e.g., in the spleen. Thus, assessment of brain-derived cathepsin S in blood samples is complicated by the fact that centrally and peripherally produced cathepsin S cannot be differentiated in the assay, which might have contributed to the negative result in the present study. Moreover, some previous reports have indicated that impaired renal function in patients with chronic kidney disease (CKD), systemic inflammation, and concomitant cardiovascular disease in CKD patients affect blood cathepsin S levels [26, 27]. Unfortunately, data on renal function or inflammatory status, which might have impacted the serum cathepsin S levels, were not available for the present study. Also, it remains unclear how the disease severity in FTD patients influences the serum cathepsin S levels. Such factors and comorbidities should be kept in mind and taken into consideration as possible when, e.g., stratifying patient groups in future biomarker studies. In addition, longitudinal sampling in the same patients would help to evaluate the effect of disease progression and severity on the examined biomarker levels.
Footnotes
ACKNOWLEDGMENTS
This study is part of the research activities of the Finnish FTD Research Network (FinFTD).
FUNDING
This study was conducted with the financial support from Academy of Finland (grant numbers 315459 and 315460), Academy of Finland and EU JPND Research SynaDeg project (grant number 351841), Sigrid Jusélius foundation, Finnish Brain Foundation, Instrumentarium Research Foundation, and Orion Research Foundation. NH is a PhD student supported by the UEF Doctoral Programme of Molecular Medicine (DPMM).
CONFLICT OF INTEREST
Barbara Borroni, Eino Solje, and Annakaisa Haapasalo are Editorial Board Members of this journal but were not involved in the peer-review process nor had access to any information regarding its peer-review.
All other authors have no conflict of interest to report.
DATA AVAILABILITY STATEMENT
The data supporting the findings of this study are available on reasonable request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.