
Abstract
Background:
Alzheimer’s disease (AD) is the most common type of neurodegenerative dementia affecting people in their later years of life. The AD prevalence rate has significantly increased due to a lack of early detection technology and low therapeutic efficacy. Despite recent scientific advances, some aspects of AD pathological targets still require special attention. Certain traditionally consumed phytocompounds have been used for thousands of years to treat such pathologies. The standard extract of Gingko biloba (EGB761) is a combination of 13 macro phyto-compounds and various other micro phytocompounds that have shown greater therapeutic potential against the pathology of AD.
Objective:
Strong physiological evidence of cognitive health preservation has been observed in elderly people who keep an active lifestyle. According to some theories, consuming certain medicinal extracts helps build cognitive reserve. We outline the research employing EGB761 as a dual target for AD.
Methods:
This study investigates various inhibitory targets against AD using computational approaches such as molecular docking, network pharmacology, ADMET (full form), and bioactivity prediction of the selected compounds.
Results:
After interaction studies were done for all the phytoconstituents of EGB761, it was concluded that all four of the phytocompounds (kaempferol, isorhamnetin, quercetin, and ginkgotoxin) showed the maximum inhibitory activity against acetylcholinesterase (AChE) and GSK3β.
Conclusion:
The highly active phytocompounds of EGB761, especially quercetin, kaempferol, and isorhamnetin, have better activity against AChE and GSK3β than its reported synthetic drug, according to molecular docking and network pharmacology research. These compounds may act on multiple targets in the protein network of AD. The AChE theory was primarily responsible for EGB761’s therapeutic efficacy in treating AD.
Keywords
INTRODUCTION
The health-care-related economic burden from neurodegenerative diseases (NDDs) has steadily increased over the last few decades [1]. Based on the most recent estimate, 50 million people worldwide suffer currently from dementia, with a yearly healthcare expenditure of more than $800 billion [2]. Among all other types of NDDs, Alzheimer’s disease (AD) particularly exhibits steeply increasing data year after year and is listed as one of the most progressive types of NDD. It is been characterized by irreversible cognitive decline including episodic memory loss, uncertainty, visuospatial concerns, and behavioral as well as psychiatric shifts [3]. Aberrant protein processing is a distinguishing feature of AD. Amyloid-β (Aβ) plaques and abnormally hyperphosphorylated tau protein get misfolded and self-accumulate in the cortical regions of brains as aggregates, known as Aβ plaques and neurofibrillary tangles (NFTs), respectively [4]. These assemblies are thought to be key players in the pathogenesis of AD and constitute the most significant histological hallmarks. However, there have been other hypothesis that have been proposed, but tau theory, amyloid cascade, and cholinergic hypothesis are the most important ones.
The cure for AD is still a challenge though there are many promising approaches with phytomedicines and gene therapy that have been developed, they are yet under exploration for targeting the wholistic AD pathological sites [5, 6].
Besides that, it has also been reported by Sintes et al. (2011) [7] that elevated GSK-3β activity leads to toxicity in cultured neurons controlling β-secretase and GSK-3 overexpression leading to cognitive impairment, NFT formation, and microtubule instability in a transgenic mouse model of AD. Many pre-clinical and initial clinical phase studies have concluded that aiming at GSK-3 pathways, other than the above-discussed causes; can lead to the development of effective targeted therapy for AD. Recent research has demonstrated the potential of GSK-3 inhibitors (GSK-3Is) to reverse the balance between neurodegeneration and neurogenesis in vitro and in vivo models [8–10]. Also, the recovery of cholinergic function is thought to be clinically advantageous since the decline in acetylcholine (ACh) levels causes cognitive and memory deterioration [11].
Moreover, the presence of acetylcholinesterase (AChE) enzyme in the synaptic cleft breaks down into acetate and choline and bind with Acetyl CoA to give ACh. During the impulse transmission ACh releases and they bind to their respective receptor on the postsynaptic neuron causing the termination by hydrolysis of ACh. Thus, there is an inability to clear ACh, resulting in cholinergic excess while in the presence of AChE inhibitors, they bind to ACh and block their action. And when the transmission of nerve impulses passes through, there is a release of ACh that binds to the receptor.
AChE is eventually involved in the deposition of Aβ plaques, and therefore, inhibiting AChE [12]. Furthermore, cholinesterase inhibitors (ChEIs) are the first-line indicative therapeutic intervention and there is always an unfulfilled necessity to develop ChEIs with enhanced therapeutic efficacy and low toxicity [13, 14]. Currently, the prescribed medications for AD are basically either cholinesterase inhibitors (tacrine, rivastigmine, galantamine, donepezil, etc.) [15] or N-Methyl-D-aspartate (NMDA) receptor antagonist (memantine) [16] and their efficacy is modest as, they enhance the cell-to-cell (neuronal cell) interaction levels by increasing the ACh concentration [17]. However, extended use of these drugs exhibits toxicity leading to hyperstimulation of cells and reduction in function which causes increased melanin, skin allergies, dizziness, and ocular toxicity [18, 19]. Thus, phytochemicals with AChE inhibitory activity are on higher exploration and have been consistently rated through computational studies with similarly high AChE binding affinity than Galantamine (synthetic inhibitor) and di-hydrotanshinone-1 (Natural Inhibitor) for the production of a new dual-binding site for heterodimeric drugs [20–23].
In our study, Gingko biloba (EGB761 standard extract), a well-known neuro potential extract in the Chinese traditional medicine system is taken as a model system [24, 25]. EGB761 is a well-defined extract of Ginkgo biloba leaves with 24% flavone glycosides (mostly quercetin, kaempferol, and isorhamnetin), 6 % terpene lactones (2.8–3.4% ginkgolides A, B, and C, and 2.6–3.2 % bilobalide), 0.8% ginkgolide B, and 3% bilobalide, as well as proanthocyanidins, glucose, and rhamnose [26].
It is known to possess neuroprotective activity and improving conditions like dementia, multiple sclerosis, glaucoma, macular degeneration, and other cognitive impairments [27–30].
In order to investigate the molecular mechanism of EGB761 in the treatment of AD, this study will use the research methods of network pharmacology and molecular docking to analyse the potential molecular mechanism of EGB761 and construct the EGB761-drug-targets-AD action network. This network will combine the analysis of the target gene with the interaction and function analysis of the target protein, as well as the discussion of their gene regulation, including the role of miRNA and transcription factors.
MATERIALS AND METHODS
In silico docking analysis of acetylcholinesterase and glycogen synthase kinase-3-beta (AChE and GSK3β)
Molecular structures of the target and ligands
3D structure of human AChE and GSK3β was experimentally stabilized with a resolution of 2.30 Å and 1.80 Å by Xray-Crystallography, PDB ID: 6O4X [31] & 1J1B [32] respectively obtained from the Protein Databank (PDB). Also, the structure of selected compounds dihydrotanshinone I (1YL- AChE natural inhibitor), galantamine (AChE drug as control), 6-bromoindirubin-3’-oxime, and olanzapine (GSK3β drugs as controls), quercetin, kaempferol, isorhamnetin, ginkgotoxin, quercetin-3-β-D-glucoside, rutin, quercitrin, bilobalide, ginkgolide A, ginkgolide B, and ginkgolide C were retrieved from the small molecule database PubChem [33].
Preparation of acetylcholinesterase (PD: 6O4X) and glycogen synthase kinase-3-beta (PDB: 1J1B)
The protein AChE and GSK3β have been prepared by the Prime program of the Schrodinger Suite. The proteins are pre-processed by adding H-atoms, forming zero-order bonds with metals, filling up the missing loops and side chains, forming disulfide bonds, eliminating water molecules that are beyond 5.00 Å of distance from het groups, and finally forming their het states utilizing Epik with 7.00±2.00 pH. On completing pre-processing, Chain A is selected. The protein is further optimized by using PROPKA at pH 7 and the water is removed beyond 3.00 Å with fewer than 3.00 Å H-bonds to the ones with non-water. Next, the proteins are minimized for stabilization to RMSD at 0.30 Å to unite heavy atoms with the OPL5_2005 force field [34, 35].
Preparation of ligands
The phytocompounds of the standardized extract of EGB761 were prepared by utilizing the Ligprep program in Schrodinger. The ligands were given with a force field of OPLS_2005 and ionization was performed at pH 7.00±2.00 using Epik. The ligands were desalted. The prepared ligands are downloaded as a maestro file [34].
Receptor grid generation
The prepared protein was then taken to form a grid around it for site-specific docking (active site targeted by galantamine) using a Receptor grid generation panel under Glide Tab. For site-specific docking, the grid is formed around the active site where galantamine and Dihydrotanshinone I have been targeted in literature. The prepared grid file is downloaded in a zip file for ligand docking [36].
Ligand docking and visualization
Ligand docking was done using the Extra Precision (XP) tool of the Glide program in the Schrodinger suite. The prepared protein and ligand were taken Ligand Docking panel of Glide [37]. The glide grid file of the protein and prepared ligand file was given as input to the Ligand docking panel to search for interactions between the protein and ligand. The poses were automatically generated by Glide. The poses were passed through several screenings of the algorithm and ultimately in the last screening, minimization of a grid to the OPLS-AA non-bonded ligand-protein interaction energy was completed. The final poses with minimum energy were then displayed in the project table [38]. The 2-D representation of ligand and protein interaction was visualized by using a ligand interaction diagram. The diagram provides the types of interaction involved with different amino acids with varied color coding. The overall analysis of the binding affinity and the interactions observed were compared to check which experimental compound can be a better ligand for AChE and GSK3β [39].
ADME of selected compounds
ADME-related properties were determined by utilizing the SwissADME program running producing truly significant descriptors, and utilize them to perform ADME forecasts, arranged by ADME-Score, drug similarity property, etc. This all data is further utilized to evaluate the pharmacokinetic profiles of the mixtures inside the library. The property demonstrates the number of physiochemical properties descriptors that are accounted outside the ideal scope of qualities like chemical formula, MW, number of heavy atoms, rotatable bonds, hydrogen bond receptors, hydrogen bond donors, molar refractivity, and TPSA. Another range is their lipophilicity characteristic, by following different methods such as physics-based methods, atomistic and knowledge-based methods, topological method, and hybrid fragmental/topological method. Moreover, to know the solubility of the compound, the properties are represented by topological and fragmental methods, Log S-ESOL Log S-Ali, and LogS (SILICOS-IT). Other properties include their pharmacokinetics involving their gastrointestinal absorption and blood-brain barrier permeation, P-glycoprotein substrate and cytochrome inhibitors, number of violations, and synthetic accessibility [40].
Quantifying ligand’s bioactivity using molinspiration tool
The bioactivity score for the selected compounds was predicted by using the Molinspiration tool. The bioactivity score depends on the characteristics of classes of drugs that target the group of proteins and exhibit their functionality like ion channels, enzymes, kinases, etc. The bioactivity score from above 0 is most preferable, 0 to –5 is moderately active and the score below –5 is inactive [41].
In silico toxicity prediction
The ProTox-II technique, which is offered as a web-based service (https://tox-new.charite.de/protoxII/), was used to forecast the toxicity to humans [42]. For the prediction of various toxicity endpoints, such as hepatotoxicity, carcinogenicity, immunotoxicity, adverse outcomes (Tox21) pathways, and toxicity targets, ProTox-II uses molecular similarity, fragment propensities, most frequent features, and (fragment similarity based cross-validation) machine learning tools.
Network pharmacology
Construction of PPI network of AChE associated proteins
The construct of protein-protein interaction (PPI) network of AChE using the string database [43] was prepared. It was then filtered for protein-protein Interaction Network (PPIN), having confidence score greater than equal to 400 [44]. Thereafter, the first interacting partners of AChE and its PPIs within interactions were used to create the complete PPIN. The network Analyzer (cytoscape tool plugin) was used to understand the topological structure and possible existence of hidden mechanisms of the network [45]. And lastly, three topological parameters (degree distribution (P(k)), betweenness centrality (CB), and closeness centrality (CC)) for PPIN were calculated [46].
Gene ontology (GO) and pathways analysis of AChE associated proteins
The GO enrichment analysis to explain the potential biological process of proteins involved in the AChE-PPI network was done. The GO functions molecular function (MF), biological process (BP), and cellular component (CC) were taken into account during the analysis [47]. The functional analysis was carried out using the ShinyGO v0.75 tool (https://bioinformatics.sdstate.edu/go/) an online webserver. Each functional enrichment analysis was taken into account for the construction of a dot plot chart that enabled good visualization of the significant terms. Furthermore, the associated pathways related to AChE network proteins were also identified and represented the top 10 significant pathways based on gene count and FDR (False Discovery Rate) value (0.05). The Wikipathways dataset was utilized for pathway analysis in ShinyGO v0.75 tool [48, 49].
Details of binding affinity and free energy (Site-specific docking) of EGB761 standard extract with AChE

Screening of gene targets of effective compounds of EGB761
The Monograph of EGB761, houses information on all of EGB761’s active constituents and was queried using the search term “EGB761" then all acquired results were confirmed in PubChem. We utilized the SwissTargetPrediction web tool (https://www.swisstargetprediction.ch/) to identify the targeted genes of the effective active compounds filtered from the EGB761’s extract. The obtained list of all the targets that EGB761’s active compounds were intended to affect [50].
Construction of compounds-target-network
The Cytoscape (V3.9.21) software was used to construct the compound-target association network [51]. The efficient bioactive compounds of EGB761 and target genes were entered into Cytoscape after sorting and categorizing the procured bioactive compounds of EGB761 and its respective target data. The connection between the network nodes was then demonstrated by connecting lines. The network “EGB761-bioactive compounds” was built using this information and the networks topology of the entire network was then analyzed using Network Analyzer (Cytoscape plugin) [51].
Screening of key targets
To identify the important gene targets in AD, initially identifying the overlapped genes between the AChE-PPI was done, followed by common genes targeting by EGB761-bioactives (Kaempferol, Isorhamnetin and Quercetin) and controls (Galantamine and Dihydrotanshinone).

Schematic representation of ligand interaction of EGB761 phytocompounds, galantamine (control) and dihydrotanshinone I (natural inhibitor)with AChE.

Graphical representation of pharmacokinetic parameters (Absorption, Distribution, Metabolism and Excretion- ADME Score) of EGB761 phytocompounds, Galantamine (control) and Dihydrotanshinone I (natural inhibitor).
Regulatory relationship of AChE and GSK3β
Identification of miRNA associated with AChE and GSK3β
From the miRWalk database [52], the miRNA-target for both the genes AChE and GSK3 β separately was identified. And we filtered the common miRNA-targets which were targeting both the genes. The miRWalk database is a consortium of three database TargetScan (conserved site context scores, version 7.1), miRDB (release 5.0) and the validated information from miRTarBase (version 7.0) [53]. The random-forest-based methods utilized by miRWalk to predict miRNA target site using software TarPmiR searching the complete transcript sequence including the 5’-UTR, CDS and 3’-UTR. In our study, we filtered and used miRNAs were targeting 3’-UTR seed regions.
Screening of AChE: Transcription factors (TFs)
TFs are the important trans-acting molecules during transcription regulation process and thus, exploring TF-target interaction is a key step to understand the regulatory mechanism in AD. TFs, may directly (TFs to gene) or indirectly (TF to miRNA to gene) regulate the gene expression. In our study, we used the Transcriptional Regulatory Relationship Unraveled by Sentence-based Text mining (TRUSST) database V2, that provides the information about the mode of regulation (activation or repression) of target genes [54].
Relationship between miRNAs-TFs
To identify the regulatory relationship between common miRNAs targeting both AChE and GSK3 β, we used TransmiR v2.0 database [55]. It manually curate TFs-miRNA regulations by using publications, included ChIP-seq-derived TFs-miRNA regulation information. Here, TF-miRNA relationship data of humans was downloaded first based on literature in tsv.gz format and then the TFs regulatory pattern (activation or repression) of the targeted miRNAs were extracted.
Construction of AChE and GSK3β regulatory network
We found a PPI between AChE and GSK3β, and further, to understand the regulatory mechanism of AChE and GSK3β at transcription and post-transcriptional level, regulatory network was constructed. Then three of the relationships networks (AChE-TFs, AChE, and GSK3 β) was used along with the common miRNAs, and miRNAs-TFs. For visualizing the network, Cytoscape v3.8.2 was used and showed transcriptional (activation or repression) regulation.
RESULTS
In silico analysis of EGB761 phytocompounds with AChE
Quercetin, kaempferol, and isorhamnetin are flavanol glycosides commonly present in fruits and flowers in polyphenol classification. Ginkgotoxin is a neurotoxin, found in seeds and leaves of Ginkgo biloba, biosynthesized on the conversion of ribulose-5-phosphate with dihydroxyacetone phosphate in the presence of PLP synthase complex, pyridoxal phosphate, and AdoMet. While Quercetin-3-β-glucoside belongs to oxoanion flavonoid which is a product of deprotonation of the hydroxy group of quercetin’s position 7 flavone moiety. Rutin is a rutinoside, a form of quercetin with the substitution of rhamnose sugar and glucose at carbon position 3. On the other hand, Quercitrin is another derivative of quercetin with substitution of alpha-L-rhamnosyl moiety and carbon position 3 with glycosidic linkage. Ginkgolides and bilobalide are terpenic lactones that are diterpenoids comprising of 20-carbon on biosynthesis from geranylgeranyl pyrophosphate and 15-carbon on biosynthesis from farnesyl pyrophosphate. The chemical structure of phytocompounds and controls with their respective PubChem IDs have been mentioned in Table 1.
Site-specific docking was performed with AChE protein (PDB ID: 6O4X) and phytocompounds from EGB761 standard extract. Site-specific docking is performed with the active/ interacting site of galantamine and dihydrotanshinone I. It is evident from the results that most of the ligands, i.e., quercetin [56, 57], isorhamnetin, ginkgotoxin [58], kaempferol [59], quercetin-3-β-glucoside, ginkgolide, rutin, and quercitrin have better binding energies than galantamine (–8.024 kcal/mol) and dihydrotanshinone I (–7.052 kcal/mol) [60]. This minimum energy confirms the stability of the complex formed. The more negative are the energies, the more stable will be the compounds. It is clear from the binding affinities that majority of the phytocompounds of EGB761 extract have more negative free energy than controls (galantamine and dihydrotanshinone I).
Additionally, other analyses were also performed after the visualization of the docked poses. Those interactions include the hydrogen bonds and the hydrophobic interactions formed between the protein and the ligands. It was observed that the residues that are involved in forming H-bonds and hydrophobic bonds were the same as those formed at the active site. Then the complex structures were checked for interactions based on their amino acid involved in the interaction of shortlisted top-scoring compounds out of them. The amino acids involved in the interaction are shown in Table.
Out of all phyto-compounds, quercetin, isorhamnetin, ginkgotoxin, and kaempferol have shown a greater number of common amino acids involved in the interaction. Overall, analysis of the three major components (binding affinity, hydrogen bonds, and hydrophobic bonds) of the docked poses showed that all the experimental compounds (isorhamnetin, kaempferol, ginkgotoxin, and quercetin) interacted favorably with AChE and can be a good inhibitor against AChE. Out of all, top dock-scoring compounds have been selected from EGB761 and then checked for ADME properties using the Molnspiration tool. From Table 1, it is very clear that the phytocompounds are showing good results when compared to control in terms of binding and free energies. Various descriptors have been used to analyze the pharmacokinetic properties of the compounds. TPSA is the topological surface area that a ligand cover on the protein surface while miLogP is the octanol-water partition coefficient. The tool checked their miLogP, natoms, molecular weight, number of OH atoms, and nrotb values. The ligands also checked for the rule of five signifying the Lipinski Rule. The ligands shortlisted from ADME properties are now checked for their bioactivity scores in the Molinspiration tool. The bioactivity scores in Table 4, clearly depict that all phytocompounds from EGB761 extract have positive bioactivity scores as enzyme and kinase inhibitors (AChE, an enzyme). It suggests that these may be significant inhibitors of AChE.
The results showed that all the three flavanol glycosides, i.e., kaempferol, quercetin, and isorhmanetin, have an active toxicity against Aryl hydrocarbon receptors, Estrogen receptor alpha, estrogen receptor ligand binding domain, and mitochondrial membrane potential. Quercetin is found to in toxicity class 3 with LD50 value of 159 mg/kg, more stable and active. While isorhamnetin and kaempferol fall under toxocity class 5 with LD50 vlaue of 5,000 mg/kg and 3,919 mg/kg. The toxicity against aryl hydrogen receptors and mitochondrial membrane potential is supported by literature wherein the exact mechanism for neuroinflammation is explained (Fig. 3). Reactive oxygen species are produced as a result of the AhR being activated by its ligands, which results in an increase in the xenobiotic metabolism enzymes (CYPs) and mitochondrial toxicity (ROS). Additionally, these enzymes interact with the arachidonic acid pathway and boost the production of a number of metabolites of arachidonic acid, including prostaglandins and EETs (epoxyeicosatrienoic acid), HETEs (hydroxyeicosatrienonic acid), and HETEs (hydroxyeicosatrienonic acid), which are sources of ROS in a variety of tissues, including the brain. Inflammasome activation results from ROS production and promotes the release of inflammatory cytokines [61]. The ROS is both produced by and targets mitochondria. ROS are produced by mitochondria in the ETC, TCA cycle enzymes, and MAO. Lipid peroxidation and the calcium signal that is dependent on inositol trisphosphate can be stimulated by the production of hydrogen peroxide in MAO or superoxide in ETC. When NADPH oxidase in PS or AD produces too many ROS, PARP is activated, consuming NAD and lowering NADH for complex I. The formation of mitochondrial ROS can hinder glucose transporters and result in the restriction of mitochondrial substrates [62].
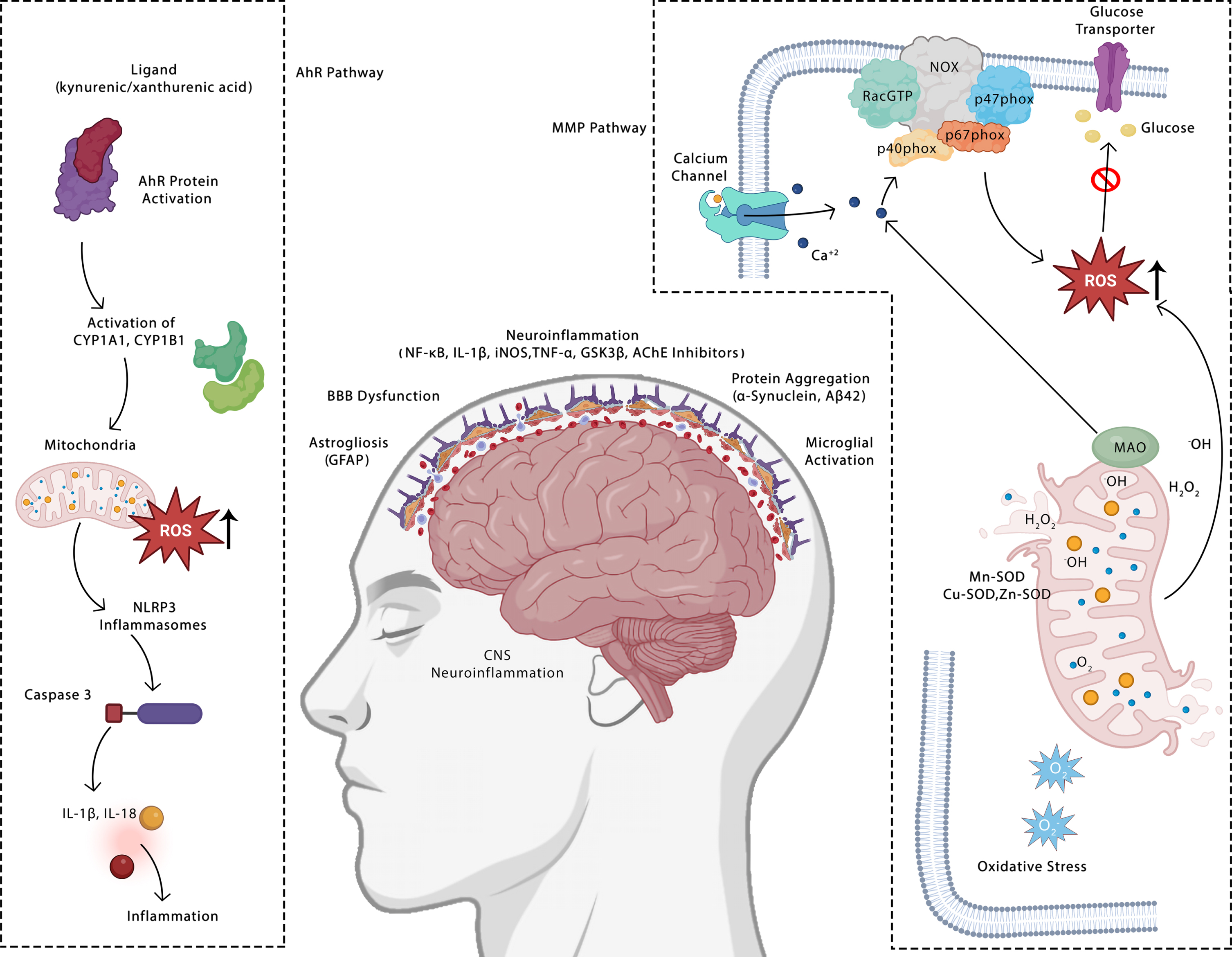
Illustration of toxicity targets of selected compounds and their mechanism for causing neuroinflammation.
AChE-PPI network and GO function analysis
The AChE-PPIN consists of 130 nodes and 1,689 edges (Fig. 4). To understand the biological importance of AChE interacting partners, we performed functional annotation of associated proteins. The AChE and its associated proteins were found to highly enriched in BP including cell to cell signaling, synaptic signaling (Fig. 5A). The MF function related to these proteins were signaling receptor binding, neurotransmitter activity, acetylcholine receptor activity in cellular components synapse, somatodritic compartment and axon (Fig. 5B, C). The pathways analysis resulted that AChE and associated proteins enriched with neuroactive ligand-receptor interaction, neurodegeneration pathways, metabolic pathways, cholinergic synapse and other pathways shown in (Fig. 5D).

Representation of protein-protein interaction network of AChE consisted 130 nodes and 1689 edges. The first, second intracting neighbouring partners of AChE showed in cyan blue color filled eclipsed shape and edges between them showed in grey line.
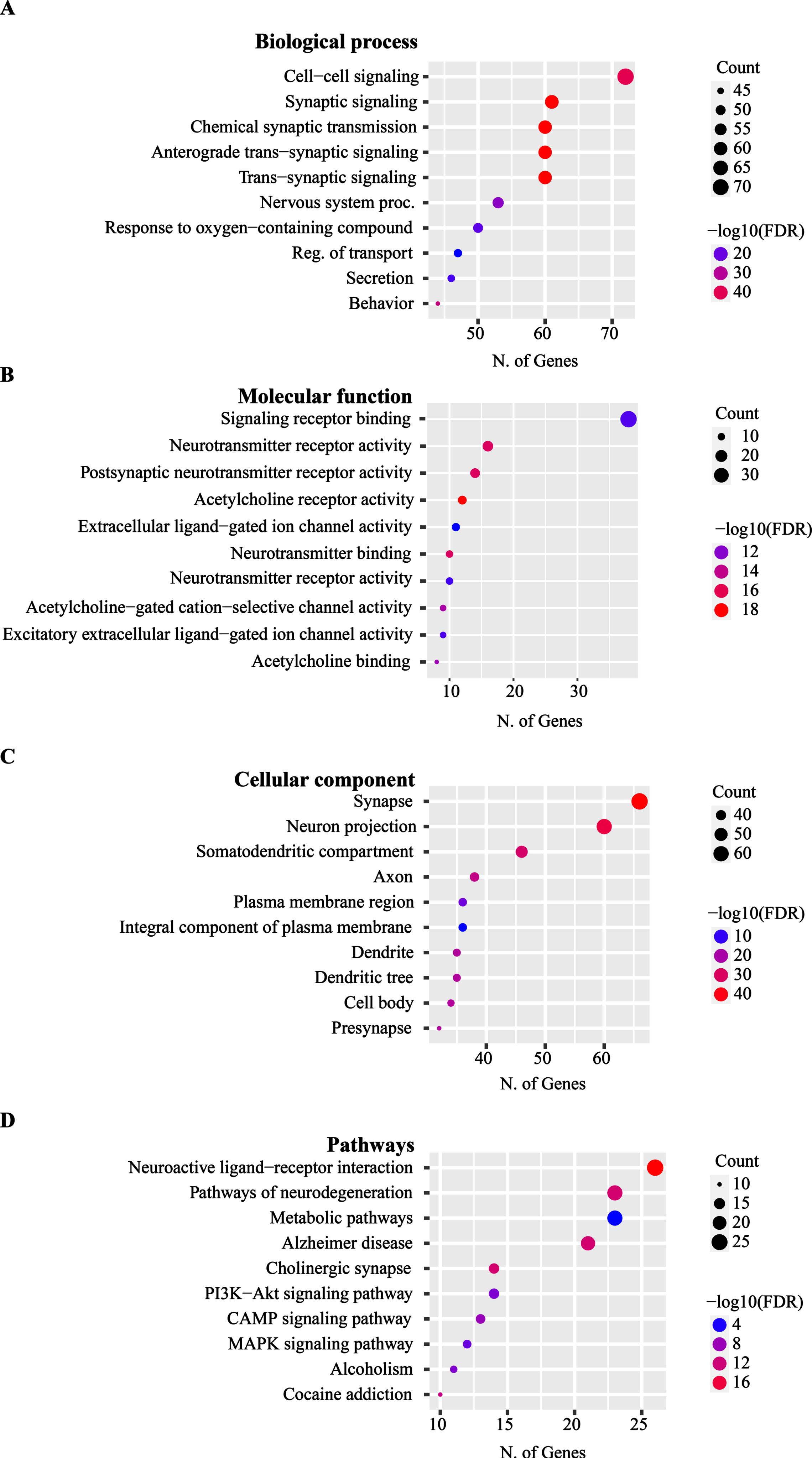
Functional annotation of AChE- interacting partners in terms of (A) biological processes (BP), (B) molecular functions (MF), (C) cellular component (CC), (D) pathway analysis of interacting partners of AChE. The FDR value were calculated on the basis of nominal p-value (0.05) from hygrometric test and fold enrichment represented the percentage of input genes related to a GO term. The size of the circle belongs to the number of AChE interacting partners belongs a particular GO term.
Screening of key gene targets by effective compounds
A total of five compounds were used for the identification of target genes. Here, we used two two control inhibitors (galantamine and dihydrotanshinonel) and three active compounds (quercetin, isorhamnetin, and kaempferol) filtered from the EGB761’s extract. We identified galantamine, dihydrotanshinonel, quercetin, isorhamnetin, and kaempferol targeted 112, 106, 104, 104, and 104 genes respectively (Fig. 6A). Overall, all the five compounds targeted 273 unique gene in which galantamine showed the largest degree followed by others. Further, we also, found seven genes (AChE, PARP1, EGFR, ESR2, GSK3β, CDK1, and CDK2) commonly targeted by all the five compounds (Fig. 6B).
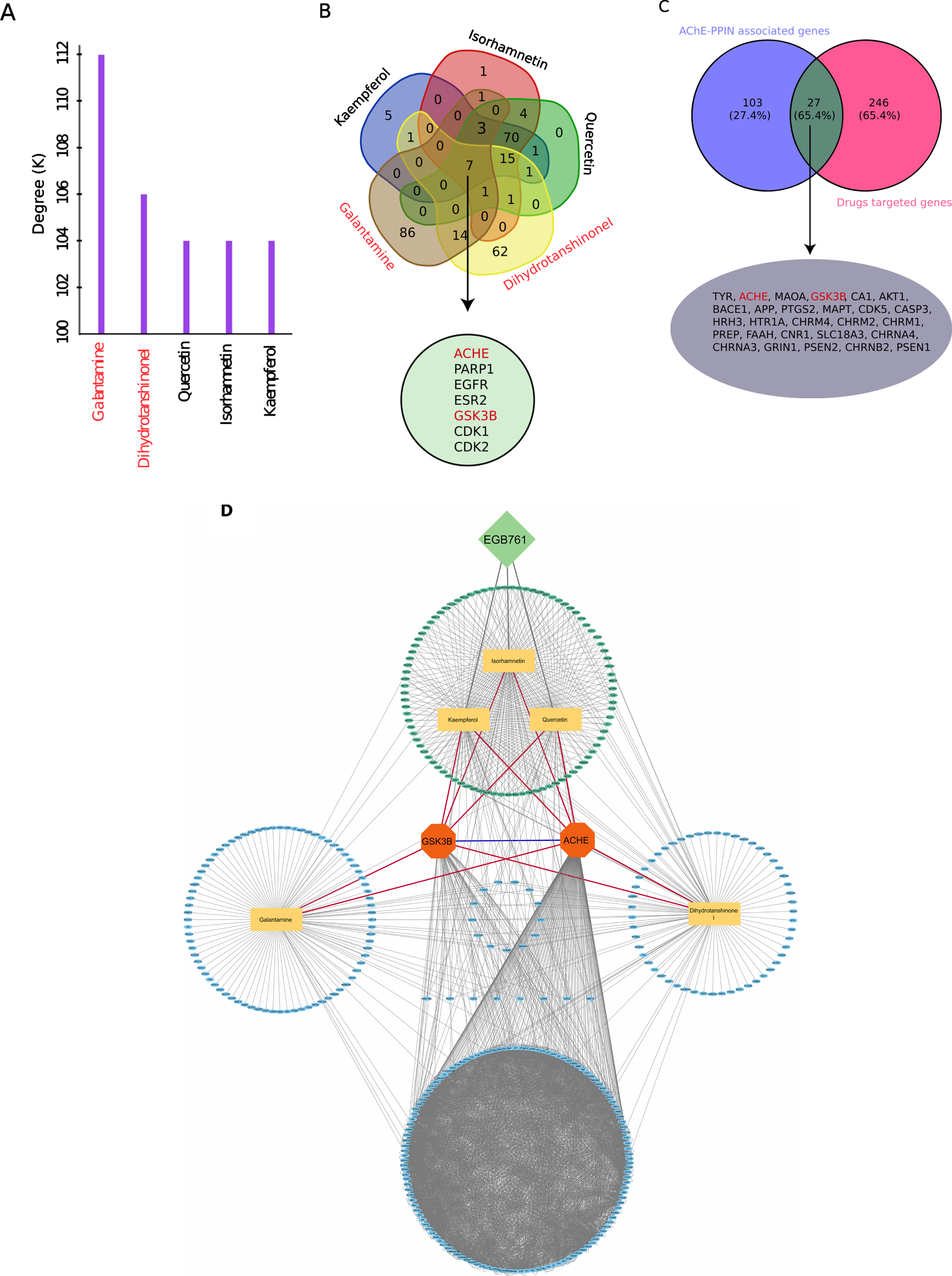
Screening of key gene targets by effective compounds and their representation in the form network. A) Statistics of degrees of effective components, X-axis represents effective components with two control inhibitors (galantamine and dihydrotanshinonel) in red text, Y-axis represents degrees. B) Venn diagram shows that 7 common targets of effective compounds. C) Venn diagram shows that 140 interacting proteins of AChE are screened, 273 gene targets of drugs, and the overlapped region indicates that there are 27 targets between them which contains AChE and GSK3β. D) Compound target network, lime-green ellipse shown the AChE-interacting protein with EGB761 compounds, blue ellipse shown the target of effective compunds and ACHE and GSK3β interacting proteins, light-orange rectangle represents the effective componds of EGB761, green square represents EGB761, and orange octagon represents two interacting (blue edge) mutiple targets (AChE and GSK3β) of effective compounds. The red edges represents common targets (AChE and GSK3β) of five effective compounds of EGB761.
A total of 129 interacting proteins obtained in AChE-PPIN and then intersection of unique genes targets of all five drugs, filtered 27 common genes further used to identify the key targets (Fig. 6C). We, observed interaction between two proteins (AChE and GSK3B) in AChE-PPIN also targeted by all the five compounds (galantamine, dihydrotanshinonel, quercetin, isorhamnetin, and kaempferol). Since, for further analysis we selected two genes (AChE and GSK3B) to identify crucial therapeutic inhibitory compounds in AD, may be consider as multi-gene target drugs (Fig. 6D).
Construction and regulatory relationship in between AChE-GSK3β-miRNA-TFs
From the miRwalk database, we identified number of miRNAs targets of both genes AChE and GSK3B were 643 and 1461 respectively. In the constructed network, we used only those miRNAs (430), which commonly targeted both the genes. By using, the TransmiR database, we got the 16 TFs (RUNX1, JUN, NFKB1, HIF1A, TP53, CEBPB, POU2F2, AR, OGG1, APEX1, TP63, MYOG, TNFSF12, and calcineurin) that regulate the transcription process of the 7 miRNAs (miR-484, miR-6797-5p, miR-320b, miR-1286, miR-1276, miR-1275, miR-198, and miR-133b), which were commonly targeting both AChE and GSK3B genes (Fig. 5). The TFs mode of regulation (activation and repression) for miRNA shown in (Fig. 7) in which NFKB1 showed the dual regulatory function (activation of miR-1296 and repression of miR-1276). TP53 were also function as activator and repressor for miR-1275 and mir-198 respectively, the miRNA (miR-1275), were also activated by another TFs, HIF1A and miR-198 by CEBPB. Further, we also identified the direct transcriptional regulatory factor (TFAP2A) regulating the AChE.
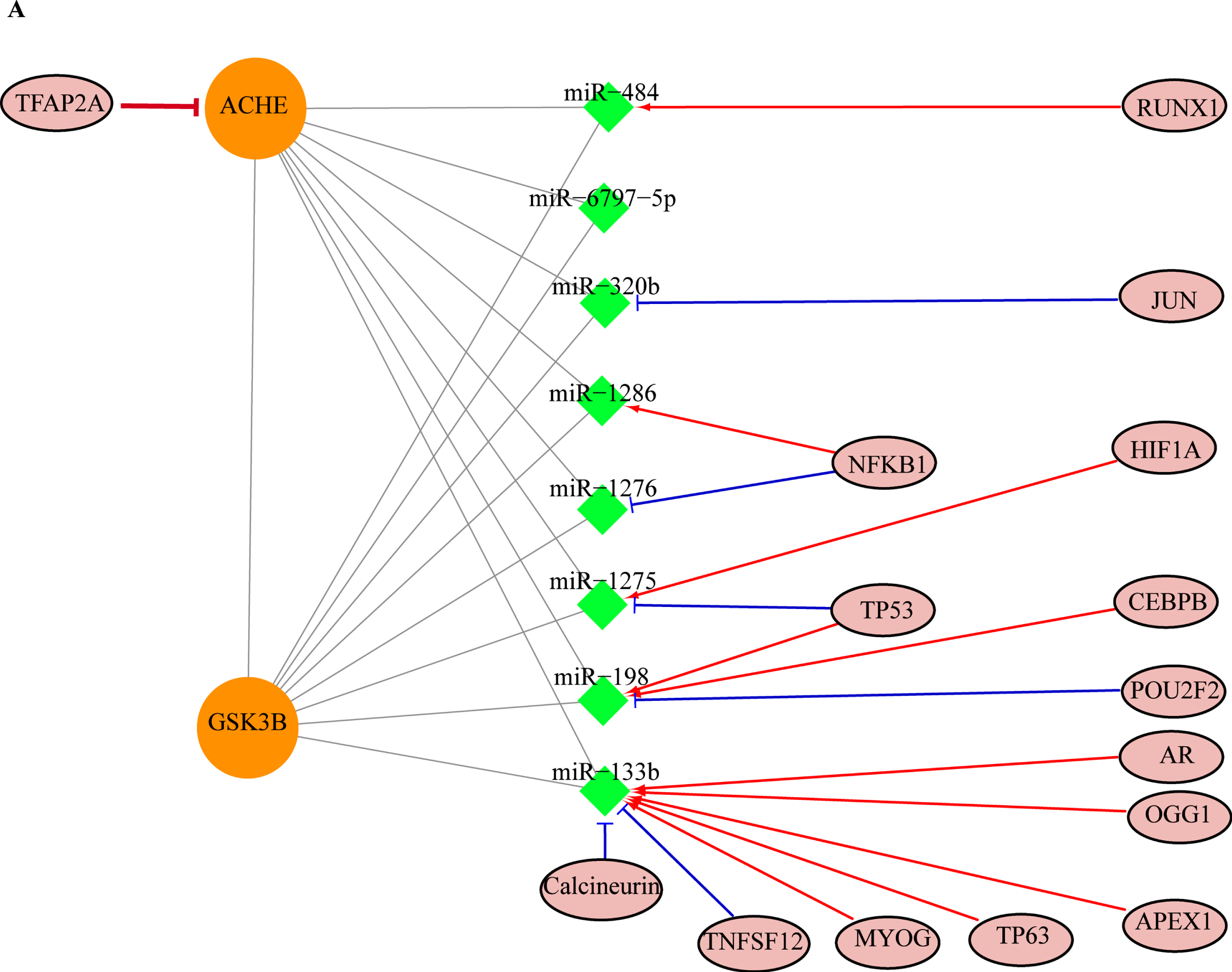
Transcritional and post-transcriptional level mode of regualtion in AChE and GSK3β. Trancriptional factors (TFs) represented in pink eclipsed filled circle, miRNA targets showed in green square. The red edge (T-shape) represents the down-regualtion process TF to AChE, red-edges (arrow) represents TF to miRNA up-regulation, blue edges (T-shape) highlights TF to miRNA downregulation, and grey edge (line) represents miRNA to AChE and GSK3β mRNA-regualtion.
In silico analysis of EGB761 phytocompounds with GSK3β
Further, on visualizing the interaction between AChE and GSK3β, molecular docking of GSK3β performed with phytocompounds of EGB761 with their respective inhibitors (6-Bromoindirubin-3’-oxime and Olanzapine), using Schrondinger. As mentioned in Table 4, the binding affinities of the phytocompounds are better than the available inhibitory drugs in market (–4.307 and –4.317 kcal/mol).
List of interacting residues of EGB761 and AChE from molecular docking studies
The amino acids plays an important role in interaction for hydrogen and hydrophobic boding between protein and the ligands. Therefore, interacting residues have been mentioned in Table 4. The common interacting residues in all the phytocompounds and controls are shown in Table 5.
The ligands from above mentioned phytocompounds are now rechecked for their ADME properties and bioactivity scores in the Molinspiration tool. The bioactivity scores of phytocompounds are mentioned in Table 6, which have positive bioactivity scores as kinase inhibitors (GSK3β- a kinase). The results suggest that these may be significant GSK3β inhibitors.
DISCUSSION
As discussed above, NDDs have affected millions of individuals globally and their underlying causes include variety of epigenetic and genetic factors [63]. Herbal medicines have proved to be an efficacious solution in treating them. Ginkgo biloba extract (EGB761) is a potent rejuvenator and a well-known Chinese medicinal extract that has been used for ages to cure a variety of CNS disorders, specifically NDDs and other stress-related issues. It has been further reported that EGB761 helps in limiting the neurite development, protects neurons from apoptosis, prevents ataxia in mice models, and averts neurodegeneration by restoring mitochondrial function [64, 65].
Bioactivity score calculation of EGB761 phytocompounds, galantamine (control) and dihydrotanshinone I (natural inhibitor) using Molinspiration tool
Details of binding affinity and free energy (Site-specific docking) of EGB761 standard extract with GSK3β

List of interacting residues of EGB761 and GSK3β from molecular docking studies

Schematic representation of ligand interaction of EGB761 phytocompounds, 6-bromoindirubin-3’-oxime, and dihydrotanshinone I (control) with GSK3β.

Graphical representation of pharmacokinetic parameters (Absorption, Distribution, Metabolism and Excretion- ADME Score) of EGB761 phytocompounds, 6-bromoindirubin-3’-oxime and Dihydrotanshinone I (control).
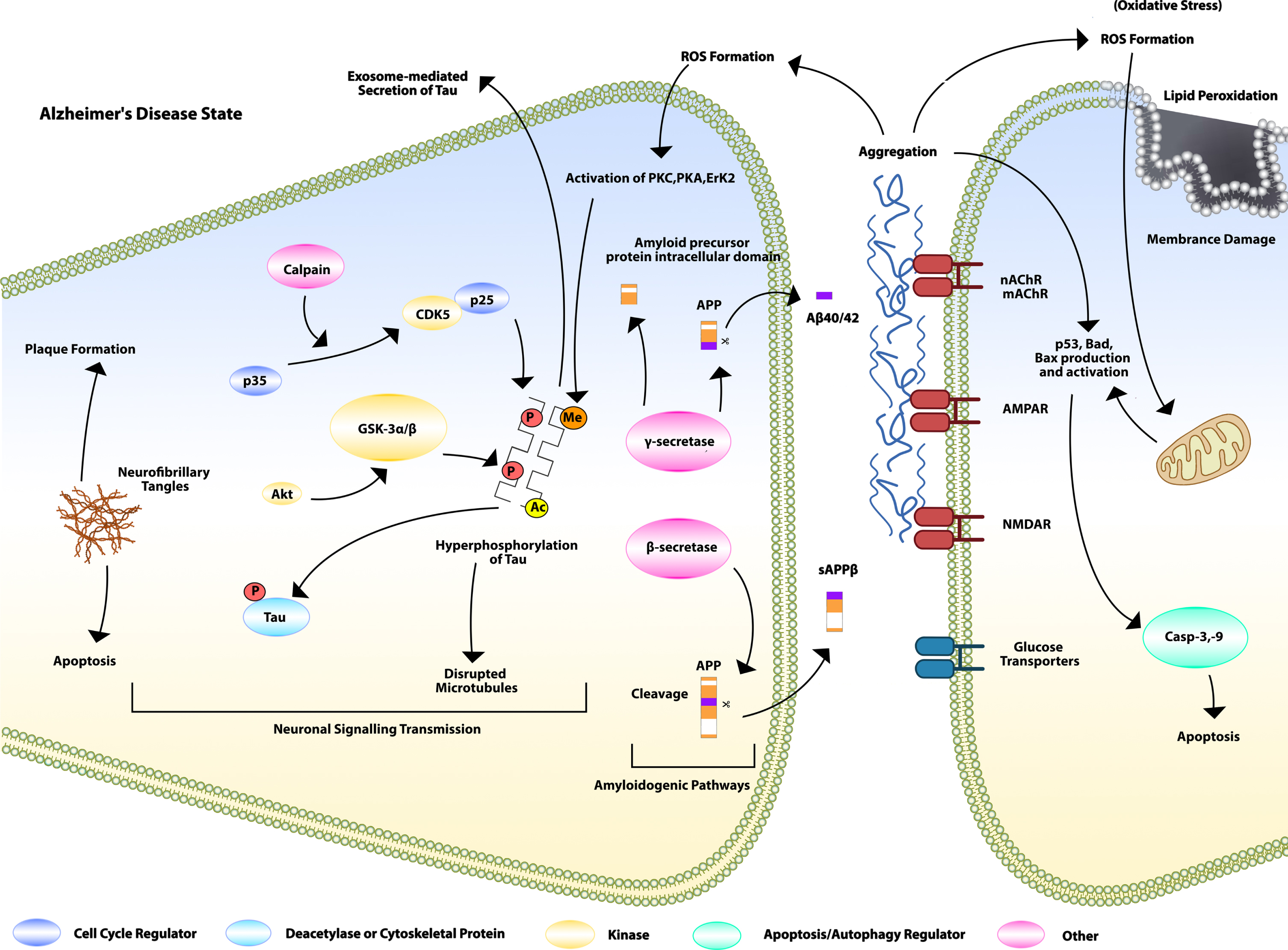
Descriptive details of the pathophysiological neuroinflammatory mechanisms by various pathways (neuronal signaling transmission, amyloidogenic, cholinergic, and NMDA pathways) initiating formation and deposition of amyloid-β in pre and post synaptic neuronal transmission leading to neurodegeneration process and further causing Alzheimer’s disease.
Bioactivity score calculation of EGB761 phytocompounds, 6-bromoindirubin-3’-oxime and Dihydrotanshinone I (control) using Molinspiration tool
In conclusion, with the AChE-PPIN analysis, it is found that the potential target of the active compounds in the treatment of AD are AChE and GSK3β and the GO functional and pathways enrichment analysis indicated that these targets mainly related to neuronal activity (mostly in synapse) and other functions. Both AChE and GSK3β were co-regulated or individually regulated by multiple common TFs via miRNA control and could be use as multi-targets for drug discovery in therapeutic intervention.
Conclusions
In conclusion, network pharmacology demonstrated that these phytocompounds might act on multiple targets in the protein network of AD, and molecular docking demonstrated that highly active phytocompounds of EGB761, particularly quercetin, kaempferol, and isorhamnetin, have better active against AChE than its reported synthetic drug. EGB761’s ability to treat AD was mostly attributed to the AChE theory. The outcomes of network pharmacology also demonstrated the role of GSK3B in the pathogenesis of AD. Furthermore, TFs and miRNAs were used to examine the regulation mechanisms of AChE and GSK3. The regulatory network revealed that 7 miRNAs target both AChE and GSK3B, and that NFKB1 and TP53 control this post-transcriptional activity by upregulating miRNAs. After then, research on how GSK3B interacts with particular phytochemicals revealed that the protein is also inhibited. Quercetin, kaempferol, and isorhamnetin were the top phytocompounds, according to network pharmacology and molecular docking, which suggested they might be crucial in the treatment of AD.
Footnotes
ACKNOWLEDGMENTS
The authors acknowledge Jaypee Institute of Information Technology, Noida, for providing the entire infrastructure to complete this project.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article.