
Abstract
Background:
Long non-coding RNAs are ubiquitous throughout the human system, yet many of their biological functions remain unknown. LINC00298 RNA, a long intergenic non-coding RNA, has been shown to have preferential expression in the central nervous system where it contributes to neuronal differentiation and development. Furthermore, previous research has indicated that LINC00298 RNA is known to be a genetic risk factor for the development of Alzheimer’s disease.
Objective:
To biochemically characterize LINC00298 RNA and to elucidate its biological function within hippocampal neuronal cells, thereby providing a greater understanding of its role in Alzheimer’s disease pathogenesis.
Methods:
LINC00298 RNA was in vitro transcribed and then subjected to structural analysis using circular dichroism, and UV-Vis spectroscopy. Additionally, affinity column chromatography was used to capture LINC00298 RNA’s protein binding partners from hippocampal neuronal cells, which were then identified using liquid chromatography and mass spectrometry (LC/MS).
Results:
LINC00298 RNA is comprised of stem-loop secondary structural elements, with a cylindrical tertiary structure that has highly dynamic regions, which result in high positional entropy. LC/MS identified 24 proteins within the interactome of LINC00298 RNA.
Conclusion:
Through analysis of LINC00298 RNA’s 24 protein binding partners, it was determined that LINC00298 RNA may play significant roles in neuronal development, proliferation, and cellular organization. Furthermore, analysis of LINC00298 RNA’s interactome indicated that LINC00298 RNA is capable of intracellular motility with dual localization in the nucleus and the cytosol. This biochemical characterization of LINC00298 RNA has shed light on its role in Alzheimer’s disease pathogenesis.
INTRODUCTION
The transcriptome is a vast array of coding and non-coding RNAs with diverse structures and functions that are known to play crucial roles in many essential biological processes. The expansive nature of the transcriptome was made evident through ENCODE and GENCODE, which found that 50%–70% of the human genome is transcribed with approximately 2% of the human genome being translated [1–3]. Through these analyses of the human genome, gene expression predominantly results in non-coding RNAs. Non-coding RNAs (ncRNAs) are classified based on their sequence length as being either small ncRNAs (<200 nt) or long ncRNAs (>200 nt) [4]. Long ncRNAs (lncRNAs) have been found in various tissues with >50,000 lncRNAs present in one or more copies per cell [5]. Additionally, lncRNAs can be transcribed from a variety of DNA sequences in the genome, including intergenic regions (lincRNAs), as well as introns and/or exons of protein-coding genes in the sense or antisense orientations [5]. lncRNAs have been found to be localized in the nucleus, cytoplasm, or have dual localization in both the nucleus and cytoplasm [5–7]. The biological function of lncRNAs have been directly linked to their cellular localization. The diverse biological functions of lncRNAs have been identified as being involved in cellular development, apoptosis, stress response, neurodegenerative disease, cancer, and cardiovascular dysfunction [6–13]. Due to the critical roles that lncRNAs play in many different biological and disease processes, further investigations into their biological functions and structures are needed to better understand the biochemical roles that lncRNAs play in the cell.
The linc00298 gene, which encodes LINC00298 RNA, was identified as a genetic risk factor for the development of Alzheimer’s disease [10]. This finding correlates with previous research on LINC00298 RNA indicating that it is preferentially expressed in the central nervous system (CNS), where it impacts development, participates in neuronal differentiation, and is expressed in neuronal induced pluripotent stem cells [14–19]. Though previous research has identified the importance of LINC00298 RNA, this lincRNA has yet to be biochemically characterized. As such, this research effort focused on developing secondary and tertiary structures of LINC00298 RNA through computational modeling as well as through circular dichroism (CD) and thermal stability spectrophotometric analysis. Additionally, the biological function, cellular localization, and interactome network of LINC00298 RNA were elucidated through liquid chromatography and mass spectrometry (LC/MS). Through these experiments, 24 protein binding partners of LINC00298 RNA were identified, which reproducibly bind to LINC00298 RNA with high fidelity.
MATERIALS AND METHODS
Materials
HiScribeTM T7 High Yield RNA Synthesis Kit E2040S (New England BioLabs, Ipswich, MA) was used to in vitro transcribe LINC00298 RNA via run-off transcription [20]. The LINC00298 RNA in vitro transcribed product was purified using the Monarch® RNA Cleanup Kit T2050 (New England BioLabs) and quantitated based on absorbance at 260 nm using a molar absorption coefficient of 24284.6 mM–1 cm–1 [21–23]. The purified LINC00298 RNA was denatured at 80°C for 1 min, followed by an addition of 1 mM MgCl2 (final concentration of MgCl2 was 10μM) and quick-cooled on ice for folding [23].
The pLINC00298 plasmid construct was created by Integrated DNA Technologies, Inc. (Coralville, IA). The linc00298 gene was synthesized as a 2,189 nt sequence containing a T7 RNA polymerase promoter sequence (5’-TAATACGACTCACTATAGGG- 3’) at the 5’ end and a HindIII restriction enzyme recognition sequence (5’ -AAGCTT- 3’) at the 3’ end of the linc00298 gene. The sequence of the linc00298 gene construct is as follows: 5’ - TAATACGACTCACTATAGGGTGCCAGTTTGAATGAAGGAAGCATGCGTGGCCTGAAGCCTCAC CGTGTTCCAGGCGTCCAGTTAAGTGCTCTGC ACATAATATCACACATTGCGCTCCACGATAA GAACAAATTCTCACATGGGAACCGAGCAAA GTGTAATGGATGTTCCCAGGAGCTGCCTATG GGATTCAAGAAAACTGTATGAGAGGAGTAAC ATTTGGGCTGGAAGCGAAAATACAAGTAAA CATATGTCAAATGGGGTCTGGAAAGCAATC CTCCACAGAAAGAAAGCAGAATCTTCTGGG CATTTCTGAGCAAGTGGACTTCCTTCAGCT GAGCACAGTCCTCAGGAGAAGCGGACAGGT GCAAAGTGTTGGAAGCCAACACTCACAGCA GTTGGGGGTTGGAGTGCTCTGGCTTCCAAC AGCATTGAAACCAACAGCATTGACTACAGC GAGTTTCTCAATCTTCAGAACACATCCCAG CCTCCCTGGGGGCTGCTAATGCACAGATTG CCGTTTCCCACTCCAAGAGTTTCTGATTCC AGAGGTCTGGGGTAGGGCTCGATAATTTGT TCTTCTAACCACCTAATGCTGTTGGTCCAG TGACCACACTTTGAGAAATGCTGTACTATG TTTTCTCATATATTTAAAAATTCTCACCTC ATTTATGGTTTAAAACTTCTAGAACAATTC TGAATAGTTTTTGTAATAGTGAGCCTCACT TTCCTGTCTCTGCCCTTAATGGGAATGTCT CTAATGATTCACCTTTCATACGGGGACTTG TTGTGGTTTACTGGGCAATTCCTTTTGGCA GGTTAAAAAAATTCCCTCTGTTTCAAGCTA AAGCCATTTACCAGGAAGAGACGCTAAGTT TTATCAGAGGGTTTTTCCAGTAACTATTGT GATAATCATGGTTTTTTTTCCTATTTATTA CGATATCAAACATGAAACTTATTTGAAGTC CTGGATTAAACGGTGCTTGTTTTCTATAAT GTTTTGTTCCTTAAATGTACTGACAAATTT AATTTATTATTTTTTTAGGATTTGAGGTAC AAGTTCATACGTGATAATGACTTACTGTTT TGTTTTCATTATTTCCTCCTTCCATCCTTT CTTGATATCCTTTTTATCTCTTGGTTTCAA AATTACACTAGCGTCATAGAATGAGTTGGA AATTAGCCCATTATTCTCCATGCCATGGAG TGTTTTGCAAAACATAAGAATGATCAGTTC CTTACATATTTGGTAGAATTTACCTTATAA AACACAGTAGGTTTTGTGTCTCTTCTGGAG TCCAGATACTGCTATGGACTGAATGTTTGT ACCCTCTAAGATTCATTAGGTTGAAATCCT AGCCTCAAAGTGGTGGTATTAGGAGGTGGG ACTTTTGGGAGGTGATCAGGCCAAGAGGGT GGAGCCCTCATGAATGGGATTAGTGCCCTT ATCAAATAAGGCAAAAGGAGCTCTTTAACC TCTCCCACCATGTGAGGACAAAGTGAAAAG ACACCATCTATGAACCAGGAAGCAGGGCCT CACCGGATGCCAAACCTGCCAGCACCTGGC CCTTGGACTTCCAGCCACCAGAACTGTGAA GAAGAAATTTCTATTGCTTATAAATACCCA GTTTACGGCATTCTGTTATAGAAGTGCCTA ATAGGCTAAGGCAAATACTATATGGTATTT AGGATTGCTTCTGTATATTTTTAAGACTTT TTTATTTTGGAATAATAATAATTCACAGAA AATTTACAGAGAAAGTCTCCACATGCCCCT CACCCCACTTCCCCTAATGTTAACATAACC GTGGTCCATTTGTCAAAACTAAGAAATGAG GATTGGTACCATACTATTGACTGAATTACA GACATTACTTGGATTTTGTCAATTGTTTTA CTAATATTCTATTATTTCTGTGTCAGATTC CAATCTGGATAACATGTTGCATTTGTTCAT CACGTCTCCTTTGTTTTTCCTGGGCTGTGA CAGTTTCTTATTCTTTTCTTGTATTGCCTG GCTTTGAATTTTGAGGAATTCTGGTTAAGT ATTTTGTAAACTGTCACTAAATTTTGCTTG TCAGATGTTTTCTCCTGGGTTAGACTGGGC TTACAGGTTTTGGGGAAGAACTCTACAAAA GGGAAATGTCTTTCTCATCTCATCTAATAAATGGATATAGGATATCAAGCTT- 3’. The synthesized linc00298 gene construct was cloned into the pUCIDT (Amp) Golden Gate plasmid, which is a standard cloning vector created by Integrated DNA Technologies, Inc. (Coralville, IA). The resulting plasmid is referred to as pLINC00298 throughout this publication.
Biotinylation of LINC00298 RNA was done using the PierceTM RNA 3’ End Biotinylation Kit (Thermo Fisher Scientific, Inc., Waltham, MA). Each biotinylation reaction contained 50 pmol of LINC00298 RNA for a total of 1000 pmol of biotinylated LINC00298 RNA product. The biotinylation reaction protocol provided with the kit was followed exactly with a 1 h room temperature incubation followed by an overnight incubation at 16°C. After the incubation period, a chloroform-isoamyl alcohol (24:1) extraction was done followed by an ethanol precipitation.
Primary mouse hippocampal neurons (Thermo Fisher Scientific, Inc., Cat # A15587) were lysed and the cell extract was used to determine the protein binding partners of LINC00298 RNA via affinity column chromatography.
DynabeadsTM MyOneTM Streptavidin T1 beads (Thermo Fisher Scientific, Inc., Cat # 65604D) were used to bind and immobilize the biotinylated LINC00298 RNA and then act as the stationary phase of column chromatography to capture protein binding partners of LINC00298 RNA from the cell lysate of primary mouse hippocampal neurons.
In vitro transcription protocol
To generate a DNA template for in vitro transcription, pLINC00298 was used to transform Escherichia coli DH5α competent cells (Thermo Fisher Scientific, Inc.). One resulting transformant was used to inoculate 5 mL Luria-Bertani medium containing 100μg/ml ampicillin (LB-Amp) and grown overnight at 37°C while shaking. A ZymoPURE Plasmid Miniprep Kit D4209 (Zymo Research, Irvine, CA) was used to isolate and purify pLINC00298. To prepare pLINC00298 for run-off in vitro transcription, pLINC00298 was digested with HindIII (Promega, Madison, WI) for 3 h at 37°C with a total volume of 80μL. Digestion of pLINC00298 by HindIII was confirmed through agarose gel electrophoresis. The resulting digested pLINC00298 was used as a template for in vitro transcription following the protocol given with the HiScribeTM T7 High Yield RNA Synthesis Kit E2040S (New England BioLabs). The volume of the in vitro transcription reaction was 100μL and was incubated at 37°C for 7 h. The in vitro transcribed LINC00298 RNA was purified following the protocol given with the Monarch® RNA Cleanup Kit T2050 (New England BioLabs) and quantitated based on absorbance at 260 nm using a molar absorption coefficient of 24284.6 mM–1 cm–1 [21–23]. LINC00298 RNA was then denatured at 80°C for 1 min, followed by the addition of 10μM MgCl2 and then placed on ice to promote RNA folding [23].
Structural characterization of LINC00298 in the presence and absence of Mg2+
Structural analysis of LINC00298 RNA was done using CD to determine the relative helical and stem-loop secondary structural characteristics of apo-LINC00298 RNA compared to LINC00298 (Mg2+). The concentration of LINC00298 RNA was 0.5μM with a MgCl2 concentration of 10μM and a total volume of 1000μL [23]. A JASCO J-815 circular dichroism spectrophotometer (Jasco, Inc., Easton, MD) set at 25°C using 50 nm/min scan speed, 0.5 s time constant with a 1 nm bandwidth was used to analyze the apo-LINC00298 RNA and LINC00298 (Mg2+) samples [23, 24]. CD scans were done in triplicate with averages taken of each scan and then plotted. The resulting plots were graphed using GraphPad Prism 9 (GraphPad Software, Inc., San Diego, CA).
Characterization of the thermal stability of apo-LINC00298 RNA and LINC00298 (Mg2+)
Both the apo-LINC00298 RNA and the LINC00298 RNA (Mg2+) were kept on ice prior to conducting the thermal stability assay. The LINC00298 RNA (Mg2+) complex was generated as previously described [23]. The apo-LINC00298 RNA and the LINC00298 RNA (Mg2+) concentrations were both 0.5μM and the concentration of MgCl2 was 10μM, all of which were kept constant throughout the temperature gradient (20°C–95°C) of the thermal stability assay. The total volume of each sample was 1000μL, with nanopure water being the solvent. A UV-Vis Spectrophotometer (UV-1800 Shimadzu, Kyoto, Japan) was used to take absorbance readings at 260 nm for each sample; readings were taken at 5°C increments. All absorption readings were taken in triplicate and subsequently graphed with GraphPad Prism 9 (GraphPad Software, Inc.).
Biotinylation of LINC00298 RNA and affinity column chromatography
Identifying the protein binding partners and therefore elucidating the biological function of LINC00298 RNA required biotinylating LINC00298 RNA and then binding it to DynabeadsTM MyOneTM Streptavidin T1 beads. The DynabeadsTM MyOneTM Streptavidin T1 beads with immobilized biotinylated LINC00298 RNA acted as the stationary phase of an affinity column chromatography experiment. As previously described, biotinylation of LINC00298 RNA was done using the PierceTM RNA 3’ End Biotinylation Kit (Thermo Fisher Scientific, Inc.). Each biotinylation reaction contained 50 pmol of LINC00298 RNA that was used to create a total of 1000 pmol of biotinylated LINC00298 RNA product. The biotinylation reaction protocol provided with the kit was followed exactly with a 1 h room temperature incubation followed by an overnight incubation at 16°C. After the incubation period, a Chloroform-Isoamyl alcohol (24:1) extraction was done followed by an ethanol precipitation. Once the biotinylated LINC00298 RNA pellet was dry, 20μL of nanopure water was added to each reaction tube.
To prepare the DynabeadsTM MyOneTM Streptavidin T1 beads, 0.5 mL (2.5 mg/mL) of beads were added to 0.5 mL of 1X Binding and Wash (B & W) Buffer (2X B & W Buffer: 10 mM Tris-HCl (pH 7.5), 1 mM EDTA, 2 M NaCl, and then centrifuged at 5,000 rpm for 1 min at 4°C. Remove the supernatant, discard, and repeat two more times for a total of three washes. The DynabeadsTM MyOneTM Streptavidin T1 beads were then washed two times with Solution A (0.1 M NaOH and 0.05 M NaCl). The supernatant was removed and discarded. Then the DynabeadsTM MyOneTM Streptavidin T1 beads were washed one time with Solution B (0.1 M NaCl), and the supernatant was removed and discarded. The biotinylated RNA was added to the DynabeadsTM MyOneTM Streptavidin T1 beads along with 40 U of RNase Inhibitor. The mixture was incubated at room temperature while shaking for 1.5 h and then at 4°C overnight while shaking. The LINC00298 RNA bound DynabeadsTM MyOneTM Streptavidin T1 beads were then centrifuged at 5,000 rpm at 4°C, and then the unbound RNA was removed with the supernatant. A total volume of 0.5 mL of RNA-Streptavidin Interaction Buffer (20 mM Tris-HCl (pH 7.5), 300 mM KCl, 0.2 mM EDTA, 0.5 mM DTT, 0.5 mM PMSF, and 40 U RNase Inhibitor) was then added to the LINC00298 RNA bound DynabeadsTM MyOneTM Streptavidin T1 beads [25]. The beads were resuspended and then centrifuged at 5,000 rpm for 1 min at 4°C, the wash was repeated two more times [25]. The beads were resuspended in 0.5 mL of RNA-Streptavidin Interaction Buffer and incubated at 4°C while shaking.
The primary mouse hippocampal neuron cells (1 mL) were centrifuged at 5,000 rpm for 5 min at 4°C, the supernatant was removed, and then the cells were resuspended in 0.5 mL Mammalian Lysis Buffer (0.1 M EDTA (pH 8.0, 0.5% SDS, and 10 mM Tris-HCl (pH 8.0) [26]. The cells were then sonicated at 35 Amp for 1.5 min with 10-s pulses. The lysed cells were centrifuged at 5,000 rpm for 30 min at 4°C [26]. The cell lysate supernatant was then applied to the LINC00298 RNA bound DynabeadsTM MyOneTM Streptavidin T1 beads, and the mixture was incubated overnight at 4°C while shaking with 40 U of RNase Inhibitor [25].
Elution of the proteins bound to the LINC00298 RNA-Steptavidin bead complex started with the centrifugation of the beads for 1 min at 5,000 rpm at 4°C [25]. The supernatant was collected, and 0.5 mL of RNA-Streptavidin Interaction Buffer was added to the beads and resuspended [25]. The resuspension was centrifuged for 1 min at 5,000 rpm at 4°C, the supernatant was collected, and repeated two more times. Finally, 150μL of 0.1% SDS was added to the pelleted beads and then were resuspended [25]. To elute the bound proteins from the LINC00298 RNA-Steptavidin bead complex, the solution was boiled for 3 min followed by a centrifugation at 5,000 rpm for 1 min at 4°C [25]. The protein elution was collected and subsequently subjected to protein electrophoresis using a 10% SDS-PAGE for protein separation. The resulting protein bands were extracted from the gel and submitted for LC/MS analysis.
Mass spectrometry for the identification of the protein binding partners of LINC00298 RNA
The protein gel fragments were first subjected to in-gel trypsin digestion, as previously described [27, 28]. As described previously, the gel fragments were cut into 2 mm2×1 mm2 rectangular pieces, then washed with 50 mM ammonium bicarbonate (pH 7.8) and 50% (v/v) acetonitrile at room temperature for 15 min and then aspirated [28]. This aspiration and wash steps were repeated one more time. Dehydration of the gel fragments with 100% (v/v) acetonitrile was conducted at room temperature for 1 min. The acetonitrile was aspirated and 50 mM ammonium bicarbonate (pH 7.8) with 10 mM dithiothreitol were added to the gel fragments and incubated at 56°C for 1 h. The buffer solution was then aspirated and 55 mM iodoacetamide was added to the gel fragments and incubated at room temperature in dark conditions for 30 min [28]. This was followed by aspiration of the buffer and then 50 mM ammonium bicarbonate (pH 7.8) and 50% (v/v) acetonitrile were added, this was again followed by aspiration and repeated two more times. Once complete, 100% (v/v) acetonitrile was added to the gel fragments and incubated at room temperature for 1 min, followed by aspiration. The next step consisted of preparing the trypsin solution, which contained 5 mg/mL sequencing grade Trypsin (Promega, Madison, WI) in 50 mM ammonium bicarbonate (pH 7.8) with 5 mM calcium chloride. The solution was then added to the gel fragments until the fragments were fully covered by the Trypsin solution. The gel fragment-Trypsin mixtures were then incubated on ice for 10 min, followed by an aspiration step. The buffer containing 50 mM ammonium bicarbonate (pH 7.8) with 5 mM calcium chloride was then used to cover the gel fragments. The gel fragment samples were then placed in a warm air incubator at 37°C for 16 h. The resulting solution from each gel fragment sample were transferred to a sterile 1.5 mL microcentrifuge tube, and 50% (v/v) acetonitrile/0.3% (v/v) formic acid was added to each tube to promote the further extraction of digested peptides from the gel fragments [28]. The sample extracts were pooled together, and the process was repeated with 75% (v/v) acetonitrile/0.3% (v/v) formic acid [28]. The resulting pooled digested peptide fragments were frozen at –80°C and then dried in a speed vac [28]. This process was followed by the resolubilization of the peptide mixtures, and then the peptide mixtures were desalted using the Stage Tip technique with 3 M Empore styrenedivinylbenzene extraction disks, as previously described [29]. The digested peptide mixtures were then dried in vacuo.
The digested peptide fragments were then analyzed and identified with LC and MS, as previously described [30].
Peptide separations were conducted using an Easy-nLC 1000 HPLC (Thermo Fisher Scientific, Inc., Waltham, MA) [28]. Samples that were ∼200 ng, were loaded onto a 30 cm×100-μm internal diameter fused silica PicoTip Emitter (New Objective, Woburn, MA), which was packed in-house with ReproSil-Pur C18-AQ (1.9μm particle, 120 Å pore; Dr. Maish GmbH Ammerbuch, Germany) at a flow rate of 1μL/min with a buffer containing 0.1% (v/v) formic acid and 2% (v/v) acetonitrile [28]. Elution of the peptides were performed using a gradient of 5–7% buffer solution B (A: 0.1% (v/v) formic acid in water and B: 0.1% (v/v) formic acid in acetonitrile) for 1 min, 7–35% solution B for 1 h, and 35–60% solution B over 5 min at a flow rate of 200μL/min [28]. The column was mounted in a nanospray source directly in line with an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific, Inc.) [28]. The spray voltage was set at 2.1 kV in positive mode [28]. The heated capillary was maintained at 275°C [28]. The acquisition method combined two scan events (i.e., a full scan and a parallel reaction monitoring (PRM) event) that target the doubly- and triply- charged precursor ions of the HVPGGGSVQIVYKPVD and VQIVYKPVD peptides without scheduling [28]. The full scan event employed a mass-to-charge ratio (m/z) 380–1,500 mass selection, an orbitrap resolution of 120,000 (at m/z 200), a target automatic gain control (AGC) value of 200,000, and maximum fill times of 100 ms [28]. The PRM event used an orbitrap resolution of 30,000 (at m/z 200), a target AGC value of 200,000, and maximum injection times 55 ms [28]. The precursor ion generated from each targeted peptide was isolated using an isolation window of 1.6-m/z [28]. Fragmentation was performed with a higher-energy collisional dissociation collision energy of 30%. MS/MS scans were collected using a scan range from 100–1,000 m/z. PRM data were collected in centroid mode [28].
To conduct the mass spectral database search, Peaks Studio 8.5 (Bioinformatics Solutions, Inc, Waterloo, Ontario, Canada) was used for interpretation of MS/MS (mass spectra) and protein inference [28, 31]. Search parameters were: Mus musculus (taxonomy ID 10090) protein sequence database from UniProt (https://www.uniprot.org/) downloaded on December 13, 2016 concatenated with the common lab contaminant database from https://www.thegpm.org/crap/; precursor mass error tolerance: 50.0 parts per million (ppm); fragment mass error tolerance: 0.1 Dalton; precursor mass search type: monoisotopic; no enzyme specificity; variable modifications: methionine oxidation and di-oxidation, cysteine carbamidomethylation, pyroglutamic acid, and protein N-terminal acetylation; maximum variable modifications per peptide: 2; false discovery rate calculation: On; spectra merge: Off; no charge state correction; and spectral filter quality: >0.65 [28].
Interpretation of the mass spectral data relied on a baseline for peptide detection. Support for the detection of peptides from each supporting MS/MS spectrum was based on: 1) a minimum of five consecutive b- or y-type peptide fragment ions, 2) 1% peptide and protein false discovery rate threshold, and 3) precursor mass accuracy <7 ppm [28].
Human and/or animal subjects were not used to conduct this research.
RESULTS
Long intergenic non-coding RNAs (lincRNA) are an under-studied group of non-coding RNAs [32]. Biochemical analyses of this group of non-coding RNAs are crucial for understanding many different biological processes and disease pathogeneses [11–13, 33–35]. As such, the biochemical analysis of LINC00298 RNA started with computationally developing a model of its secondary structural elements (Fig. 1). Figure 1 indicates that LINC00298 RNA has significant and complex secondary structure, which is expected due to the large size of LINC00298 RNA (2,165 nt). The RNAfold 2.4.18 software platform was used as the computational tool to render the secondary structure of LINC00298 RNA based on positional entropy [36]. Positional entropy of each nucleotide in LINC00298 RNA was depicted in Fig. 1 through a color gradient, where red indicates low positional entropy and blue indicates high positional entropy [36]. Based on the predicted secondary structure, LINC00298 RNA is highly dynamic as indicated by numerous areas of high entropy (blue), areas of moderate entropy (green), and some areas with low positional entropy (red/yellow). Though LINC00298 RNA has regions with high positional entropy, the predicted free energy of the secondary structure of LINC00298 RNA is –617.82 kcal/mol, which may be due to the stabilizing effects of the helical elements within stem-loop secondary structural motifs.

Secondary structural prediction of LINC00298 RNA. The computational secondary structural prediction of LINC00298 RNA was based on the thermodynamic ensemble prediction model available on the RNAfold WebServer platform using RNAfold 2.4.18 [36]. The free energy of the thermodynamic ensemble was –617.82 kcal/mol. The bases are color coded as determined by the positional entropy, the lowest entropy on the color gradient is red and the highest entropy is blue [36].
The computationally rendered tertiary structure of LINC00298 RNA was predicted by the 3dRNA/DNA modeling program (Fig. 2A–C) [37]. The 3dRNA/DNA modeling program predicted five structures for LINC00298 RNA, of these five, the lowest energy (lowest 3dRNA score) structure was chosen [37]. The PyMol Molecular Graphics System, Version 2.0 was used to manipulate the structure of LINC00298 RNA and POV-ray, Version 3.7 was used to create the final rendered structure of LINC00298 RNA. The computationally predicted tertiary structure of LINC00298 RNA shows that the RNA is elongated and cylindrical in shape, as indicated by the structural depictions of LINC00298 RNA on the x, y, and z axes. Figure 2A-C are presented using a color spectrum that designates the sequence position of the nucleotides within LINC00298 RNA’s primary structure.
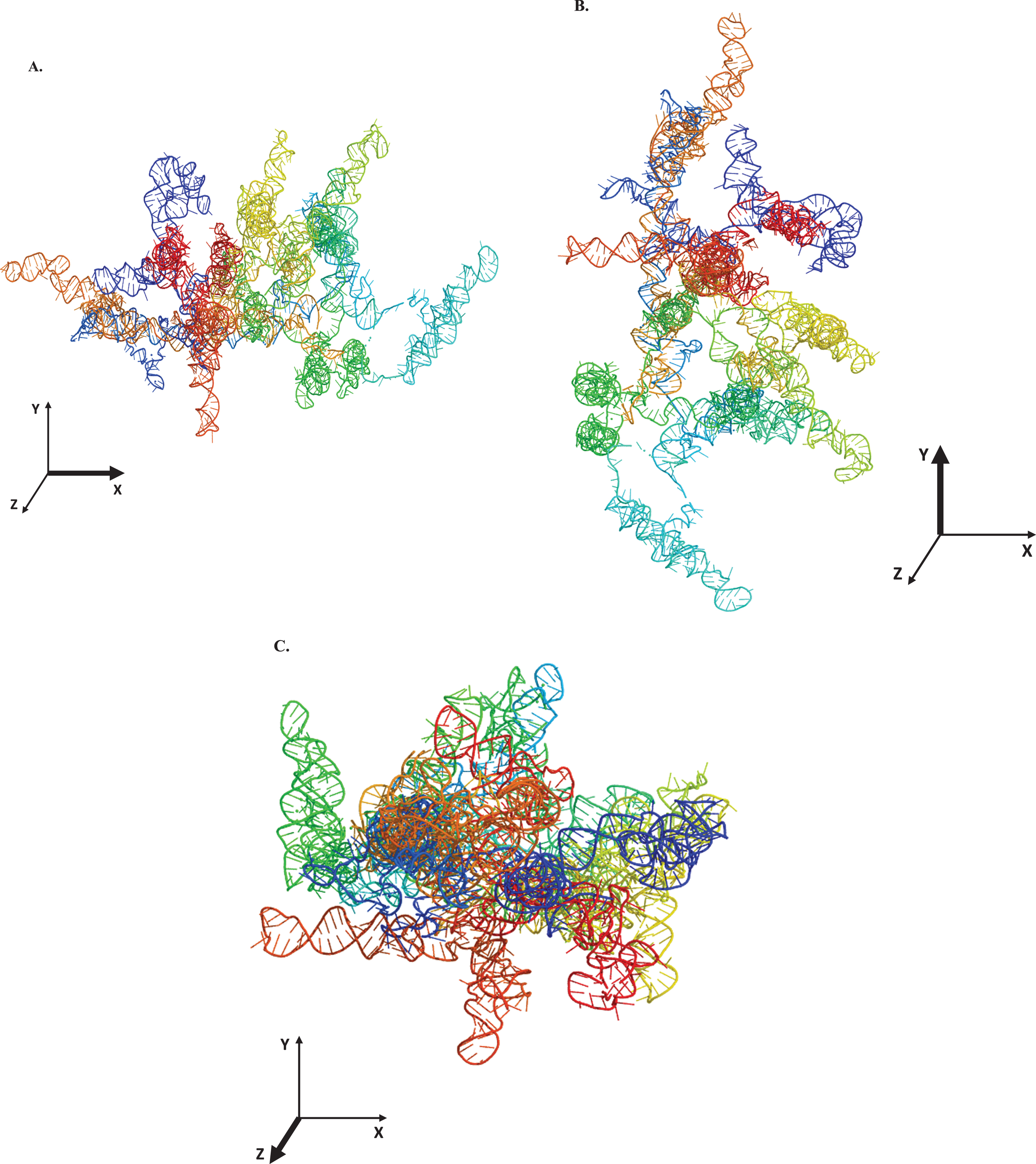
Tertiary structural prediction of LINC00298 RNA. The tertiary structure was predicted using the 3dRNA/DNA modeling program [37]. A. The LINC00298 RNA visualized along the x-axis. B. The LINC00298 RNA visualized along the y-axis. C. The LINC00298 RNA visualized along the z-axis. All structures were manipulated with The PyMol Molecular Graphics System, Version 2.0 (Schrodinger, LLC., New York City, NY). The final rendering of the structures was done using POV-Ray, Version 3.7 (Persistence of Vision Raytracer Pty. Ltd., Victoria, Australia). The structural color spectrum is indicative of the nucleotide position within the sequence. The nucleotides 1–406 are blue, 407–536 are teal, 537–1071 are green, 1071–1261 are yellow, 1261–1671 are orange, and 1672–2165 are red.
Empirical structural analysis of LINC00298 RNA was done using CD to compliment the computational structural renderings of LINC00298 RNA. Helical structures in RNA are similar to A-form DNA helices and therefore absorb maximally near 260 nm in CD spectra [23, 38–40]. Additionally, stem-loop secondary structures are commonly formed in RNA and can be identified on CD spectra by a negative peak at around 245 nm and a positive peak in the range of 265 nm–285 nm [40]. However, due to the large size and complex tertiary structure of LINC00298 RNA, CD spectral analysis showed shifts in the negative peak spanning from around 240 nm–280 nm and a positive peak spanning from around 285 nm–300 nm (Fig. 3). Shifts in CD spectra occur upon tertiary structure formation as helical secondary structural elements affect the absorption of stem loop secondary structural elements [41]. Additionally, changes in pH, ion concentration, and temperature can all cause shifts in CD spectra [41].
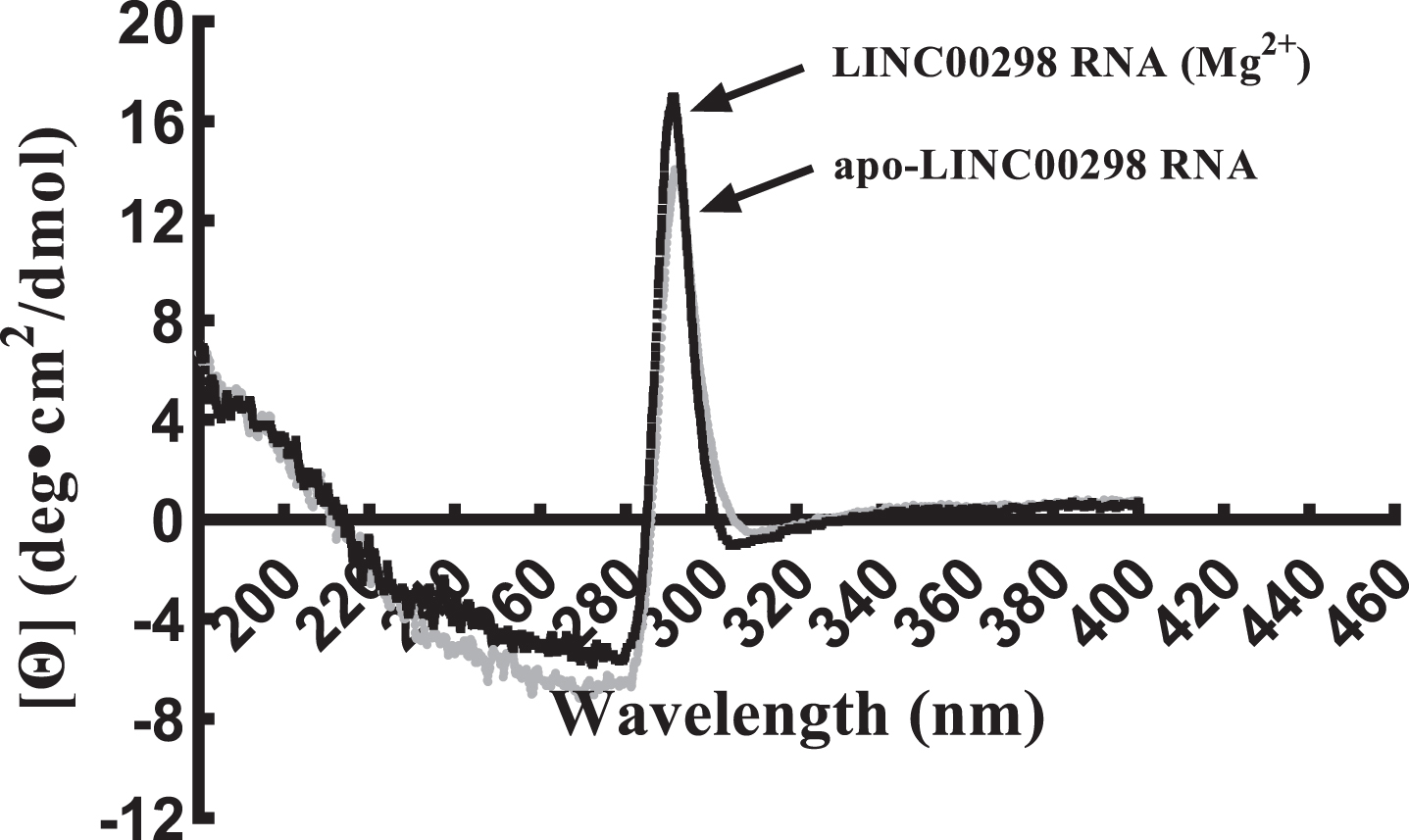
Circular Dichorism spectrum of structural changes between apo-LINC00298 RNA and LINC00298 (Mg2+). Circular Dichorism spectrum of apo-LINC00298 RNA, (–); and LINC00298 RNA (Mg2+), (–). The LINC00298 RNA concentration was 0.5μM and the MgCl2 concentration was 10μM in nano-pure water. Both LINC00298 RNA samples were scanned in triplicate using a JASCO J-815 circular dichroism spectropolarimeter set at 25°C using 50 nm/min scan speed, 0.5 sec time constant and a 1 nm bandwidth [24].
Two CD spectra were taken to analyze the relative helical and stem-loop content of apo-LINC00298 RNA and LINC00298 RNA (Mg2+) (Fig. 3). The CD spectrum of apo-LINC00298 RNA is gray and the CD spectrum of LINC00298 RNA (Mg2+) is black. The LINC00298 RNA (Mg2+) spectrum indicates higher helical and stem-loop secondary structural content relative to apo-LINC00298 RNA (Fig. 3). This result would be expected due to the important role divalent cations (Mg2+) play in stabilizing the tertiary structure of RNA by reducing like-charge repulsions of the negatively-charged phosphodiester bonds within the backbone of RNA [41, 42].
Characterization of the tertiary structures of LINC00298 RNA through thermal stability studies were carried out to determine the structural stability of apo-LINC00298 RNA relative to LINC00298 RNA (Mg2+). The thermal stability of each RNA was plotted as a change in absorption at 260 nm versus temperature. The absorption plot for apo-LINC00298 RNA is gray and the absorption plot for LINC00298 RNA (Mg2+) is black. Figure 4 indicates that there were absorption differences between apo-LINC00298 RNA and LINC00298 RNA (Mg2+), which may be due to differences in the tertiary structures of these two RNAs. Since divalent cations have a stabilizing effect on the secondary and tertiary structures of RNA, LINC00298 RNA (Mg2+) would therefore be expected to have a lower absorption at 260 nm than apo-LINC00298 RNA, which is evident in Fig. 4 [23, 41]. Reducing like-charge repulsions within phosphodiester bonds allows for the closer associations of RNA bases, resulting in a more compact tertiary structure for LINC00298 RNA (Mg2+) [23]. Absorption patterns for both apo-LINC00298 RNA and LINC00298 RNA (Mg2+) increase as the temperature increases. Additionally, absorption patterns for both RNAs show fluctuations, which is indicative of dynamic structural changes that both RNAs experience as the temperatures increase [41]. This result would be expected since tertiary structure formation does impact absorption, which would be more pronounced for a large and complex RNA [41].

Thermal-stability differences for apo-LINC00298 RNA and LINC00298 (Mg2+). UV/Vis spectrum at 260 nm of apo-LINC00298 RNA, (–); and LINC00298 RNA (Mg2+), (–) measuring the denaturation of LINC00298 RNA under these two states. The LINC00298 RNA concentration was 0.5μM and the MgCl2 concentration was 10μM.
To determine the biological function of LINC00298 RNA, affinity column chromatography and LC/MS were used. Biotinylated LINC00298 RNA was tethered to streptavidin beads while cell lysate from hippocampal neurons passed over the beads to immobilize the protein binding partners of LINC00298 RNA, which were then identified by LC/MS. Identification of the protein binding partners of LINC00298 RNA was essential in elucidating LINC00298 RNA’s biological function. In order to identify the legitimate protein binding partners of LINC00298 RNA, common protein contaminates had to be removed from the LC/MS data [43]. Additionally, a lower threshold of a 10% coverage value for identified proteins was set [44]. Figure 5A tabulates the protein binding partners of LINC00298 RNA that were identified with LC/MS and analyzed with Thermo Scientific Proteome Discoverer version 3.0. The 24 proteins listed in the table were identified with high confidence among the replicates of the LC/MS experiments. Figure 5B shows that LINC00298 RNA binds numerous 60 S and 40 S ribosomal proteins, as well as multiple ATP Synthase protein subunits, Heterogeneous nuclear ribonucleoproteins, and Histones. Furthermore, LINC00298 RNA binds to ADP/ATP Translocase 2, Cofilin-1, Dihydropyrimidinase-related protein 2, Eukaryotic initiation factor 4A-I, Heat Shock Protein (HSP) 90-beta, Prohibitin 1, Alpha-enolase, and Elongation factor 2.
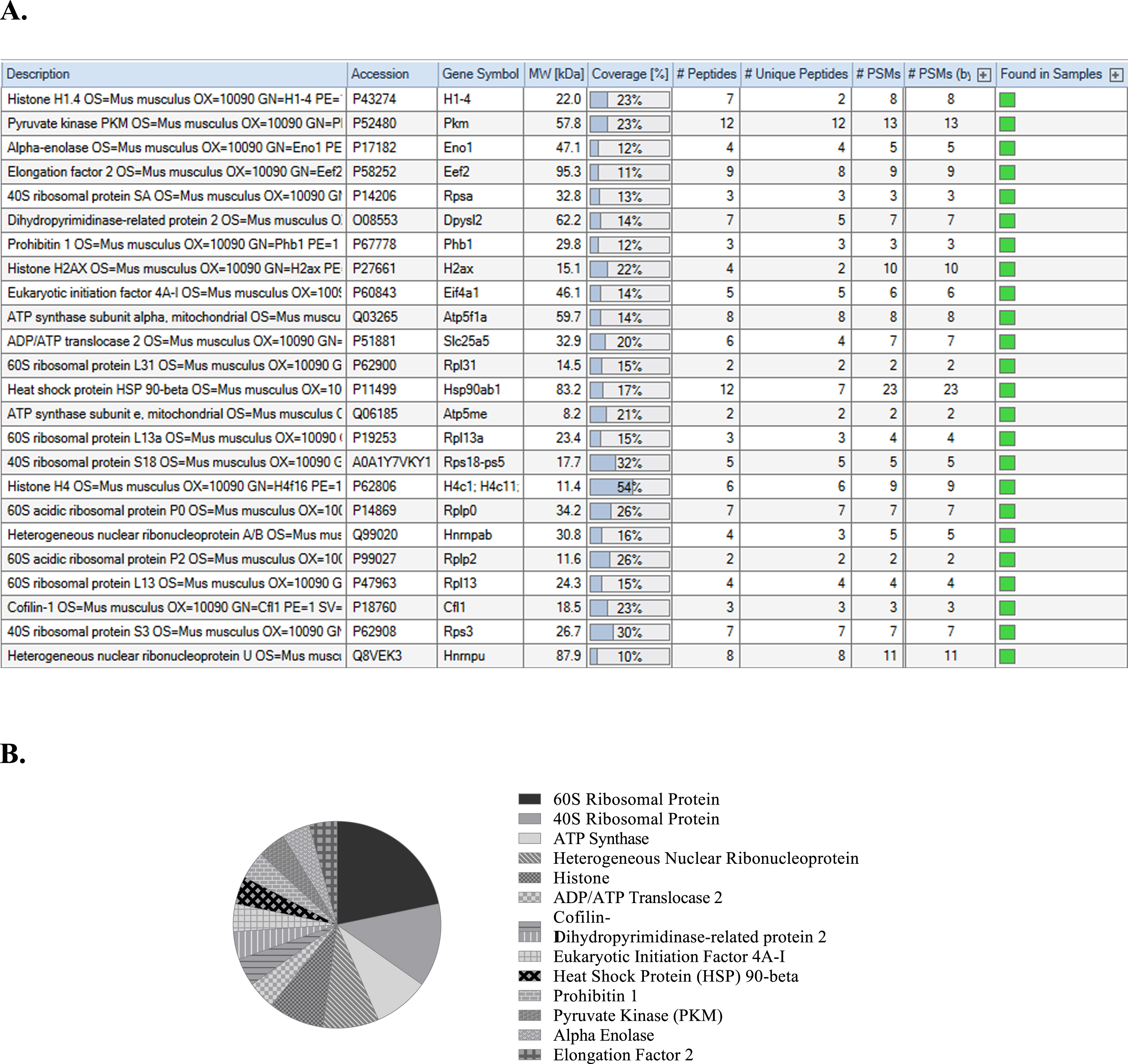
Identification of the protein binding partners of LINC00298 RNA through LC/MS analysis. A) Protein identification table generated by Thermo Scientific Proteome Discoverer version 3.0 from replicated LC/MS data [69]. B) Pie chart indicating the frequency of related protein types that are binding partners of LINC00298 RNA as indicated by Thermo Scientific Proteome Discoverer version 3.0 analysis [69].
The next step in analyzing the protein binding partners of LINC00298 RNA, is to determine the biological processes that are facilitated by these proteins, which will shed light on the biological function of LINC00298 RNA. Due to LINC00298 RNA’s strong predilection in binding to 40 S and 60 S ribosomal proteins, it therefore is logical that Protein Metabolism is the predominant biological process in Fig. 6. Protein Metabolism is also facilitated by Cofilin-1, Eukaryotic initiation factor 4A-I, Prohibitin 1, Elongation factor 2, Pyruvate kinase, and Histone H1.4. Cell Organization and Biogenesis are carried out by Histone H1.4, Pyruvate kinase, Dihydropyrimidinase-related protein 2, Prohibitin 1, ADP/ATP Translocase 2, Heat Shock Protein HSP 90-beta, Histon H4, Cofilin-1, Heterogeneous nuclear ribonucleoprotein U, 40 S and 60 S ribosomal proteins. Developmental Processes is the next major biological process, which is facilitated by Pyruvate kinase, Alpha-enolase, Dihydropyrimidinase-related protein 2, Prohibitin 1, Histone H2AX, Heat Shock Protein HSP 90-beta, Cofilin-1, Heterogeneous nuclear ribonucleoprotein U, 60 S ribosomal subunits L13 and L13a. Proteins that participate in Transport include: Dihydropyrimidinase-related protein 2, ATP synthase subunit alpha, ADP/ATP Translocase 2, ATP synthase subunit e, 60 S acidic ribosomal protein P0, Cofilin-1, and Heterogeneous nuclear ribonucleoprotein U. The proteins involved in stress response include: Prohibitin 1, Histone H2AX, Heat Shock Protein HSP 90-beta, 60 S ribosomal protein L13a, Cofilin-1, and 40 S ribosomal protein S3. The proteins involved in RNA Metabolism and Transcription include: Pyruvate kinase, Alpha-enolase, Prohibitin 1, Heterogeneous nuclear ribonucleoprotein A/B, and Heterogeneous nuclear ribonucleoprotein U. The proteins involved in DNA metabolism include: Prohibitin 1, Histone H2AX, Heat Shock Protein HSP 90-beta, and 40 S ribosomal protein S3. The proteins involved in the biological processes of Cell Cycle or Cell Proliferation include: Histone H2AX, Cofilin-1, 40 S ribosomal protein S3, and Heterogeneous nuclear ribonucleoprotein U. Finally, the protein involved with Signal Transduction included Prohibitin 1.

Biological processes of the protein binding partners of LINC00298 RNA. Data analysis by Thermo Scientific Proteome Discoverer version 3.0 indicated that the protein binding partners of LINC00298 have a diverse range of biological processes in which they are involved. Data represented in this graph is based on replicated LC/MS experiments.
As a further step in the analysis of the LC/MS data, Thermo Scientific Proteome Discoverer version 3.0 was used to identify the Molecular Functions of each of the protein binding partners of LINC00298 RNA, as seen in Fig. 7. Overwhelmingly, the predominant Molecular Function was Nucleic Acid Binding activity. The proteins that have Nucleic Acid Binding activity include: Histone H1.4, Pyruvate kinase, Alpha-enolase, Elongation factor 2, Histone H2AX, Eukaryotic initiation factor 4A-I, Heat Shock protein HSP 90-beta, 60 S ribosomal protein L13a, 40 S ribosomal protein S18, Histone H4, 60 S acidic ribosomal protein P0, Heterogeneous nuclear ribonucleoprotein A/B, 60 S ribosomal protein L13, 40 S ribosomal protein S3, and Heterogeneous nuclear ribonucleoprotein U. Cytoskeletal Activity was the second predominant Molecular Function. The proteins that had Cytoskeletal Activity included: Elongation factor 2, Dihydropyrimidinase-related protein 2, Heat Shock protein HSP 90-beta, Heterogeneous nuclear ribonucleoprotein U, Cofilin-1, and 40 S ribosomal protein S3. The proteins that had the Molecular Functions of Signal Transduction Activity or Receptor Binding included: Prohibitin 1, ATP synthase subunit alpha, Heat Shock protein HSP 90-beta, and Cofilin-1. The proteins that had Transporter Activity included: ATP synthase subunit alpha, ADP/ATP translocase 2, and ATP synthase subunit e. The proteins that had Translation Activity included Elongation factor 2 and 40 S ribosomal protein SA. The Heat Shock protein HSP 90-beta was identified as having Enzyme Regulator Activity, and Pyruvate kinase was identified as having Kinase Activity.

The molecular functions of the protein binding partners of LINC00298 RNA. Through an analysis using Thermo Scientific Proteome Discoverer version 3.0 software, the molecular functions of each of the protein binding partners of LINC00298 RNA were identified. Data represented in this graph is based on replicated LC/MS experiments.
To determine the cellular localization of LINC00298 RNA, Thermo Scientific Proteome Discoverer version 3.0 was used to identify the cellular components associated with each of LINC00298 RNA’s protein binding partners. Figure 8 indicates that the protein binding partners of LINC00298 RNA are predominantly associated with the nucleus and the cytosol. Those that are associated with the nucleus included: Histone H1.4, Pyruvate kinase, Alpha-enolase, 40 S ribosomal protein SA, Prohibitin 1, Histone H2AX, ATP Synthase subunit alpha, 60 S ribosomal protein L31, Heat Shock protein HSP 90-beta, Histone H4, 60 S acidic ribosomal protein P0, Heterogeneous nuclear ribonucleoprotein A/B, Heterogeneous nuclear ribonucleoprotein U, 60 S ribosomal protein L13, Cofilin-1, and 40 S ribosomal protein S3. Those that are associated with the cytosol included: Pyruvate kinase, Alpha-enolase, Elongation factor 2, 40 S ribosomal protein SA, Dihydropyrimidinase-related protein 2, 60 S ribosomal protein L31, Heat Shock protein HSP 90-beta, 60 S ribosomal protein L13a, 40 S ribosomal protein S18, 60 S acidic ribosomal protein P0, Heterogeneous nuclear ribonucleoprotein A/B, Heterogeneous nuclear ribonucleoprotein U, 60 S acidic ribosomal protein P2, Cofilin-1, and 40 S ribosomal protein S3. The proteins associated with Translational Apparatus included: Elongation factor 2, 40 S ribosomal protein SA, Eukaryotic initiation factor 4A-I, 60 S ribosomal protein L31, 60 S ribosomal protein L13a, 40 S ribosomal protein S18, 60 S acidic ribosomal protein P0, 60 S acidic ribosomal protein P2, 60 S ribosomal protein L13, and 40 S ribosomal protein S3. The proteins associated with the Plasma membrane included: Alpha-enolase, Elongation factor 2, 40 S ribosomal protein SA, Dihydropyrimidinase-related protein 2, Prohibitin 1, ATP synthase subunit alpha, mitochondria, Heat Shock protein HSP 90-beta, Cofilin-1, and 40 S ribosomal protein S3. The proteins associated with the Cytoskeleton included: Dihydropyrimidinase-related protein 2, Histone H2AX, ADP/ATP Translocase 2, Heterogeneous nuclear ribonucleoprotein U, Cofilin-1, and 40 S ribosomal protein S3. The proteins associated with the mitochondria included: Prohibitin 1, ATP synthase subunit alpha, mitochondria, ADP/ATP Translocase 2, ATP synthase subunit e, mitochondria, Cofilin-1, and 40 S ribosomal protein S3. The proteins associated with the ER/Golgi cellular components included: Pyruvate kinase, 60 S acidic ribosomal protein P0, 60 S ribosomal protein L13, and 40 S ribosomal protein S3. The proteins associated with the Non-structural Extracellular components included: Heat Shock protein HSP 90-beta, and Cofilin-1. Finally, the protein that is associated with the cellular components of the Extracellular Matrix is 40 S ribosomal protein SA.
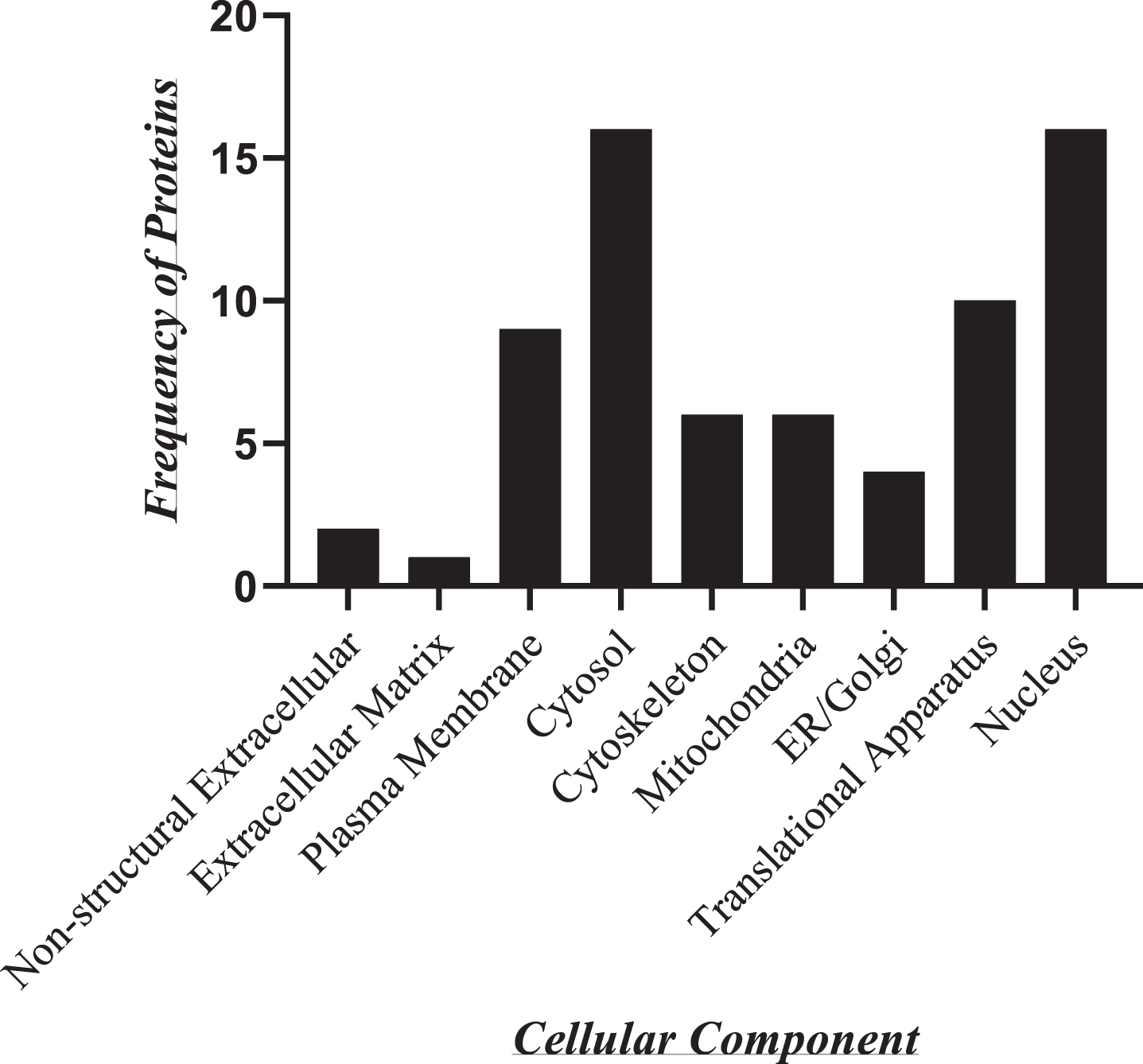
Cellular components associated with the protein binding partners of LINC00298 RNA. As determined by a Thermo Scientific Proteome Discoverer version 3.0 analysis, the identified protein binding partners of LINC00298 RNA are associated with specific cellular components and compartments. Data represented in this graph is based on replicated LC/MS experiments.
After determining the cellular components that are associated with LINC00298 RNA’s protein binding partners, an interactome was mapped, as shown by Fig. 9. The interactome was based on an analysis of the cellular components associated with each protein binding partner of LINC00298 RNA, as indicated by Thermo Scientific Proteome Discoverer version 3.0, as well as through analysis of previous research on lncRNA localization [7]. Through mapping the interactome of LINC00298 RNA, it was determined that it has dual localization in the nucleus and cytosol. Likewise, numerous proteins within the interactome also have dual localization in the nucleus and cytosol, as indicated by the italicized names of the proteins within the interactome map diagram.

LINC00298 RNA’s interactome mapped within the nucleus and cytosol. Mapping of the interactome of LINC00298 RNA was based on the cellular localizations of the protein binding partners of LINC00298 RNA as determined through analysis using Thermo Scientific Proteome Discoverer version 3.0 software, as well as previous research findings. Italicized protein names are indicative of proteins that can be found in both the nucleus and the cytosol.
Lastly, Fig. 10 is comprised of LC/MS spectra of the protein binding partners of LINC00298 RNA that occur in high frequency with high Percent Coverage, as well as a high number of peptides identified. Each of the spectra in Fig. 10 is of a representative peptide from each of the high frequency protein groups that bind to LINC00298 RNA as indicated in Fig. 5A and 5B. Figure 10A is the LC/MS spectrum of the 60 S acidic ribosomal protein P0, which had 26% Coverage, and seven unique peptides identified. The LC/MS spectrum is of the peptide that is composed of residues 113–124, which was identified with high confidence and with 0 missed cleavage sites. Figure 10B is the LC/MS spectrum of the 40 S ribosomal protein S3, which had 30% Coverage, and seven unique peptides. The LC/MS spectrum is of the peptide that is composed of the residues 215–227, which was identified with high confidence with 0 missed cleavage sites. Figure 10C is the LC/MS spectrum of the ATP synthase subunit e protein, which had 21% Coverage, and two unique peptides. The LC/MS spectrum is of the peptide that is composed of the residues 57–71, which was identified with high confidence, and with 0 missed cleavage sites. Figure 10D is the LC/MS spectrum of the Heterogeneous nuclear ribonucleoprotein A/B protein, which had 16% Coverage, and three unique peptides. The LC/MS spectrum is of the peptide that is composed of the residues 161–174, which was identified with high confidence, and with 0 missed cleavage sites. Figure 10E is the LC/MS spectrum of the Histone H4 protein, which had 54% Coverage, and six unique peptides. The LC/MS spectrum is of the peptide that is composed of the residues 47–60, which was identified with high confidence, and with 0 missed cleavage sites.

LC/MS spectra of the protein binding partners of LINC00298 RNA that occur in high frequency. Each spectra is of a representative peptide from each of the high frequency proteins identified by LC/MS. A) LC/MS spectra of the 60 S acidic ribosomal protein P0 peptide of residues 113–124 that was identified with high confidence with 0 missed cleavage sites. B) LC/MS spectra of the 40 S ribosomal protein S3 peptide consisting of the residues 215–227 that was identified with high confidence with 0 missed cleavage sites. C) LC/MS spectra of the ATP synthase subunit e peptide consisting of the residues 57–71 that was identified with high confidence with 0 missed cleavage sites. D) LC/MS spectra of the Heterogeneous nuclear ribonucleoprotein A/B peptide consisting of the residues 161–174 that was identified with high confidence with 0 missed cleavage sites. E) LC/MS spectra of the Histone H4 peptide consisting of the residues 47–60 that was identified with high confidence with 0 missed cleavage sites.
DISCUSSION
Analysis of the human genome through ENCODE and GENCODE have indicated that approximately 50%–70% of the human genome is transcribed with only 2% of the genome being translated [1–3]. Through these remarkable analyses of the human genome, it became clear that more research needed to be conducted to better understand the biological roles of non-coding RNA transcripts. The goal of this research project was to better understand the structural nature and biological functions of the long non-coding RNA, LINC00298. The role of LINC00298 RNA in Alzheimer’s disease underscores the importance of biochemically characterizing this non-coding RNA that is preferentially transcribed in the CNS [10, 15]. As such, primary mouse hippocampal neurons were used as the biological system of choice to elucidate the biological function of LINC00298 RNA, while computational modeling software was used to develop secondary and tertiary structures of LINC00298 RNA. The computational secondary structural prediction of LINC00298 RNA was accomplished using RNAfold 2.4.18 [36]. Through developing a computationally predicted secondary structure of LINC00298 RNA, it was determined that LINC00298 RNA is primarily comprised of stem-loop secondary structural elements. Based on the positional entropy, the helical portions of the stem-loop secondary structural elements, which typically assume an A-form helical confirmation, have lower positional entropy while the more disordered loop confirmations have higher positional entropy, giving the overall structure a predicted free energy of –617.82 kcal/mol. Thus, the secondary structure of LINC00298 RNA has numerous loop confirmations, giving rise to dynamic and flexible RNA, but the stem confirmations provide structural stability with lower positional entropy, making LINC00298 RNA an excellent binding partner for other macromolecules. Typically, RNA docking sites are flexible and often are loop structures, which allow for the malleability needed to create stable interactions between the RNA and its binding partners [45, 46]. Additionally, binding partners of RNAs can cause a cascade of structural remodeling events in the targeted RNA, which when formed, rely on the structural flexibility of the RNA [45]. The secondary structure of LINC00298 RNA indicates numerous large loop structural configurations that have higher positional entropy values, which correlates with greater malleability and therefore could serve as docking sites for macromolecular binding partners.
Further structural analysis using the 3dRNA/DNA modeling program to computationally develop a tertiary structure of LINC00298 RNA, complements LINC00298 RNA’s predicted secondary structure [37]. The predicted tertiary structure of LINC00298 RNA is inherently flexible, with an elongated, cylindrical structure that has numerous stem-loop structural motifs extending from the structure’s core. This structural arrangement would maximize the interactions possible, which could promote docking of many different macromolecular binding partners.
Experimentally, structural analysis of LINC00298 RNA was done using CD and thermal stability assays. CD is a technique that can be used to identify secondary structural motifs in RNA [23]. Helical structures in RNA are similar to A-form DNA helices and therefore absorb maximally near 260 nm in CD spectra [23, 38–40]. Additionally, stem-loop secondary structures are commonly formed in RNA and can be identified on CD spectra by a negative peak at around 245 nm and a positive peak in the range of 265 nm–285 nm [40]. Since the computationally predicted secondary structure of LINC00298 RNA is predominantly comprised of stem-loop structural motifs, it would be reasonable to expect that the CD spectra would also indicate stem-loop structural spectral patterns. Stem-loop CD spectral patterns in isolation and in the absence of tertiary structure, would be expected to have a negative peak at around 245 nm and a positive peak in the range of 265 nm–285 nm [40]. However, due to the large and complex tertiary structure of LINC00298 RNA, the expected CD spectral peaks characteristic of stem-loop structures would be expected to shift [41]. Indeed, the CD spectral analysis of LINC00298 RNA shows a shift in the negative peak spanning from around 240 nm–280 nm and a positive peak spanning from around 285 nm–300 nm. This indicates that LINC00298 RNA is comprised primarily of stem-loop structures that are part of a larger tertiary structure with helical secondary structural motifs that have influenced the CD spectral output.
Furthermore, CD analysis was used to compare the relative secondary/tertiary structural content of apo-LINC00298 RNA and LINC00298 RNA (Mg2+). It has been well established that divalent cations impact secondary structure formation in RNAs, and specifically, Mg2+ ions are needed to bind and stabilize RNA’s negatively charged phosphodiester backbone [23, 48]. By neutralizing the like-charge repulsion of the phosphodiester backbone through binding to Mg2+, RNAs can assume more compact and stable three-dimensional structures [23, 48]. Comparative CD analysis of apo-LINC00298 RNA and LINC00298 RNA (Mg2+) confirmed that when LINC00298 RNA is bound to Mg2+, there is a greater peak intensity of the CD spectrum, indicating the formation of a more ordered and stable structure. Likewise, the Thermal Stability assays indicate that when LINC00298 RNA is bound to Mg2+, there are less spectral fluctuations, which is indicative of a more stable structure. With this said, both spectra have fluctuations, highlighting the dynamic nature of LINC00298 RNA. The Thermal Stability experiments support the computationally rendered structures of LINC00298 RNA, which show numerous stem-loop structural motifs that have greater flexibility and higher positional entropy. Therefore, the overall structural analysis of LINC00298 RNA concluded that the structure is malleable, complex, and can accommodate docking of numerous binding partners.
Determining the biological function of LINC00298 RNA was the other focus of this research effort. Prior to this research endeavor, the specific biological function(s) of LINC00298 RNA remained unknown [10]. However, it is known that LINC00298 RNA has specific expression in the CNS and that its expression is associated with Alzheimer’s disease as well as other brain diseases [10, 15–18]. Additionally, it is known that LINC00298 RNA’s expression is associated with development, neuronal differentiation, and may play a role in regulating neuronal plasticity [10]. Therefore, elucidating the biological function(s) of LINC00298 RNA was of primary importance to begin to shed light on its role in Alzheimer’s disease pathogenesis as well as its role in other important biological and/or disease processes. To begin to understand the biological role LINC00298 RNA may play, identifying its protein binding partners was a necessary step to develop LINC00298 RNA’s interactome network and its cellular localization within hippocampal neuronal cells. To this end, affinity column chromatography was carried out with LINC00298 RNA immobilized on the stationary phase, and cell lysate from primary mouse hippocampal neuron cells comprising the mobile phase. Affinity column chromatography allowed for the binding partners of LINC00298 RNA to be eluted and identified through LC/MS. The high confidence, reproducible binding partners of LINC00298 RNA included 24 proteins, as tabulated in Fig. 5A. LINC00298 RNA binds with numerous 60 S and 40 S ribosomal proteins, which indicates that LINC00298 RNA impacts protein translation processes, thereby affecting the biological processes of Protein Metabolism, Cell Organization and Biogenesis, as well as Development. LINC00298 RNA’s role in protein translation processes, as well as its role in Alzheimer’s disease, may provide a connection between protein synthesis and forming, renewing, and extinguishing long-term memories [10, 49]. Additionally, this research finding correlates with recently reported research, which highlights the important role protein translation plays in the pathogenesis of Alzheimer’s disease [49]. To further strengthen our assertion that LINC00298 RNA is involved in protein translation processes, it was found that LINC00298 RNA also binds to Eukaryotic initiation factor 4A-I and Elongation factor 2, which are both part of the protein translation machinery.
LINC00298 RNA also impacts the biological processes of Cell Organization and Biogenesis, which do encompass protein translation processes, but also includes chromatin organization as well as cellular machinery and compartmental organization. LINC00298 RNA binds Histone H1.4, Histone H4, and Histone H2AX, which are involved in the organization of DNA by forming the nucleosome subunits of chromatin. LINC00298 RNA also binds Dihydropyrimidinase-related protein 2, which regulates neuronal cell organization by promoting axonal growth and has been implicated in the pathogenesis of Alzheimer’s disease [50, 51]. Additionally, LINC00298 RNA binds to Cofilin-1, which is also known to be part of the pathogenesis of Alzheimer’s disease through its role in regulating actin cytoskeleton [52]. Lastly, LINC00298 RNA binds to Prohibitin 1, which is known to regulate mitochondrial function, but it also maintains the structural integrity of myelin in Schwann cells [53, 54]. These identified binding partners of LINC00298 RNA that are involved in Cell Organization and Biogenesis play important roles in maintaining crucial CNS cellular structures, and impacting neuronal cellular health, thereby shedding light on LINC00298 RNA’s role in the CNS.
As previously stated, LINC00298 RNA has been associated with Developmental biological processes [10]. Further evidence that supports this assertion can be garnered through analyzing its binding partners: Pyruvate kinase, Alpha-enolase, Dihydropyrimidinase-related protein 2, Prohibitin 1, Histone H2AX, Heat Shock Protein HSP 90-beta, Cofilin-1, Heterogeneous nuclear ribonucleoprotein U, and 60 S ribosomal proteins L13 and L13a. Many of these binding partners’ functions have already been discussed and are known to be involved in metabolic pathways, cell organization, and maintaining cell and/or macromolecular structure integrity. As such, it is a natural extension of the functionality of these protein binding partners to also be involved in developmental processes. Within this list of LINC00298 RNA’s binding partners, there are a few worth highlighting, which at first glance may not be intuitively involved with developmental processes. Pyruvate kinase and Alpha-enolase are both glycolytic enzymes, and have both been identified as multifunctional enzymes, whose biological roles, among many other things, have been to help regulate cell proliferation [55, 56]. Additionally, Heterogeneous nuclear ribonucleoprotein U is known to participate in RNA splicing, but recent studies have shed light on Heterogeneous nuclear ribonucleoprotein U’s role in CNS development and maintaining neuronal cell integrity [57]. Thus, through the binding interactions of LINC00298 RNA with the aforementioned proteins, it is clear that LINC00298 RNA plays an important role in developmental processes, which is supported by previous research findings [10].
LINC00298 RNA was found to bind proteins involved in Transport biological processes, namely, ATP synthase subunit alpha, ATP synthase subunit e, and ADP/ATP Translocase 2. Though it is possible that LINC00298 RNA plays a role in ATP synthesis and translocation within the mitochondria, it is unlikely given previous research on LINC00298 RNA as well as the research findings presented herein [10]. As more research is conducted on the biological roles of ATP synthase and ADP/ATP Translocase 2, diverse functions are being uncovered, which include energy metabolism and growth in axons, synthesis of neuronal extracellular ATP, regulating neuronal intracellular pH as well as playing a role in neurodegenerative diseases [58–60]. Therefore, it is unlikely that LINC00298 RNA is associated with signification transport processes, but it is much more likely that LINC00298 RNA associates with ATP synthase and ADP/ATP Translocase 2 proteins to regulate neuronal cell function, promote neuronal growth, and to maintain the integrity of neuronal cellstructures.
Several of the protein binding partners of LINC00298 RNA have been associated with Stress Response, one is the Heat Shock Protein HSP 90-beta. The Heat Shock Protein HSP 90-beta is a well-studied chaperone protein that facilitates protein folding and therefore protein maturation [61]. The Heat Shock Protein HSP 90-beta is also known to stabilize aggregation-prone proteins as well as to keep proteins in activation-competent conformations [61]. Additionally, Heat Shock Protein HSP 90-beta has been implicated in reducing neuronal cell apoptosis and thus protecting against Alzheimer’s disease [62]. As such, LINC00298 RNA’s interaction with Heat Shock Protein HSP 90-beta may impact neuronal apoptotic pathways, which may be important for the pathogenesis of Alzheimer’s disease.
LINC00298 RNA also binds to proteins involved in RNA Metabolism and Transcription, specifically, Heterogeneous nuclear ribonucleoprotein A/B, and Heterogeneous nuclear ribonucleoprotein U, which are both known to participate in RNA splicing [57, 63]. Furthermore, these proteins are known to participate in RNA transcription and mRNA nucleo-cytoplasmic export as well as DNA Metabolic biological processes that include DNA replication, repair, and telomere maintenance [63]. In addition to these important roles, Heterogeneous nuclear ribonucleoprotein A/B, and Heterogeneous nuclear ribonucleoprotein U have both been identified as playing important roles in neurodegenerative diseases [57, 64]. Heterogeneous nuclear ribonucleoprotein A/B is known to facilitate splicing of the APP gene that encodes the amyloid-β (Aβ) peptide, which when expressed in Alzheimer’s disease can lead to Aβ aggregates in the CNS [64]. Due to the many functions of Heterogeneous nuclear ribonucleoprotein A/B, and Heterogeneous nuclear ribonucleoprotein U and their roles in neurodegenerative diseases, it therefore is not a surprise that LINC00298 RNA binds to these Heterogeneous nuclear ribonucleoproteins to impact RNA and DNA metabolism as well as transcription processes.
DNA metabolism is also impacted by Prohibitin 1, which is known as a multifunctional protein that is involved in cell proliferation, development, signal transduction, as well as the aforementioned biological roles in maintaining mitochondrial function and impacting the structural integrity of myelin [53, 65]. Prohibitin 1 plays a critical role in the PI3K/Akt and Ras/MAPK/ERK signaling pathways, which have widespread effects in the cell, impacting cellular metabolism, proliferation, and development [65]. Since Prohibitin 1 is involved in the PI3K/Akt signaling pathway, it is biologically linked to the Histone H2AX protein, which can be phosphorylated through this signaling pathway, thereby impacting neural stem cell proliferation and self-renewal [66]. Furthermore, both of these proteins bind to LINC00298 RNA, which is also known to impact cell proliferation and development [10]. Through further investigating LINC00298 RNA’s binding interactions with Prohibitin 1 and Histone H2AX, the mechanisms by which LINC00298 RNA does regulate cell proliferation and development may come to light.
Through analysis of the biological functions of LINC00298 RNA’s binding partners, it becomes clear that LINC00298 RNA plays important roles in protein translation as well as neuronal development and proliferation. The molecular functions of LINC00298 RNA’s protein binding partners mirrors their biological functions. As indicated by Fig. 8, the predominant molecular function of the binding partners of LINC00298 RNA is Nucleic Acid Binding Activity. This result aligns with the predominant biological functions of protein translation, cellular development, and cell proliferation. Thus, LINC00298 RNA’s predominant molecular function may also be Nucleic Acid Binding Activity or to facilitate nucleic acid binding.
Identification of biological and molecular functions is even more meaningful when paired with cellular components and compartmentalization to provide context to the specific biological or molecular function. As previously discussed in the Results section, we have identified the cellular components that are associated with each of the binding partners of LINC00298 RNA. Due to the large number of protein binding partners of LINC00298 RNA, it is not surprising that there are many different Cellular Components associated with this group of proteins. However, based on previous research findings, it is unrealistic that a lncRNA would associate or be transported through the cell to many different cellular components [5, 6]. To date, the localizations of lncRNAs have been limited to the nucleus and the cytoplasm, with very few lncRNAs having dual distribution between the nucleus and the cytoplasm [5–7]. Since it has been established that LINC00298 RNA is preferentially expressed in the CNS and is known to play a role in development, neuronal differentiation, and gene expression, it is highly likely that LINC00298 RNA is biologically active in the nucleus [10]. Herein, we have provided additional evidence that LINC00298 RNA is localized to the nucleus through its binding interactions with the following nuclear proteins: Histone H1.4, Histone H2AX, Histone H4, Pyruvate kinase, Alpha-enolase, 40 S ribosomal proteins SA and S3, 60 S ribosomal proteins L31 and L13, 60 S acidic ribosomal protein P0, Prohibitin 1, ATP Synthase subunit alpha, Heat Shock Protein HSP 90- beta, Heterogeneous nuclear ribonucleoprotein A/B, Heterogeneous nuclear ribonucleoprotein U, and Cofilin-1. Additionally, we propose that LINC00298 RNA is also biologically active in the cytosol due to its strong binding associations with protein translation machinery and other cytosolic proteins. The cytosolic proteins that LINC00298 RNA binds to includes: Elongation factor 2, Eukaryotic initiation factor 4A-I, 40 S ribosomal proteins SA, S18, and S3, 60 S ribosomal proteins L31 and L13a, 60 S acidic ribosomal proteins P0 and P2, Dihydropyrimidinase-related protein 2, Pyruvate kinase, Alpha-enolase, Heat Shock protein HSP 90-beta, Heterogeneous nuclear ribonucleoprotein A/B, Heterogeneous nuclear ribonucleoprotein U, and Cofilin-1. Though there is substantial overlap between the list of nuclear proteins and the list of cytosolic proteins, there are some cytosolic proteins that are exclusively found in the cytosol, which includes: Elongation factor 2, Eukaryotic initiation factor 4A-I, 40 S ribosomal protein S18, 60 S ribosomal protein L13a, and 60 S acidic ribosomal protein P2. The strong binding interactions between LINC00298 RNA and protein binding partners that are exclusively found in the cytosol adds substantial evidence that LINC00298 RNA is also localized to the cytosol. Therefore, we propose the dual localization of LINC00298 RNA in the nucleus and in the cytosol.
Furthermore, it is important to address LINC00298 RNA’s interactions with ATP synthase subunits alpha and e, as well as with ADP/ATP Translocase 2, which are all found in the mitochondria. As previously stated, ATP synthase and ADP/ATP Translocase 2 have diverse functions, which include energy metabolism and growth in axons, synthesis of neuronal extracellular ATP, regulating neuronal intracellular pH as well as playing a role in neurodegenerative diseases [58–60]. As such, ATP synthase and ADP/ATP Translocase 2 are not exclusively found in the mitochondria but can also be localized to the nucleus and cytosol. Additionally, the translation or partial translation of these protein complexes is known to take place in the cytosol [67, 68]. Thus, LINC00298 RNA has ample opportunity to interact with ATP synthase and ADP/ATP Translocase 2 while in the nucleus, cytosol, and during their respective translation processes. Therefore, we propose that the localization of LINC00298 RNA in the mitochondria is unlikely, but rather, LINC00298 RNA is localized in the nucleus or cytosol while interacting with either ATP synthase or ADP/ATP Translocase 2.
In conclusion, data presented herein provides evidence that LINC00298 RNA’s biological function is to participate, regulate, and/or facilitate protein translation as well as neuronal development, proliferation, and cellular organization. Additionally, analysis of the data indicates that LINC00298 RNA has dual localization in the nucleus and the cytosol. Lastly, LINC00298 RNA’s structure is comprised of stem-loop secondary structural motifs and that these secondary structural elements interact to form a large and complex tertiary structure that is highly dynamic to facilitate and maximize binding interactions with 24 different protein binding partners.
Footnotes
ACKNOWLEDGMENTS
This work was supported by Bemidji State University, Inter Faculty Organization, and the McNair Scholars Program. We would like to thank the faculty and staff at the Center for Mass Spectrometry and Proteomics at the University of Minnesota for processing our samples using LC/MS. Additionally, we would like to thank Dr. Stefan Vetter in the School of Pharmacy at North Dakota State University for his assistance with Circular Dichroism.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and/or its supplementary material.