
Abstract
Background:
Deprivation of extracellular serotonin has been linked to cognitive decline and neuropsychiatric disturbances in Alzheimer’s disease (AD). However, despite degeneration of serotonin-producing neurons, whether serotonin release is affected in AD-sensitive brain regions is unknown.
Objective:
This study investigated the impact of mitochondrial dysfunction in decreased hippocampal serotonin release in AD amyloidosis mouse model 5xFAD mice.
Methods:
Electrochemical assays were applied to examine hippocampal serotonin release. We also employed multidisciplinary techniques to determine the role of oligomeric amyloid-β (Aβ) in hippocampal mitochondrial deficits and serotonin release deficiency.
Results:
5xFAD mice exhibited serotonin release decrease and relatively moderate downregulation of serotonergic fiber density as well as serotonin content in the hippocampal region. Further experiments showed an inhibitory effect of oligomeric amyloid-β (Aβ) on hippocampal serotonin release without affecting the density of serotonergic fibers. Pharmaceutical uncoupling of mitochondrial oxidative phosphorylation (OXPHOS) disrupted hippocampal serotonin release in an ex vivo setting. This echoes the mitochondrial defects in serotonergic fibers in 5xFAD mice and oligomeric Aβ-challenged primary serotonergic neuron cultures and implicates a link between mitochondrial dysfunction and serotonin transmission defects in AD-relevant pathological settings.
Conclusion:
The most parsimonious interpretation of our findings is that mitochondrial dysfunction is a phenotypic change of serotonergic neurons, which potentially plays a role in the development of serotonergic failure in AD-related conditions.
INTRODUCTION
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder, arising from a perplexing mix of the aging process, protein aggregates, and genetic risks [1]. Disturbed homeostasis of a variety of neurotransmitters such as glutamate, acetylcholine, and monoamines constitutes a pathological characteristic of this neurodegenerative disease and offers therapeutic opportunities for the mitigation of AD symptoms [2, 3]. Previous clinical and neuropathological studies have proposed an association of serotonergic failure with cognitive decline as well as neuropsychiatric symptoms including mood disturbances and behavioral changes in patients with AD [4–11]. Reduced serotonin and its metabolite, 5-hydroxyindoleacetic acid (5-HIAA) [4–8], as well as diminished serotonergic terminals demonstrated by reduced serotonin transporter (SERT) clusters [9, 10] have been repeatedly identified in multiple brain loci including the hippocampus in patients in various stages of AD, indicating compromised serotonergic transmission in this neurodegenerative disorder. Whether suppressed extracellular serotonin signaling plays a role in the pathophysiology of AD is complicated by the multifaceted effects of serotonin on cognition-related synaptic transmission in the hippocampus [12–16]. However, this concern has been seemingly settled by the symptom-alleviating effects of selective serotonin reuptake inhibitors (SSRIs) and serotonin receptor modulators in AD patients and animal models [17–23]. Moreover, serotonin-enhancing agents lower the production of amyloid-β (Aβ) [17, 24], a key mediator of AD [25], and demonstrate promising effects in preventing the development of AD especially in individuals with depressive symptoms [22, 26]. The increasing recognition of disrupted serotonergic neurotransmission in AD has thus highlighted serotonin failure as a bona fide AD pathology and raised a critical and yet-unaddressed scientific question about the mechanisms of extracellular serotonin insufficiency in this neurological disorder. In the central nervous system (CNS), serotonin is predominantly produced by the raphe nuclei and subsequently released to brain regions innervated by serotonergic fibers including AD-sensitive brain regions such as the frontal and cingulate cortices and hippocampus [27]. Impaired serotonin release in multiple brain regions including the hippocampus has been reported in rodent models of depression [28, 29]. Given the role of serotonin failure in the development of psychological symptoms, it was postulated that patients with AD, especially those reporting psychological disturbances may also suffer from compromised serotonin release in affected brain regions [30]. However, despite reported raphe nuclei degeneration and loss of serotonin-producing neurons in AD patients [31–33], whether serotonin release from serotonergic fibers is affected in AD-sensitive brain regions in patients or animal models has never been previously investigated.
Like many other neurotransmitters, the packaging and release of serotonin is an energy-consuming process [34]. By virtue of their energy-producing capacity, mitochondria are the major source of ATP in neurons and play a crucial role in neurophysiology [35]. Previous studies have consistently found mitochondrial defects in AD brains and animal models of AD-like pathology [36–38]. Of note, due to the scarce distribution of mitochondria and lack of mitochondrial repair mechanisms in axons, it is a well-documented notion that axonal mitochondria are at a greater risk to develop functional defects as compared with their siblings in the soma, thus rendering presynaptic transmission vulnerable to AD-related toxic molecules including Aβ [38, 39]. In this context, it is logical to assume that serotonergic fibers in AD-sensitive brain regions may undergo mitochondrial defects, which may contribute to compromised serotonin release, leading to extracellular serotonin deficiency. So far, the argument for a link between mitochondrial dysfunction and defects in serotonin release in AD-related conditions suffers from lack of experimental evidence and thus warrants our current investigation.
In this study, we found decreased hippocampal serotonin release alongside mitochondrial abnormalities in hippocampal serotonergic fibers in 5xFAD mice, a mouse model of AD-like brain amyloidopathy and cognitive impairment [40]. Consistent with the deleterious impact of oligomeric Aβ on mitochondrial bioenergetics in cultured serotonergic neurons, further ex vivo assays showed an inhibitory impact of oligomeric Aβ on hippocampal serotonin release, which was recapitulated by inducing the uncoupling of mitochondrial respiration. These findings implicate a connection between mitochondrial dysfunction and hippocampal serotonin release defects in AD-related conditions and offer us a better understanding of the role of mitochondrial dysfunction in the pathogenesis of this neurological disorder.
MATERIALS AND METHODS
Mouse subjects and ELISA test
Studies on mice were approved and performed under the guidelines of the University of Kansas Institutional Animal Care and Use Committee (IACUC). 5xFAD mice (B6SJL-Tg(APPSwFlLon,PSEN1*M146L*L286V)6799Vas/Mmjax) were purchased from Jackson Laboratory and crossed with B6SJLF1/J. Genotypes of animals were confirmed using PCR. Both male and female mice were used in the experiments. Mouse hippocampal serotonin was measured using a commercial ELISA kit (Eagle Biosciences, #SUE39-K01).
Serotonin release measurement
Chemicals and reagents
Serotonin (5-hydroxytryptamine, 5-HT) hydrochloride was purchased from Sigma-Aldrich (St. Louis, MO). Mouse aCSF was made with 127 mM NaCl, 25 mM NaHCO3, 1.25 mM NaH2PO4, 2.5 mM KCl, 25 mM D-glucose, and 1 mM MgCl2 in purified (18.2 MΩ) H2O and bubbled with 95% O2/5% CO2 for 20 min before adding 2 mM CaCl2. The pH was adjusted to 7.3 and aCSF buffers were stored at 4°C until use. 1 mM stock solutions of 5-HT were prepared in 0.2 M HClO4 and was remade weekly. Fresh calibration solutions of 5-HT were prepared each day.
Carbon-fiber microelectrodes
CFMEs were fabricated as previously described. Briefly, a vacuum pump was used to aspirate 7μm diameter carbon fibers (Goodfellow Cambridge LTD, Huntingdon, UK) into glass capillaries (1.2 mm D.D. and 0.68 mm I.D., 4 in long; A-M Systems, Inc., Carlsborg, WA) which were then pulled using a PE-22 heated coil puller (Narishige Int. USA, East Meadow, NY) to form two electrodes. Exposed carbon fibers were trimmed to a length of 50–100μm. To seal the CFMEs, the tips were dipped in epoxy resin (EPON resin 815C and EPIKURE 3234 curing agent, Miller-Stephenson, Danbury, CT) for 45 seconds and cured at 100°C for 1 h. CFMEs were treatment via an isopropanol soak for at least 20 min prior to initial use.
All CFMEs were pre-calibrated against standard solutions of 5-HT, introduced to a flow cell through a six-port valve (Valco Instruments Company Incorporated, Houston, TX, USA). The flow solution, which consisted of mouse aCSF without D-glucose, was pumped at a rate of 2 mL min–1 using a syringe pump (Chemyx Inc, Stafford, TX, USA). Injections were conducted in triplicate to determine the 5-HT concentration per nanoamp of current foreach CFME.
Electrochemical instrumentation
The electrochemical system was comprised of a CFME working electrode backfilled with 0.5 M potassium acetate and a chlorided Ag/AgCl wire as a reference electrode. Potentials were applied to CFME working electrodes using a ChemClamp potentiostat and headstage (Dagan, Minneapolis, MN). For all fast-scan cyclic voltammetry (FSCV) experiments, the extended serotonin waveform was used to apply potentials which scanned from +0.2 V to +1.3 V to –0.1 V to +0.2 V with a scan rate of 1000 V/s and a holding potential of +0.2 V. Data were collected and analyzed using TarHeel CV (Department of Chemistry, University of North Carolina at Chapel Hill) software, a custom-made breakout box, and two National Instruments computer interface cards (PCI 6052 and 6711, National Instruments, Austin, TX). All experiments were conducted in a grounded Faraday cage, and all data were collected at a frequency of 10 Hz and low pass filtered at 3 kHz.
Stimulated release slice experiments
For brain slice experiments, mice were anesthetized by isoflurane inhalation and euthanized by cervical dislocation. The brain was removed and immediately placed in ice-cold oxygenated aCSF for 1 min. The cerebellum and part of the cortex was removed using a razor blade and the brain was glued to a plate. A vibratome (Leica Microsystems, Bannockburn, IL) was used to create 300μm coronal brain slices containing the CA1 region of the hippocampus, one of which was transferred to a recording chamber (Warner Instruments, Hamden, CT) which was perfused with oxygenated aCSF held at 34°C. Additional viable brain slices were maintained in a slice saver containing continually oxygenated aCSF maintained at room temperature until they were used for experiments.
Evoked serotonin release was achieved by electrical stimulation. Two tungsten micro-stimulus electrodes (Microprobes, Gaithersburg, MD, USA), fixed at a 150μm separation distance, were used to deliver 30 biphasic current pulses (350μA, 1 ms duration at 30 Hz). CFMEs were positioned between the stimulus electrodes in the CA1 region of the hippocampus. For each release measurement, a 30 s collection file was conducted, and stimulation was triggered after 5 s of quiet time, and files were collected every 10 min. The maximum 5-HT oxidation current after stimulation was recorded.
For pharmacology experiments, nonTg mice brain slices were used to collect 3 or 4 release measurements (pre-drug) prior to changing the perfusion solution to either 500 nM oligomerized amyloid-β or 10μM carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, Sigma-Aldrich, #C2920). After 20 min of treatment, release measurements continued until the 5-HT signal stabilized (post-drug). The pre-drug measurements were used to calculate the average 5-HT release in nonTg mice. 5-HT release was determined similarly for 5xFAD mice by conducting measurements in triplicate in the CA1. To account for the heterogeneity between 5xFAD mice and therefore reduce the number of animals needed, triplicate measurements were taken in one or two locations of the CA1 in multiple slices, and these values were averaged together to calculate the concentration of release for a single N value.
Amyloid-β oligomer preparation
Aβ1–42 peptide (GenicBio) films were prepared as previously described [41]. Aβ1–42 peptide was diluted in 1,1,1,3,3,3,-hexafluoro-2-propanol (HFIP, Sigma-Aldrich) to 1 mM. The clear solution was aliquoted in microcentrifuge tubes and dried in the fume hood overnight. Aβ1–42 peptide films were dissolved in DMSO to 5 mM and resuspended in cold Ham’s F-12. The solution was then incubated at 4°C for 24 h to form Aβ oligomer.
Immunocytochemistry
Frozen mouse brain sections were prepared according to previous protocol [42]. Mouse brains were dissected and fixed in 4% paraformaldehyde (Sigma-Aldrich, #158127) overnight at 4°C. Primary cultured raphe neurons were fixed in 4% paraformaldehyde for 30 min at 37°C. Brain slices or cultured raphe neurons were treated with blocking buffer (5% goat or donkey serum (Sigma-Aldrich), 0.3% Triton X-100 (Fisher Scientific) in PBS, pH 7.4) and probed with primary antibodies accordingly: rabbit-anti-serotonin (Sigma-Aldrich, #S5545, 1:400), rabbit-anti-serotonin transporter (abcam, #ab25358, 1:400), mouse-anti-ATP5B (Santa Cruz, #sc-55597, 1:500). After overnight incubation with primary antibodies, the brain slices or neurons were probed with proper cross-adsorbed secondary antibodies conjugated to Alexa Fluor 488, Alexa Fluor 594, or Alexa Fluor 647 (Thermo Fisher Scientific, 1:500). Cell nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI). Images were collected on a Nikon confocal microscope. Serotonin intensity, serotonergic fiber density, and mitochondrial volume and density were analyzed using Nikon-Elements Advanced Research software. Mitochondrial density was calculated as number of mitochondria per μm serotonergic fiber.
Serotonergic neuron culture and treatment
Serotonergic neurons were cultured as previously described with modifications [43]. Raphe nuclei were dissected from day 0 pups and kept in cold Hanks’ balanced salt solution (HBSS, Corning) until digestion. Tissues were digested in trypsin (Gibco) at 37°C for 15 min to dissociate neurons. Mouse serotonergic neurons were cultured using neuron culture medium (Neurobasal A with 2% B27 supplement, 0.5 mM L-glutamine) on poly-D-lysine- (Sigma-Aldrich) coated Lab-Tek chamber slides (Nunc, #177445) in an incubator (37°C, 5% CO2). Neuron cultures were supplemented with 25 ng/ml brain-derived neurotrophic factor (BDNF) (Millipore Sigma, #GF029) on day in vitro (DIV) 2 to help serotoninergic neuron differentiation. Serotonergic neurons were treated with 500 nM oligomerized Aβ or vehicle on DIV 12 and assays were performed 24 h after the treatment.
Mitochondrial membrane potential, density, and volume measurement
Serotonergic neurons were incubated with 200 nM tetramethylrhodamine methyl ester (TMRM, Sigma-Aldrich) as previously described [44]. Mitochondria were labeled with 50 nM MitoTracker deep red (Invitrogen). After 30 min of incubation in an incubator (37°C, 5% CO2), neurons were washed with prewarmed HBSS and imaged on a Nikon Ti2 fluorescent microscope with on-stage incubator (Oko). The intensity of TMRM was analyzed using Nikon-Elements Advanced Research software. Mitochondrial density was calculated as number of mitochondria determined by MitoTracker deep red staining per μm. The volume of each single mitochondrion was analyzed by Nikon-Elements Advanced Researchsoftware.
Cultured serotoninergic neuron ATP measurement
ATP in serotonergic neurons was labeled with 10μM BiTracker ATP-Red liver cell dye (Millipore Sigma, #SCT045) for 30 min at 37°C, in a 5% CO2 incubator. After a careful wash with prewarmed HBSS, neurons were imaged using Nikon Ti2 fluorescent microscope with on-stage incubator (Oko). ATP intensity was analyzed using Nikon-Elements Advanced Research software.
Statistical analysis
Statistical analyses were performed using the GraphPad Prism 8 software. Unpaired or Paired student t-test were used when appropriate in data analysis. The results are expressed as means±SEM. Significance was concluded when the p value was less than 0.05. The statistical significance was indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
RESULTS
Hippocampal serotonin release is impaired in 5xFAD mice
To determine whether hippocampal serotonin release is vulnerable to amyloidosis, we subjected hippocampal slices from 9-10-month-old 5xFAD mice and their age- and gender-matched nontransgenic (nonTg) littermates to electrochemical assays for serotonin release. At 9-10 months old, 5xFAD mice demonstrate heavy brain Aβ deposition and AD-like cognitive and psychological symptoms [45, 46]. The stratum radiatum (SR) of the hippocampal CA1, an AD-sensitive region [47], was selected for the measurement. In comparison with their counterparts from nonTg mice, hippocampal slices from 5xFAD mice exhibited decreased serotonin concentrations in response to electrical stimulation (Fig. 1 A, B; nonTg 14.6±2.185 nM versus 5xFAD 8.536±1.306 nM, p = 0.0299), indicating impaired hippocampal serotonin release in 5xFAD mice. To determine whether the abundance of hippocampal serotonergic fibers is decreased in 5xFAD mice, we examined the density of serotonergic fibers in the hippocampal CA1 region by immunolabeling serotonin transporters (SERT), a specific serotonergic fiber marker [48]. In comparison with their nonTg littermates, 5xFAD mice displayed a lower density of SERT-immunoactive fibers in the SR region (Fig. 1 C, D; nonTg 0.127±0.004μm/μm2 versus 5xFAD 0.109±0.006μm/μm2, p = 0.0308), indicating loss of serotonergic fibers in 5xFAD mice at the tested age. However, we noticed that the moderate change of serotonergic fibers (∼14% Fig. 1C) was not in alignment with the greater suppression of hippocampal serotonin release (∼42% Fig. 1A) in 5xFAD mice. To determine whether reduced hippocampal serotonin release is associated with diminished serotonin content in 5xFAD mice, we performed an ELISA assay for serotonin in hippocampal homogenates as well as immunolabeling for serotonin in serotonergic fibers. The results showed no difference in hippocampal serotonin content between 5xFAD and nonTg mice, (Fig. 1E) in line with comparable serotonin immunoreactivity within serotonergic fibers in 5xFAD mice (Fig. 1 F, G). Although lowered serotonergic fibers may contribute to decreased serotonin release, the relatively preserved serotonergic fibers and serotonin content seem to implicate impairment in stimulation-elicited serotonin release from presynaptic terminals.
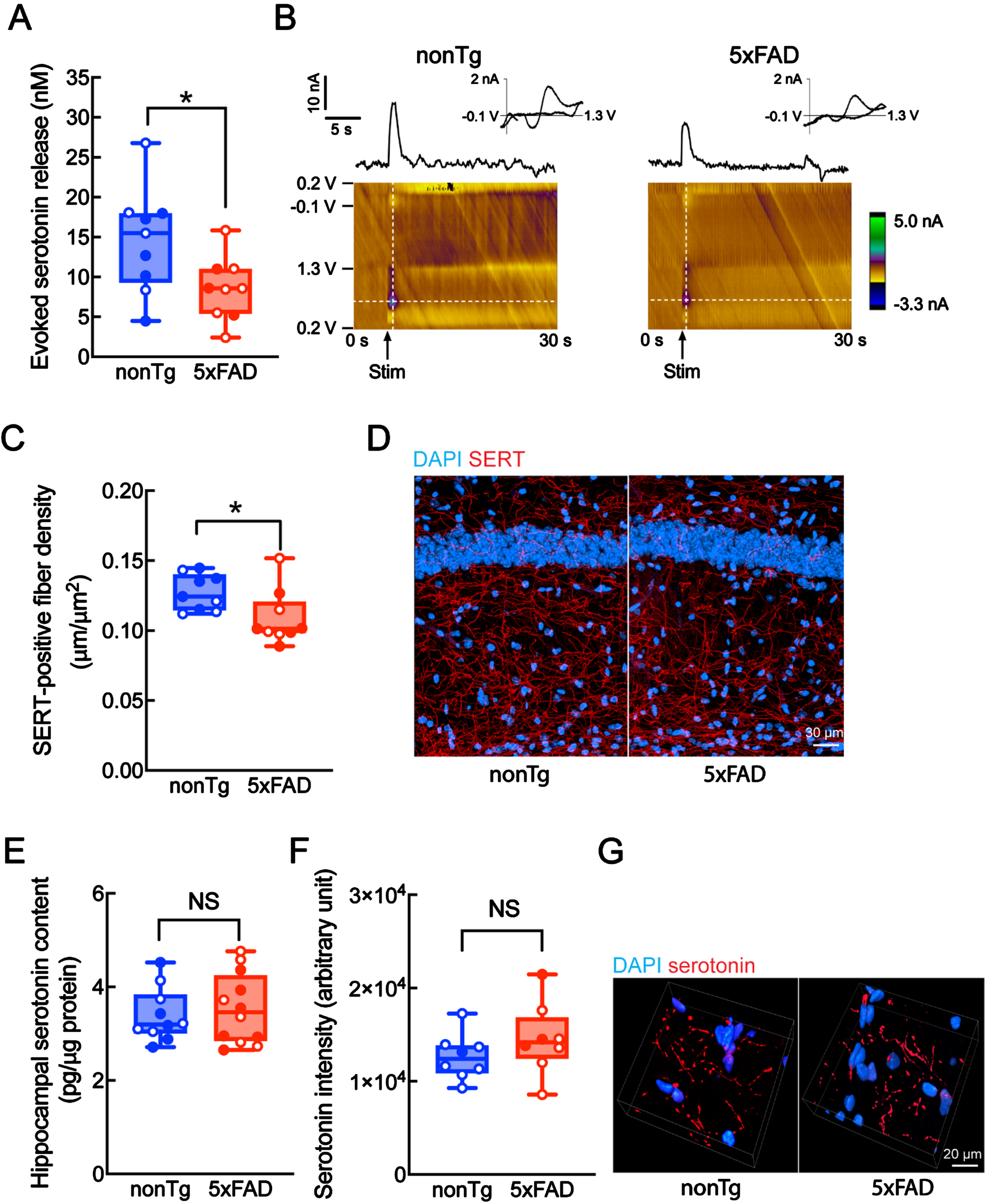
Reduced hippocampal serotonin release in 9-10-month-old 5xFAD mice. A) Voltammetric determination of hippocampal serotonin release in nonTg and 5xFAD mice. n = 9 in each group. Females: open circles, males: filled circles. Unpaired student t-test. *p < 0.05. B) Representative voltage versus time (top) and color plots (bottom) for serotonin release measurement. C,D) Immunolabeling of serotonergic fibers with serotonin transporter (SERT) antibody in the hippocampal CA1 SR region of nonTg and 5xFAD mice. n = 9 in each group. Females: open circles, males: filled circles. Unpaired student t-test. *p < 0.05. D) Representative images of SERT labeling. Cell nuclei were labeled with DAPI. Magnification at 400x. Scale bar = 30μm. E) Hippocampal serotonin concentration in nonTg and 5xFAD mice determined by ELISA. n = 10 nonTg mice, and 12 5xFAD mice. Females: open circles, males: filled circles. Unpaired student t-test. NS, not significant. F, G) Immunolabeling of serotonin in the hippocampal CA1 SR region of nonTg and 5xFAD mice. n = 9 in each group. Females: open circles, males: filled circles. NS, not significant. *p < 0.05. G) Representative images of serotonin labeling. Magnification at 400x. Scale bar = 20μm.
Oligomeric Aβ inhibits hippocampal serotonin release
5xFAD mice are an Aβ-based mouse model of AD [40]. To determine whether Aβ has a direct impact on hippocampal serotonin release, we examined serotonin release in hippocampal slices from nonTg mice pre- and post-perfusion of oligomeric Aβ1–42 at 500 nM. The selection of the dose of oligomeric Aβ1–42 is based on previous observations of acute Aβ toxicity on neuronal mitochondrial function in in vitro settings [38, 50]. In comparison with the baseline level, treatment with oligomeric Aβ1–42 reduced hippocampal serotonin release (Fig. 2 A, B). In contrast, oligomeric Aβ1–42-challenged hippocampal slices displayed no change in density of SERT-immunoreactive fibers (Fig. 2 C, D) or total serotonin content (Fig. 2 E). These ex vivo findings implicate a direct impact of oligomeric Aβ in blunting hippocampal serotonin release, at least, in the tested conditions.

Impeded hippocampal serotonin release with oligomeric Aβ treatment. A) Voltammetric determination of the impact of oligomeric Aβ1–42 on hippocampal serotonin release. NonTg mouse brain slices were perfused with 500 nM Aβ for 20 min to examine the influence on serotonin release in the hippocampal CA1 SR region. n = 3. Females: open circles, males: filled circles. Paired student t-test. **p < 0.01. B) Representative voltage versus time (top) and color plots (bottom) for serotonin release measurement. C, D) Immunolabeling of serotonergic fibers in oligomeric Aβ-treated nonTg acute mouse brain slices. n = 6 in each group. Females: open circles, males: filled circles. Unpaired student t-test. NS, not significant. D) Representative images of SERT labeling. Magnification at 400x. Scale bar = 30μm. E) Serotonin content determined by an ELISA assay. n = 4 in each group. Females: open circles, males: filled circles. Unpaired student t-test. NS, not significant.
Oligomeric Aβ induces mitochondrial defects in serotonergic fibers
In view of the importance of normal mitochondrial function to the release of neurotransmitters including serotonin [34], we sought to determine whether mitochondria in serotonergic fibers are impaired in AD-related conditions. Serotonergic fibers and mitochondria in the hippocampal CA1 SR region were determined by staining for SERT and a specific mitochondrial marker, ATP synthase β subunit [51], respectively. Further analysis showed decreased mitochondrial density (Fig. 3 A, C) in hippocampal serotonergic fibers in 5xFAD mice, implicating deficits in mitochondrial delivery and docking. Moreover, further observation of reduced mitochondrial volume (Fig. 3 B, C) also suggests mitochondrial fragmentation in 5xFAD serotonergic fibers. These mitochondrial changes in 5xFAD mice suggest a deleterious impact of Aβ toxicity on axonal mitochondrial motility and dynamics in serotonergic neurons. Altered mitochondrial morphology frequently accompanies mitochondrial functional changes [52]. To this end, we extended the observations to the influence of Aβ on mitochondrial bioenergetics in serotonergic neurons in an in vitro setting. Serotonergic neuron cultures were treated with in the 500 nM oligomeric Aβ or vehicle. The purity of serotonergic neuron culture was determined by > 90% cells with axonal expression of SERT (Supplementary Figure 1), a specific indicator of serotonergic terminals [53]. The axons were identified by their unique morphology. Mitochondrial membrane potential (mΔΨ), a sensitive indicator of mitochondrial bioenergetics state, was determined by the staining of tetramethylrhodamine methyl ester (TMRM) [51]. In comparison with their counterparts in the vehicle-treated group, Aβ-exposed axonal mitochondria exhibited decreased intensity of TMRM staining (Fig. 3 D, E), indicating mΔΨ collapse. Consistent with decreased mitochondrial membrane potential, compromised neuritic mitochondrial ATP production in oligomeric Aβ-treated serotonergic neurons was demonstrated (Fig. 3 F, G) by diminished staining of BiTracker ATP-Red live cell dye, a specific fluorescent ATP indicator targeting mitochondria [54]. The lowered mitochondrial ATP production (Fig. 3 F, G) was in line with reduced cellular ATP content (Fig. 3 H) in Aβ-challenged serotonergic neuron cultures, which further supports a pivotal role of mitochondria as a major source of ATP in serotonergic neurons. Moreover, in comparison with the vehicle-treated group, axonal mitochondrial density was decreased (Fig. 3 I, K) as was axonal mitochondrial volume (Fig. 3 J, K) in oligomeric Aβ-exposed serotonergic neuron cultures, mimicking the changes in 5xFAD mice. Together, the results suggest axonal mitochondrial vulnerability in Aβ-rich milieus.

Mitochondrial deficits in oligomeric Aβ-enriched serotonergic fibers and neurons. A-C) Immunolabeling of serotonergic fiber-associated mitochondria with ATP5B antibody in the hippocampal CA1 SR region of nonTg and 5xFAD mice. Mitochondria density (A) and volume (B) in serotonergic fibers were analyzed using NIS elements software. n = 4 nonTg and 5 5xFAD mice. Females: open circles, males: filled circles. Unpaired student t-test. *p < 0.05, **p < 0.01. C) Representative images of serotonergic fiber mitochondria labeling. (I) Original images, (II) mitochondria inside (multicolor) serotonergic fibers (grey), and (III) mitochondria in serotonergic fibers labeled with different colors to identify each mitochondrion. Magnification at 400x. Scale bar = 10μm. D, E) Mitochondrial membrane potential labeled with TMRM fluorescent dye for vehicle and 500 nM Aβ treated serotonergic neurons. n = 32 each group. Unpaired student t-test. **p < 0.01. E) Representative images of TMRM labeling. Magnification at 400x. Scale bar = 20μm. F, G) Mitochondrial ATP labeled with BiTracker ATP-Red live cell dye for vehicle and 500 nM Aβ treated serotonergic neuron cultures. n = 31 for vehicle, 29 for Aβ treatment. Unpaired student t-test. ***p < 0.001. G) Representative images of ATP staining. Magnification at 400x. Scale bar = 20μm (inset, scale bar = 5μm). H) Serotonergic neuron ATP content measurement in both vehicle and 500 nM Aβ treatment groups. n = 3 each group. Unpaired student t-test. **p < 0.01. I-K) Serotonergic neuron mitochondria labeled with MitoTracker fluorescent dye in vehicle or 500 nM Aβ treated neurons. Mitochondria density (I) and volume (J) in serotonergic neurons were analyzed using NIS elements software. n = 13 for vehicle, 15 for Aβ1–42 in mitochondria density analysis; n = 200 for vehicle, 119 for Aβ1–42 in mitochondria volume analysis. Unpaired student t-test. **p < 0.01, ***p < 0.001. K) Representative images of MitoTracker labeling. Magnification at 400x. Scale bar = 4μm.
Hippocampal serotonin release is compromised by disrupted mitochondrial bioenergetics
If mitochondrial fitness is crucial for serotonin release, we would expect to see that disruption of mitochondrial bioenergetics has a deleterious influence on serotonin release. To this end, we exposed hippocampal slices from nonTg mice to carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), a mitochondrial oxidative phosphorylation (OXPHOS) uncoupler [55], and performed electrochemical assays for hippocampal serotonin release. As expected, in comparison with the baseline, FCCP-insulted hippocampal slices displayed decreased serotonin release in response to electrical stimulation (Fig. 4 A, B), supporting a link between mitochondrial deficits and serotonin dysregulation.
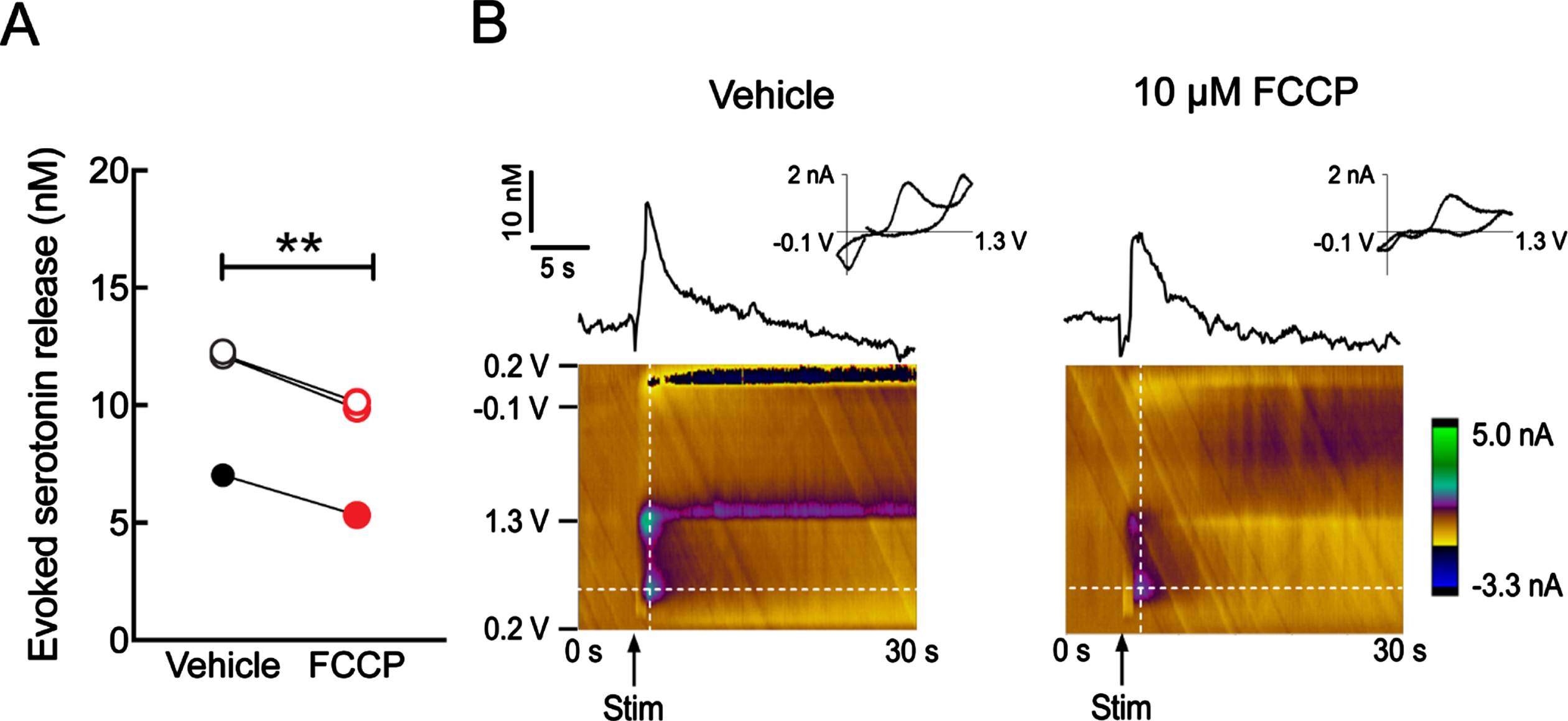
Reduced hippocampal serotonin release with FCCP treatment. A) Voltammetric determination of the impact of mitochondrial deficits on hippocampal serotonin release. NonTg mouse brain slices were perfused with 10μM FCCP for 20 minutes to examine the influence on serotonin release in the hippocampal CA1 SR region. n = 3. Females: open circles, males: filled circles. Paired student t-test. **p < 0.01. B) Representative voltage versus time (top) and color plots (bottom) for serotonin release measurement.
DISCUSSION
Although the precise mechanisms of the etiopathogenesis of AD remain elusive, neurotransmitter perturbations in AD-sensitive brain regions including the hippocampus have been proposed to contribute to the development of this devastating neurodegenerative disorder [2, 56–58]. Consistent with the importance of serotonergic signaling to the modulation of hippocampus-related reward behavior [59], mood and anxiety [60, 61], learning and memory [62–64] as well as hippocampal neurogenesis [65], serotonin-enhancing and serotonin receptor-modulating agents have demonstrated their therapeutic potential in the prevention and treatment of AD [17–23]. However, the complex functional responses of various serotonin receptors and their differential vulnerability in AD [66–70] have complicated the therapeutic effects of SSRIs and rendered difficulties in targeting multiple subtypes of serotonin receptors for the treatment of this neurological disorder. In addition, the anticholinergic properties of several SSRIs such as paroxetine require cotreatment with cholinesterase inhibitors otherwise they may induce worsened cognition in patients [71–73]. In this context, preservation of serotonin metabolism as well as the release and recycling of this neurotransmitter has its unique advantage in preventing the development of serotonin failure and addressing neuropsychiatric symptoms due to serotonin deficiency in patients with AD. In this study, we have established a link between mitochondrial dysfunction and impairments in serotonin release in AD-relevant pathological settings. Previous studies predominantly focused on AD-associated mitochondrial changes in pyramidal neurons in the neocortex and hippocampus [74–77]. Current extension of mitochondrial dysfunction to serotonergic neurons supports mitochondrial vulnerability in multiple neuron types in AD and further strengthens the mitochondrial cascade hypothesis of this disease [78]. To this end, these findings implicate a therapeutic potential of mitochondria-boosting agents for the mitigation of serotonergic failure in AD-related conditions and warrant further in-depth investigation in our future studies.
Of note, we have observed decreased serotonergic fibers in the hippocampi of 5xFAD mice, suggesting degeneration of serotonergic neurons, corroborating previous neuropathological and neuroimaging studies on AD patients [9, 31–33]. However, a previous study showed increased hippocampal serotonergic fibers in a triple transgenic mouse model of AD [79]. Such a discrepancy in hippocampal serotonergic fibers may arise from the difference in the severity of brain damage between the two types of mouse models as 5xFAD mice demonstrate both extracellular and intraneuronal Aβ deposition that is more pathologically relevant to brain amyloidopathy in AD patients [40]. In contrast to decreased hippocampal serotonergic fiber density in 5xFAD mice, we did not observe any change in the density of serotonergic fibers in oligomeric Aβ-challenged hippocampal slices. These findings indicate the deleterious effect of chronic Aβ toxicity on the structural integrity of serotonergic neurons and further support a direct impact of Aβ on serotonin release regardless of the changes in serotonergic fiber density. Indeed, early brainstem lesions including the atrophy of the raphe nuclei have been increasingly reported in AD [80, 81], which promotes the appraisal of the indicative capacity of neuropsychological signs in predicting AD onset. A relationship between AD and depression is implicated by the overlapping neurodegenerative changes between the two conditions. Furthermore, there is an increased risk for older individuals with depression to develop AD [82, 83]. So far, the molecular mechanisms of the development of depression symptoms in AD remain elusive. Serotonin insufficiency in AD and depression [82, 83] and the inhibitory effect of serotonin-promoting drugs against Aβ production [17] appear to underpin a critical role of serotoninergic failure in the development of AD. In view of the importance of mitochondrial function to neurophysiology [35] and the observed mitochondrial dysfunction in Aβ-challenged serotonergic fibers, we cannot ignore the possibility that mitochondrial defects not only contribute to compromised serotonin release but are also involved in the degeneration of the raphe nuclei in AD-related conditions. It should be noted that only moderate changes in serotonergic fibers and serotonin content were detected in 5xFAD mice at the tested age. This could result from the lack of tauopathy in 5xFAD mice and thereby do not model the neurofibrillary tangles that are found in degenerative serotonergic neurons in AD patients [31]. We will employ animal models demonstrating both amyloidosis and tauopathy in our future mechanistic studies on mitochondrial dysfunction and serotonergic degeneration in AD-relevant pathological settings.
Another critical question that merits discussion is the cause of impaired serotonin release in AD-related conditions. Although the precise mechanisms of serotonin release have not been fully described, it is a well-documented notion that adequate provisions of ATP are prerequisite for the integral steps in serotonin release including the vesicular monoamine transporter (VMAT)-mediated packaging of serotonin into secretory vesicles and kinesin-mediated synaptic vesicle transport [84–87]. Therefore, there is little doubt that reduced ATP production due to mitochondrial dysfunction contributes to impaired serotonin release, which is supported by our mitochondrial uncoupling experiments. However, deficits in kinesins have been repeatedly identified in AD-related conditions [88–91]. Although there is a lack of information on the functional status of kinesin-related motor proteins in serotonergic neurons in AD, the decreased mitochondrial distribution in hippocampal serotonergic fibers in 5xFAD mice serves as evidence of impaired axonal transport. Therefore, it cannot be excluded that decreased serotonin release also results from defects in kinesin-related motor proteins in serotonergic neurons in AD-related conditions. Furthermore, APOE4, a genetic risk for AD [92], inhibits VMAT2 [93], a subtype of VMAT responsible for serotonin packing into secretory vesicles [94], implicating an association of VMAT dysregulation with AD. Moreover, serotonin receptors, 5-HT1A, 5-HT1B, and 5-HT1D on the presynaptic terminals of serotonergic neurons modulate serotonin release [95, 96]. We therefore cannot exclude the impact of functional alterations of these serotonin receptors on serotonin release in AD-related conditions. In this context, the contribution of factors other than suppressed mitochondrial bioenergetics to the development of serotonin failure in AD should not be neglected.
Lastly, a prominent change of mitochondria in hippocampal serotonergic fibers in 5xFAD mice is decreased volumes, suggesting defects in mitochondrial morphological control towards fragmentation. These findings agree with the observation of increased neuronal mitochondrial fission in AD patients and animal models [97, 98]. However, previous studies found a paradoxical increase in the volume of fragmented mitochondria in the soma of AD hippocampal neurons, probably due to disrupted cristae organization [97, 99]. A possible explanation of such a discrepancy in the size of fragmented mitochondria in the soma and axon is that axonal mitochondria with severe morphological and functional defects in whole or in part by fission return to the soma via retrograde axonal transport for mitochondrial repair and degradation. Indeed, neuronal mitochondrial redistribution from the axons to the soma has been detected in AD brains [99], and many previous studies including ours consistently report reduced axonal mitochondrial density and decreased axonal mitochondrial volume in Aβ-rich milieus [38, 101]. Therefore, it would be of great interest to determine whether the mitochondria in the serotonergic neurons also exhibit altered cristae architecture and redistribution in AD-related conditions in our future study. The answer to this question will help to fully depict mitochondrial damage in serotonergic neurons in AD.
In summary, in this study, we found impaired hippocampal serotonin release in AD-related conditions. Although the detailed mechanisms causing impairment in serotonin release in AD are not fully elucidated, our observations suggest that mitochondrial dysfunction contributes to the development of disrupted serotonin transmission in this neurodegenerative disorder. Several limitations of the current study should be mentioned. It remains unclear whether mitochondrial dysfunction plays a major role in driving serotonin release defects in AD-related conditions. In addition, serotonin has a positive impact on mitochondrial biogenesis and bioenergetics [102]. It is possible that diminished serotonin levels not only affect mitochondrial regulation in non-serotonergic neurons but also impose an adverse impact on mitochondria in serotonergic fibers, which forms a vicious cycle, leading to exaggerated serotonin failure. Lastly, whether hippocampal serotonin release defects have a temporal relationship with brain amyloidosis and develop early in 5xFAD mice remains unclear. These outstanding questions form the groundwork for our future investigation. Regardless of these limitations, this proof-of-concept study provides a novel mechanism of serotonin failure in AD and offers therapeutic opportunities by targeting mitochondrial dysfunction in serotonergic neurons for the treatment of AD. Moreover, further investigation on mitochondrial dysfunction in serotonergic neurons will add support for the mitochondrial hypothesis of AD for a better understanding of the pathogenesis of this neurological disease.
FUNDING
This work was supported by research fundings from NIH (R01AG053588, R01AG059753 and R01AG075108 to HD), Higuchi Biosciences Center research grant to HD, Brightfocus Foundation research grant A20201159S to HD, NIH P30 AG072973 to the University of Kansas Alzheimer’s Disease Research Center’s Research Education Component, and REC Fellowship to JT.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
Data sharing is not applicable to this article as no datasets were generated or analyzed during this study.