
Abstract
Alzheimer’s disease (AD) is the most common form of dementia in the elderly. AD is a multifactorial disease, affected by several factors including amyloid-β42 oligomers, self-assembled tau, microbiota molecules, etc. However, inflammatory components are critical to trigger AD. Neuroinflammatory pathology links glial activation by “damage signals” with tau hyperphosphorylation, as explained by the Neuroimmunomodulation Theory, discovered by the ICC laboratory. This theory elucidates the onset and progression of several degenerative diseases and concept of “multitarget” therapy. These studies led to the rationale to identify inflammatory targets for the action of bioactive molecules or drugs against AD.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is the most prevalent cause of dementia in people older than 65 [1]. One of the major drawbacks of AD is currently the lack of an effective therapy. During the last several years, much effort has been made to develop a therapy to control the cognitive decline associated with AD. The principal target of this therapy has been amyloid-β (Aβ), which is now, due to the evidence provided, failing due to the lack of results and misinformation [2]. Furthermore, it is now demonstrated that senile plaques have no influence on cognitive decline as no correlation was found [3, 4]. However, soluble Aβ1 - 42 oligomers can act as an activator of microglia [5, 6], as a danger signal to promote an inflammatory microenvironment, similar to advanced end glycan products (AGEs) [7].
This sheds light on the tau theory. Hyperphosphorylation of tau leads to conformational changes that are directly related to cognitive decline. The cognitive decline, at the same time, is associated with neurodegeneration related to neuroinflammatory processes mediated by danger signals [8–12]. This connection of glial activation with the inflammatory cascade has been known as The Neuromodulation Theory and is the basis for the search of new therapeutic components for AD (see Fig. 1).
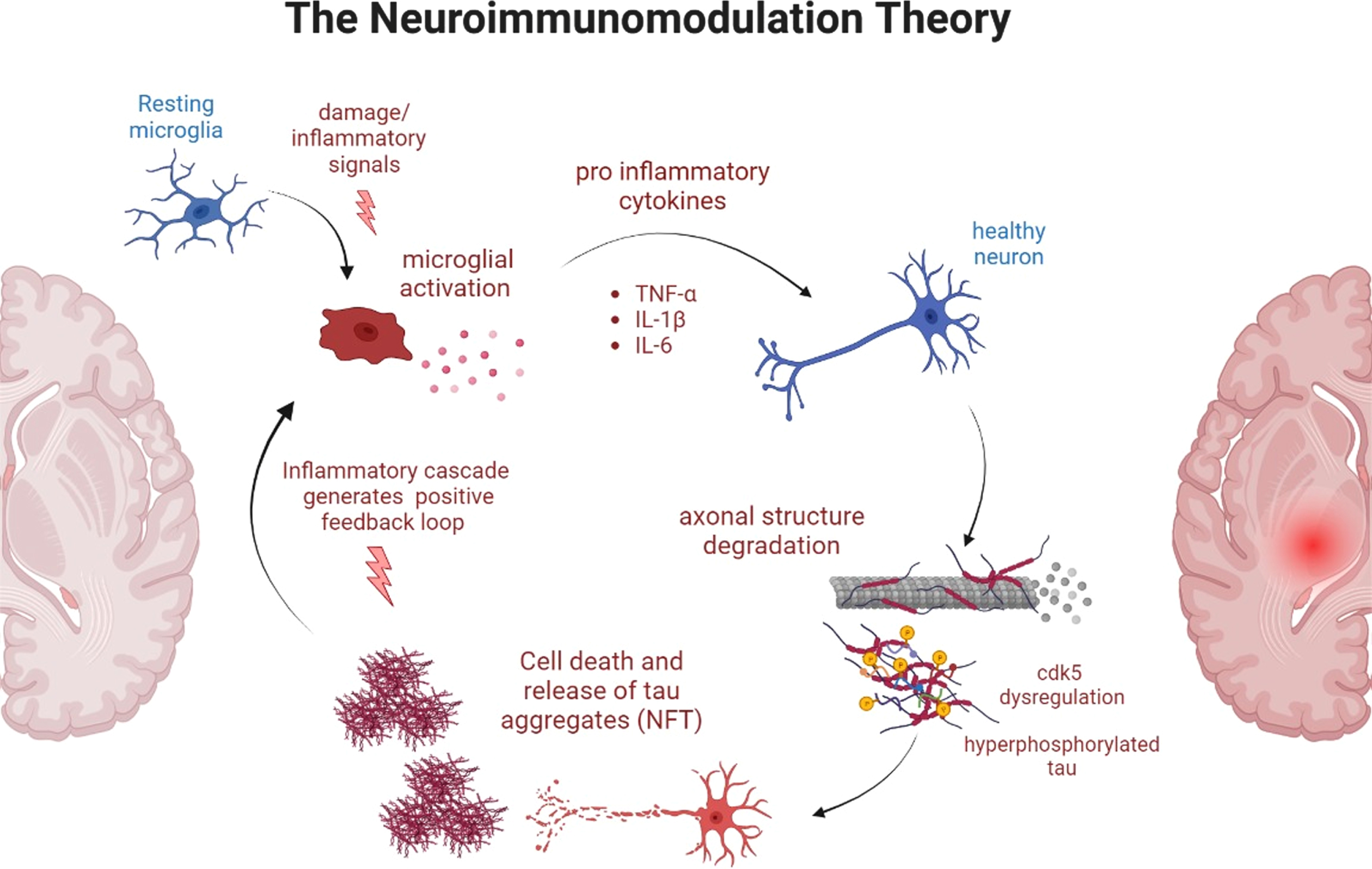
Representative diagram of the Neuroimmunomodulation Theory. In this unifying theory, damage signals (such as AGEs, fragments of hyperphosphorylated tau, oxidative radical, soluble Aβ peptides, etc.) activate the resting microglia in the brain, which activates several inflammatory signaling cascades that leads to the production of pro-inflammatory cytokines. In a healthy neuron, this triggers a dysregulation of CDK5 kinase which in turn hyperphosphorylates tau and this tau, after the neurodegeneration takes place, acts as a damage signal in other microglia in a positive feedback loop.
THE NEUROIMMUNOMODULATION THEORY: A BRIDGE INTEGRATING THE SEVERAL FACTORS OF AD
The unifying neuroimmunomodulation theory represents the common point between the several etiological origins of AD, hyperphosphorylated tau generating neurofibrillary tangles and soluble Aβ peptide just to mention a few (see Fig. 1). According to this theory, proinflammatory cytokines are released after the activation of the microglia due to damage signals (e.g., soluble Aβ, paired helical filament fragments, oxidative molecules, etc.) which in turn activates signaling cascades that leads to hyperphosphorylation of tau in a cyclic process [13, 14]. Thus, tau neurofibrillary tangles activate the microglia in a positive feedback process (see Fig. 1).
This theory is one of the most important milestones in AD research, providing a novel view in the field, which up until recently was based mostly on Aβ peptide misfolding and senile plaque appearance. Contrarily, it has been demonstrated that senile plaques are also present in the cognitively normal elderly population.
Another connecting point between the neuroimmunomodulation theory and the etiopathology of AD is related to the glucose metabolism impairment observed in AD patients. It has been observed that advanced glycation end products (AGEs) can act as danger signals that activate microglia [15]. Additionally, uncontrolled diabetes leads to a high production of AGEs, which might be one of the etiological origins of AD [16]. Furthermore, AD due to glucose metabolism impairment could now be considered as diabetes type 3 [17].
POTENTIAL BENEFITS OF THE APPLICATION OF THIS THEORY IN THE CLINIC
An additional benefit from this neuroimmunomodulation theory is that it provides a solid basis for the generation of multitarget bioactive compounds beneficial to treat AD, and also early-detection biomarkers.
Multitarget bioactive compounds
As stated in the neuroimmunomodulation theory, the neuroinflammatory processes are key in the generation and progression of AD, and the sources of the inflammatory signals are the ones that should be controlled to ameliorate the neuroinflammation and consequently, neuroinflammation and neurodegeneration. As an example, several studies have demonstrated the effects of nutraceutical compounds on AD [18, 19]. The common mechanism of action is the anti-inflammatory properties, but also other beneficial properties such as: anti-tau aggregates, antioxidants, and gut-microbiome modifying compounds, just to mention a few [20–22]. In that regard, it is worth pointing out Andean shilajit has anti-inflammatory properties, along with anti-tau aggregation properties [23, 24]. A novel formula that includes Andean shilajit and vitamin B complex has proven to ameliorate neuropsychiatric symptoms associated with AD [25].
Early detection biomarkers
One of the major modifications of the neuroimmunomodulation theory is that it opens novel approaches since it is based on danger signals that activates microglia. As an example, oligomeric tau is employed on a novel blood-based biomarker, the Alz-Tau® [26], which has been validated by five clinical trials. Why is tau currently rising as a potential biomarker? Because tau has one of the best correlations with cognitive decline. Furthermore, it was demonstrated that platelet tau (the basis for the Alz-Tau® biomarker) correlates with brain atrophy [27]. Gold standards, such as cerebrospinal fluid measures and PET scans, also have modified the measurements to evaluate tau for the same reason.
BRAIN HOMEOSTASIS AND THE BLOOD-BRAIN BARRIER
Immunological changes in AD are not confined to the central nervous system, and AD-associated systemic immune changes appear to affect brain homeostasis. In AD, the permeability of the blood-brain barrier (BBB) is increased due to the decreased expression of the tight junction molecules in vascular endothelial cells [28]. Moreover, an increase of adhesion molecules (ICAM-1, VCAM-1) is observed in the AD BBB endothelium, potentially contributing to immune cell migration [29]. Emerging evidence supports that T and B cell infiltration into the brain exacerbates neuroinflammation and accelerates neuronal death. Although the significance of T cells in AD pathogenesis is still debated, elevated peripheral Th1 and Th17 cells have been associated with the release of proinflammatory molecules [28]. Higher levels of cytokines (IL-6 and IL-1b), and chemokines (CCL5, CXCL9) have been detected in the AD periphery and brain [29]. CD8+ and CD4+ T cells also have been found in the brain parenchyma of AD patients which could contribute to AD progression by acting on neurons and functional properties of microglia through the release of proinflammatory factors [30]. Besides, B cell infiltration seems to contribute to immunoglobulin deposits [31], acting also as a modulator of neuroinflammation; hence the importance of research into key players of neuroimmunomodulation to develop therapeutic interventions in AD.
DISCUSSION
In this article, our aim was to shed light to some of the etiological origins of AD and how the neuroimmunomodulation theory is a bridge integrating them (see Fig. 2).

Alzheimer’s disease as multifactorial disease. One single factor is not sufficient to trigger Alzheimer’s disease. All the etiopathological hallmarks converge in the neuroinflammatory process, direct or indirectly, and continue to exacerbate the pathology due to the cyclic events taking place.
AD has long gone from the molecular hallmarks, in particular with the Aβ hypothesis. However, tau is also a molecular hallmark, and it has a better correlation with cognitive decline and clinical observations.
AD is a multifactorial disease and should be considered as such. The latter is one of the main reasons why current therapeutic approaches have been unsuccessful, as the majority have been directed against Aβ. The neuroimmunomodulation theory includes the majority (if not all) of the etiological origins of AD that lead to a common path: neuroinflammation triggered by activation of microglia and also astrocytes. The latter, consequently, leads to neurodegeneration that becomes the onset of AD.
Thus, all these multifactorial components converge at some point in which a neuroinflammatory microenvironment is triggered. Thus, all the molecular hallmarks, such as soluble Aβ, tau, or AGEs should be considered as danger signals, independently of their correlation with the cognitive decline. However, the correlation with cognitive decline is key to choose a proper target in the search of an early detection biomarker and effective potential therapies. And until now, tau is the one best candidates in that regard. Could there be other targets? No doubt, but by aiming at multiple targets, the best results can be obtained to develop an effective therapy. The multitarget therapy is currently one of the best options in AD.
CONCLUSION
AD is a multifactorial disease with a complex etiopathogenesis. The amyloid theory alone is not sufficient to explain the physiopathology. In that regard, the neuroimmunomodulation theory converges several etiological origins of AD, such as amyloid and tau aggregation, glucose metabolism impairment, etc., by stating that the main onset of AD is the neuroinflammatory process that takes place after the chronic activation of the microglia. All the etiological components are now part of this theory as damage signals that activate the microglia, which is the starting point for the neuroinflammatory processes that eventually lead to neurodegeneration. Those danger signals might be a useful tool as an early detection biomarker, but the correlation with cognitive decline should be considered first.
In that regard, potential therapies should now focus on this novel approach as the multitarget therapy, currently one of the best options for AD patients.
Footnotes
ACKNOWLEDGMENTS
This paper is in homage to Prof. Ricardo Maccioni, MD, PhD for his outstanding contributions to elucidate the causes of Alzheimer’s disease, and the paradigm of neuroinflammation, a major breakthrough in Alzheimer’s disease research.
We thank the Ricardo B. Maccioni Foundation and the International Center for Biomedicine for their support of this work.
FUNDING
The research done for this paper was supported by the Ricardo B. Maccioni Foundation and by a Corfo grant 21CYEM-199974.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
Data sharing is not applicable to this article as no datasets were generated or analyzed during this study.