
Abstract
Alzheimer’s disease (AD) is the most common form of dementia and a public health problem. It exhibits significant oxidative stress and redox alterations. The antioxidant enzyme systems defend the cellular environment from oxidative stress. One of the redox systems is the thioredoxin system (TS), which exerts decisive control over the cellular redox environment. We aimed to review the protective effects of TS, which include thioredoxin (Trx), thioredoxin reductase (TrxR), and NADPH. In the following, we discussed the physiological functioning and the role of the TS in maintaining the cellular redox-homeostasis in the AD-damaged brain. Trx protects the cellular environment from oxidative stress, while TrxR is crucial for the cellular detoxification of reactive oxygen species in the brain. However, TS dysregulation increases the susceptibility to cellular death. The changes in Trx and TrxR levels are significantly associated with AD progression. Though the data from human, animal, and cellular models support the neuroprotective role of TS in the brain of AD patients, the translational potential of these findings to clinical settings is not yet applied. This review summarizes the current knowledge on the emerging role of the TrxR-Trx system in AD.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a common and heterogeneous neurodegenerative dementia, which is associated with memory impairment and deterioration of cognitive functions [1–3]. AD accounts for 60% to 80% of the total 50 million dementia patients globally [4]. Although the molecular etiology of AD is still elusive, amyloid-β (Aβ) plaques and neurofibrillary tangles (NFTs) are considered the molecular hallmarks of AD [3, 5]. In addition to memory impairment and cognitive decline, AD is characterized by neuroinflammation, synaptic dysfunction, and progressive neuronal loss in key brain regions, such as the hippocampus and cortex. These pathophysiological changes lead to the clinical symptoms of dementia and ultimately, a profound impact on patients’ quality of life. However, aging remains the major pathological risk factor for AD [6, 7]. Oxidative stress (OS) is a major contributor to the development and progression of AD. In particular, an imbalance between the production of reactive oxygen species (ROS) and the cellular antioxidant defense (AOD) mechanisms leads to oxidative damage to biomolecules such as lipids, proteins, and DNA [8]. This process can disrupt cellular homeostasis and contribute to neurodegeneration in the brain of AD patients. The thioredoxin (Trx) is an antioxidant protein that plays a significant role as a crucial regulator of the cellular redox-homeostasis environment in AD. Therefore, this review provides a comprehensive overview of the role of the Trx and thioredoxin reductase (TrxR) antioxidant enzymes in AD. Thus, examined the evidence for the protective effects of Trx and TrxR in maintaining cellular redox balance in the AD-affected brain. Understanding the role of thioredoxin system (TS) in AD pathogenesis may have implications for the development of new therapeutic strategies to prevent or slow the progression of the disease.
THE ROLE OF OXIDATIVE STRESS IN THE PATHOGENESIS OF AD
OS has a detrimental role and significant contributing factor in the etiology and pathogenesis of AD. The imbalance in the cell redox homeostasis, systematic ROS production, and impaired AOD lead to OS [9]. These oxidative damages play a pivotal role in cellular dysfunction, including neurons, which has been seen and observed in aging and neurodegenerative disorders, including AD. It has been shown that OS and free radical damage correlated with histopathological hallmarks of AD, i.e., Aβ and NFTs [10, 11]. Amyloid plaques are formed by the accumulation of Aβ protein, which is believed harm neurons. OS can increase the expression of amyloid-β protein precursor (AβPP) which induces abnormal Aβ deposition, leading to the formation of amyloid plaques [12]. Furthermore, OS can cause damage to the mitochondrial membrane, resulting in the accumulation of amyloid peptide oligomers, which further contribute to its progression [11]. NFTs are formed by the abnormal accumulation of tau protein, which leads to the disruption of the microtubule network that supports neuronal structure and function. OS can contribute to the hyperphosphorylation of tau protein [13], leading to its abnormal aggregation into NFTs. Apart from OS, microglial cells also induce the production of ROS in neurons, aggravating tau protein causes synaptic loss [14]. Microglial cells are responsible for removing cellular debris, dead cells, and pathogens. In AD pathogenesis, microglial cells become activate, which is followed by increasing production of ROS, as a response to the presence of abnormal protein aggregates, such as tau protein [15]. Produced by activated microglial cells, ROS can lead oxidative damage to nearby neurons, including those containing tau protein, promoting the formation of NFTs, which are a hallmark of AD. In addition, synaptic loss can occur as a result of this process, which further contributes to the cognitive decline seen in AD [16].
Although ROS production occurs normally during oxidative metabolism in mitochondria-containing cells, but to a lower extent, imbalanced, i.e., overwhelmed ROS production that exceeds the capacity of AOD systems leads to OS [9]. In the brain, there is an abundant lipid content, high rate of oxidative metabolic activity, high-energy consumption, and the intense chemically diverse ROS production causes OS, increasing susceptibility to the brain and its functional integrity by changing the neural redox balance [17]. Moreover, the brain tissues have a low cell-repairing capacity and relatively low levels of antioxidants. Thus, OS and mitochondrial dysfunction adversely affect the brain and significantly lead to aging and neurodegenerative disorder, in particular AD. The brain tissues are vulnerable to oxidatively damaged nucleic acid, protein, carbohydrate, or lipids are detected in the pathogenesis and development of AD [18–21]. In abundant oxygen concentration, the ROS leaking from the electron transport chain within mitochondria occurs, which is associated with disease-dependent mitochondrial dysfunction disrupted metal homeostasis and diminished AOD mechanisms that ameliorate the risk of AD [20, 22]. Therefore, OS is the common pathological feature that contributes to the mitochondrial dysfunction and pathogenesis of AD.
ANTIOXIDANT ENZYMES AND AD
In normal cellular processes, balanced cellular redox homeostasis is crucial for normal physiological functioning and intra- or extracellular signaling. In neurodegenerative diseases, including AD, the increase in OS induces neural loss and synaptic dysfunctions [23]. The cells deploy antioxidant mechanisms to scavenge the cytotoxicity mediated by ROS provide protection against OS-induced cell damage. In normal cellular settings, redox enzymes protect the cellular environment from OS. The cellular defense mechanism against OS includes a network of antioxidant enzymes and antioxidants, which delay or inhibit the oxidation of biomolecules, thus, protect cells against cytotoxicity mediated by ROS, maintaining intracellular redox status [24]. The brain is rich in lipids, with the fact that certain regions of the brain are particularly sensitive to oxidative lipid damage, i.e., lipid peroxidation, and contain more AOD enzymes, such as catalase, glutathione peroxidase, and glutathione reductase [25, 26], i.e., substantia nigra have high lipid peroxide levels [27–29]. Differential lipid peroxidation expressed throughout different brain regions exert vulnerability to the negative consequences of free radical production [30]. Therefore, understanding the role of antioxidant enzymes, glutathione reductase, glutathione peroxidase, TrxR, catalase, peroxiredoxins, etc., in AD and maintaining balanced cellular redox homeostasis is crucial.
Moreover, increased immunoreactivity of NFTs for antioxidant enzymes is associated with the neuropathologic lesions of AD. OS may lead to the abnormal phosphorylation and accumulation of tau protein, which then forms NFTs. The presence of antioxidant enzymes in these tangles could indicate a protective response of the neurons to the OS and the accumulation of toxic protein aggregates. For instance, superoxide dismutase-1 was found to be increased in NFT vulnerable neurons [31], such as the hippocampus, neocortex, cerebral hemispheres, and brain stem. In addition, heme oxygenase-1, which is associated with mitochondrial breakdown in AD, is also increased in vulnerable neurons [25]. The increased expression of antioxidant enzymes in AD, including superoxide dismutase-1, heme oxygenase-1, glutathione peroxidase, and catalase, reflects the activation of the cellular AOD. These antioxidant enzymes play a crucial role in protecting neurons from OS-induced damage by neutralizing free radicals and ROS that can damage cellular macromolecules like lipids, proteins, and DNA. Thus, oxidative defense in the brain is essential for the brain to perform normal tasks.
Vitagenes role in AD
Neurodegenerative and metabolic disorders are characterized by a decline in biosynthetic and repair activities, leading to a compromised proteome integrity [32]. However, the presence of protective gene known as vitagenes, which regulate the aging process, plays a significant role in influencing the link between stress, protein homeostasis, and lifespan of an organism [33]. The maintenance of optimal long-term health conditions relies on a complex network of longevity assurance processes, which are regulated by vitagenes [34]. Vitagenes encompass a range of proteins, including heat shock proteins, Trx/TrxR, sirtuins (Sirt), and uncoupling proteins, which are significant components of vitagene-mediated defense mechanisms. These vitagene products are key players in cellular pathways associated with programmed cell death, providing protection against OS [35]. Vitagene family members also counteract the neurotoxic effects of nitric oxide and reactive nitrogen species [36]. Furthermore, Cornelius et al. [37] reported significantly higher levels of Sirt-1 and Sirt-2 in AD lymphocytes, accompanied by increased expression of Trx and uncoupling proteins in AD patients. These elevated protein expressions appear to be a consequence of an intense oxidant environment, which may have implications for the pathogenesis of AD [37].
THIOREDOXIN SYSTEM
TS, a cellular disulfide reductase system, includes two antioxidant oxidoreductases: Trx and TrxR, a coenzyme electron donor: α NADPH, and a Trx-interacting protein (TXNIP) [38, 39]. TS plays a crucial role in the regulation of redox signaling pathways and maintaining cellular redox balance. TS has thiol-disulfide oxidoreductase activity and it regulates the dithiol/disulfide balance by providing electrons to thiol-dependent peroxidases to remove ROS [38]. The dithiol/disulfide balance is a critical component of cellular redox homeostasis. Various enzymes and proteins, including the TS, tightly regulate the balance between reduced (thiol) and oxidized (disulfide) forms of thiol-containing biomolecules [40]. The proper maintenance of this balance is crucial for redox signaling pathways that play a significant role in various cellular processes, including cell differentiation, proliferation, and survival [41]. The TS plays a critical role in maintaining a reducing environment in mammalian cells by transferring electrons and reducing target proteins through highly conserved thiol groups, from nicotinamide adenine dinucleotide phosphate through TrxR to Trx [17]. However, an imbalance in the thiol/disulfide balance can lead to increased ROS levels, resulting in OS and damage to cellular components [42]. TS acts as a reducing agent by donating electrons to other proteins, thereby reducing disulfide bonds and maintaining the thiol/disulfide balance. This process helps to regulate the activity of various enzymes involved in redox signaling pathways and protects against OS-induced damage [43]. Furthermore, the Trx system is involved in various biological processes, including DNA synthesis, protein folding, and redox signaling. The system is also involved in the regulation of various cellular processes such as apoptosis, proliferation, and differentiation [44].
The TS is an evolutionarily conserved system found in all living cells due to its antioxidant activity and fundamental role in cellular redox homeostasis. The evolution of the TS system has been extensively studied, and it has been shown to have undergone several changes throughout evolution [45]. For example, the number of Trx genes and their isoforms vary in different organisms, and the expression patterns of Trx genes can be regulated in response to different stimuli [46, 47]. These evolutionary changes may have implications for studying the TS system in different organisms, as they may affect the activity and specificity of the system. For example, the TS in plants has been found to play a crucial role in protecting against OS caused by environmental factors such as high light intensity and pollutants [46, 48, 49]. In recent years, the role of TS has been well studied in various diseases, such as cancers [39], aging [50], and neurodegenerative disorders, particularly AD [51]. In cancer, TS has been shown to have a crucial role in regulating cell proliferation, survival, and metastasis, making it a potential target for cancer therapy [52]. In aging, the TS has been found to play a role in protecting against age-related diseases by maintaining cellular redox homeostasis [53]. In neurodegenerative disorders, particularly AD, TS has been implicated in the pathology of the disease. Studies have shown that TS activity is decreased in the brains of AD patients, leading to increased OS and neuronal damage [54]. Studying the TS in different organisms can also reveal differences in its regulation and function. For example, in some bacteria, TS has been found to play a role in regulating the cell cycle and DNA repair [55]. Understanding these differences can provide important insights into the diverse functions of the TS and its potential for therapeutic targeting in various diseases.
Thioredoxin
Trx (ubiquitous, small reductase, and oxidoreductase) are endogenous antioxidants, small proteins with a catalytic cysteine residue [56] (Fig. 1, Table 1). It is an essential cellular redox homeostasis regulator, characterized by Laurent et al. [57] in Escherichia coli, and later on all organisms, ranging from microbes to Homo sapiens. Trx was identified as reducing the substrate of ribonucleotide reductase, an enzyme required for DNA synthesis [57] and then some peroxiredoxins, molecules for peroxides reduction [58]. Two types of Trx exist, cytoplasmatic Trx-1 and mitochondrial Trx-2. Different subcellular localization, allow each Trx to perform specific functions in the respective space within the cell. Though they share same conservation of specific amino acids, Trx-2 has a few additional amino acid residues compared to Trx-1. These structural differences can result in different properties and interactions with other molecules or proteins. Despite the differences, both Trx-1 and Trx-2 have a similar function in maintaining the redox balance and participate in numerous biological processes such as redox control, cell growth and proliferation, and stress response. Trx-1 and Trx-2 are substrates for their respective TrxR, TrxR1 and TrxR2, operating in the cytoplasm and mitochondria, respectively. Antioxidant role of Trx refers to scavenging of ROS, a reducing protein disulfide bonds, and providing an electron reducing equivalents to peroxiredoxin, which reduces hydrogen peroxide into water. Trx reduces ROS-induced cell damage during mitochondrial respiration by reducing oxidized forms of proteins [59]. Previous studies suggest the protective role of Trx within the cellular environment in many conditions, including cerebral ischemia [60], cancer [39], cardiovascular disorders [61], endoplasmic reticulum stress, and mitochondria-mediated neuronal apoptosis [62], as well as neurodegenerative disorder (AD and Parkinson’s disease) [63, 64]. Trx exerts neuroprotective effects by inhibiting endoplasmic reticulum stress-mediated neural apoptosis.
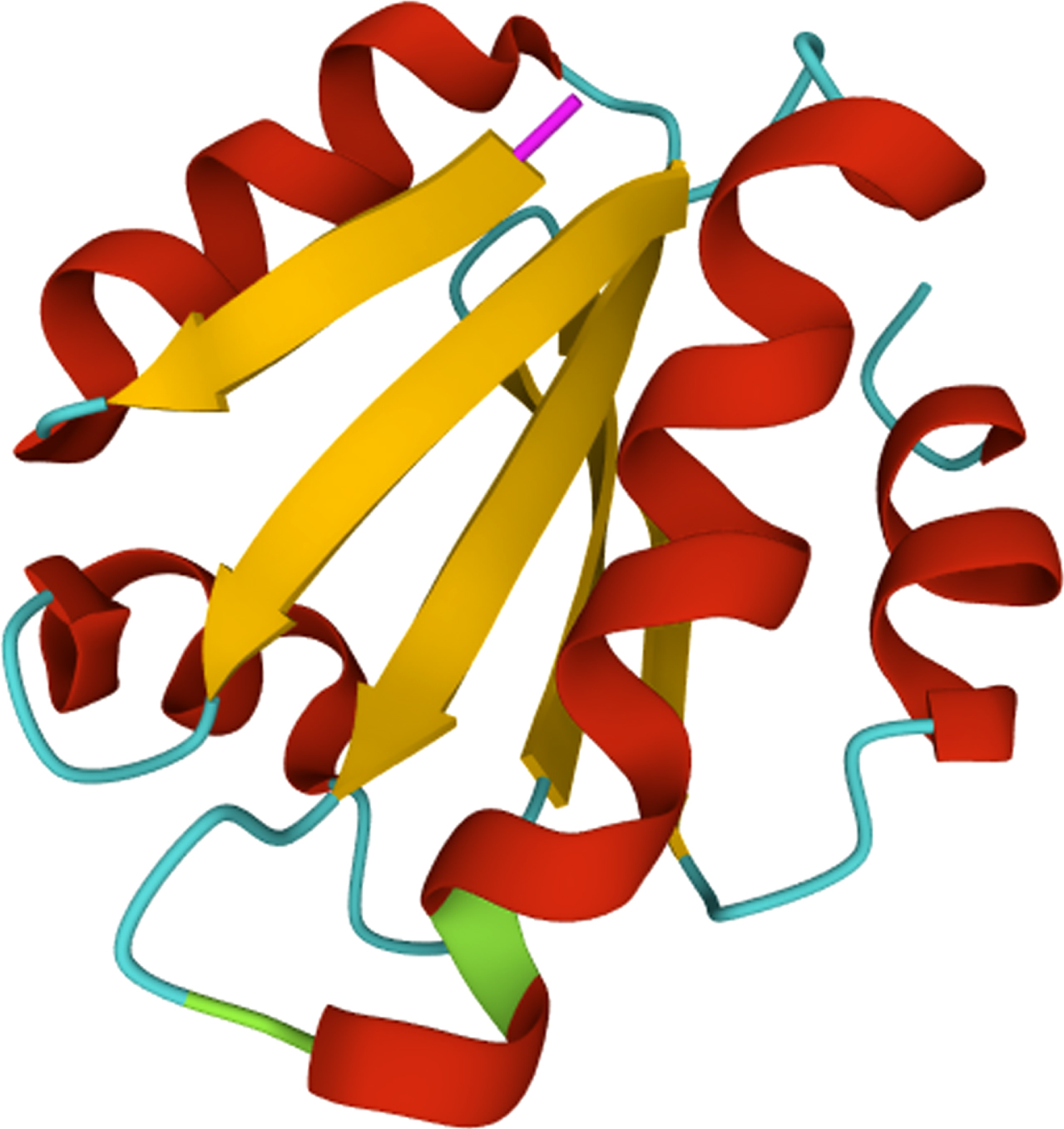
The high resolution 3D structure of human thioredoxin (PDB: 1TRW) [100]. Red color represents α-helix; yellow color represents β-sheets; green color represents actives sites; whereas, pink color represents the start codon, i.e. initiator methionine.
Thioredoxin reductase
In the TS, Trx, TrxR, and NADPH form reduced disulfide bonds in the cells. TrxR is an antioxidant enzyme, a selenoprotein essential for cellular defense against ROS in the brain. Utilizing NADPH as a donor of reducing equivalents (electron plus proton), it catalyzes the reduction of disulfide, oxidized form of Trx (Trx-S2), to reduced, sulfhydryl form Trx-(SH)2 [60, 65], first identified in bovine tissue by Holmgren and Luthman [66]. It requires a selenocysteine residue to reduce the active site disulfide of Trx (Fig. 1). TrxR is a substrate-specific enzyme [67]. TrxR, as the only Trx reducing enzyme, catalyze the reaction very efficiently to maintain the cellular redox milieu.
Isoforms of thioredoxin reductase
Three isoforms of TrxR have been identified so far, including cytosolic TrxR1, mitochondrial TrxR2, and testicular TrxR3 [44, 56]. Evidently, they have distinct tissue distribution, localization, and functions. Thioredoxin reductase-1 is the most extensively studied isoform and is widely expressed in various tissues, including the brain [68]. It plays a critical role in maintaining cellular redox homeostasis by reducing Trx1, which is involved in multiple cellular processes, including cell proliferation, differentiation, and survival [69]. Thioredoxin reductase-2 is localized in mitochondria and participates in the AOD of the organelle. It reduces Trx2, which is essential for maintaining mitochondrial redox balance and regulating mitochondrial metabolism [70]. TrxR2 has been shown to play a protective role in mitochondrial dysfunction and OS-induced apoptosis [71]. Therefore, TrxR1 and TrxR2 are essential for maintaining brain activity (Table 1). In addition, TrxR3 is a unique isoform that is specifically expressed in testis and spermatozoa. Its exact function is still unclear, but it has been suggested to play a role in protecting spermatozoa from OS-induced damage [72].
Comparison of the structural and functional features of human thioredoxin and its isozymes, thioredoxin reductase 1 (TrxR-1) and thioredoxin reductase 2 (TrxR-2)
NEUROPROTECTIVE ROLE OF TS IN AD
TS plays a critical role in maintaining redox homeostasis in the brain, which is essential for normal brain function. Several studies have reported that TrxR plays a crucial role in protecting against neurological disorders [54, 73]. For instance, in cerebral ischemia, Trx-1 has been shown to protect against neuronal cell death by reducing OS [74]. In Parkinson’s disease, TrxR deficiency has been associated with increased OS and neuronal death [75, 76]. Moreover, TrxR has been implicated in the pathogenesis of other neurological disorders, including Huntington’s disease, amyotrophic lateral sclerosis, and multiple sclerosis [54]. In these disorders, TrxR deficiency has been linked to increased OS, inflammation, and neuronal damage. In AD, TrxR has been found to be downregulated, leading to increased OS and neuronal damage (Table 2).
Summary of studies describing the expression levels of thioredoxin in Alzheimer’s disease
AD, Alzheimer’s disease; Aβ, amyloid-β; CSF, cerebrospinal fluid; Trx, thioredoxin; TrxR, thioredoxin reductase; TXNIP, Trx-interacting protein; WT, wild type.
Aβ peptides are one of the main neuropathological components in AD, causing severe neurotoxicity. It also has been implicated that Aβ-induced neurotoxicity also affects cellular redox homeostasis. Aβ peptides impair mitochondrial redox activity and ameliorates the ROS generation that leads to neuronal cell death [77]. The TS has a crucial defending role against cytotoxicity mediated by ROS overprotection. In AD, the TS also exhibits neuroprotective effects against Aβ toxicity [63, 78]. Lovell et al. [78] demonstrated significant decrease of Trx and increase of TrxR in the brain of AD patients (Fig. 2). The highest TrxR activity and the lowest Trx concentration were observed in the amygdala [78]. Similarly, Akterin et al. [79] reported the decreased Trx expression in the CA1 hippocampal region and frontal cortex. Similarly, the decreased TrxR level is also observed in the frontal cortex [80]. However, another study reports that Trx levels in the cerebrospinal fluid (CSF), from early-stage AD, markedly increase [37, 81]. The elevated Trx-1 and reduced Trx-2 levels were observed in the hippocampus tissue section, CSF, and plasma of AD patients in the early stages of AD compared to mild cognitive impairment [81]. Moreover, overexpression of mitochondrial TrxR-2 reduces oxidized Trx-2 and significantly decreases Aβ peptides, inhibiting its deposition in a transgenic nematode model of AD [82]. The exact mechanism by which Trx levels increase in the CSF is not fully understood. However, it might be that increased Trx expression in the brain is a protective response to the increased OS and inflammation associated with AD. Trx helps to maintain the redox balance in cells, and increased Trx levels in the CSF may be a compensatory mechanism to counteract the effects of OS and inflammation. Therefore, OS-associated inflammation is confirmed by an increase in Trx levels in the CSF in AD.
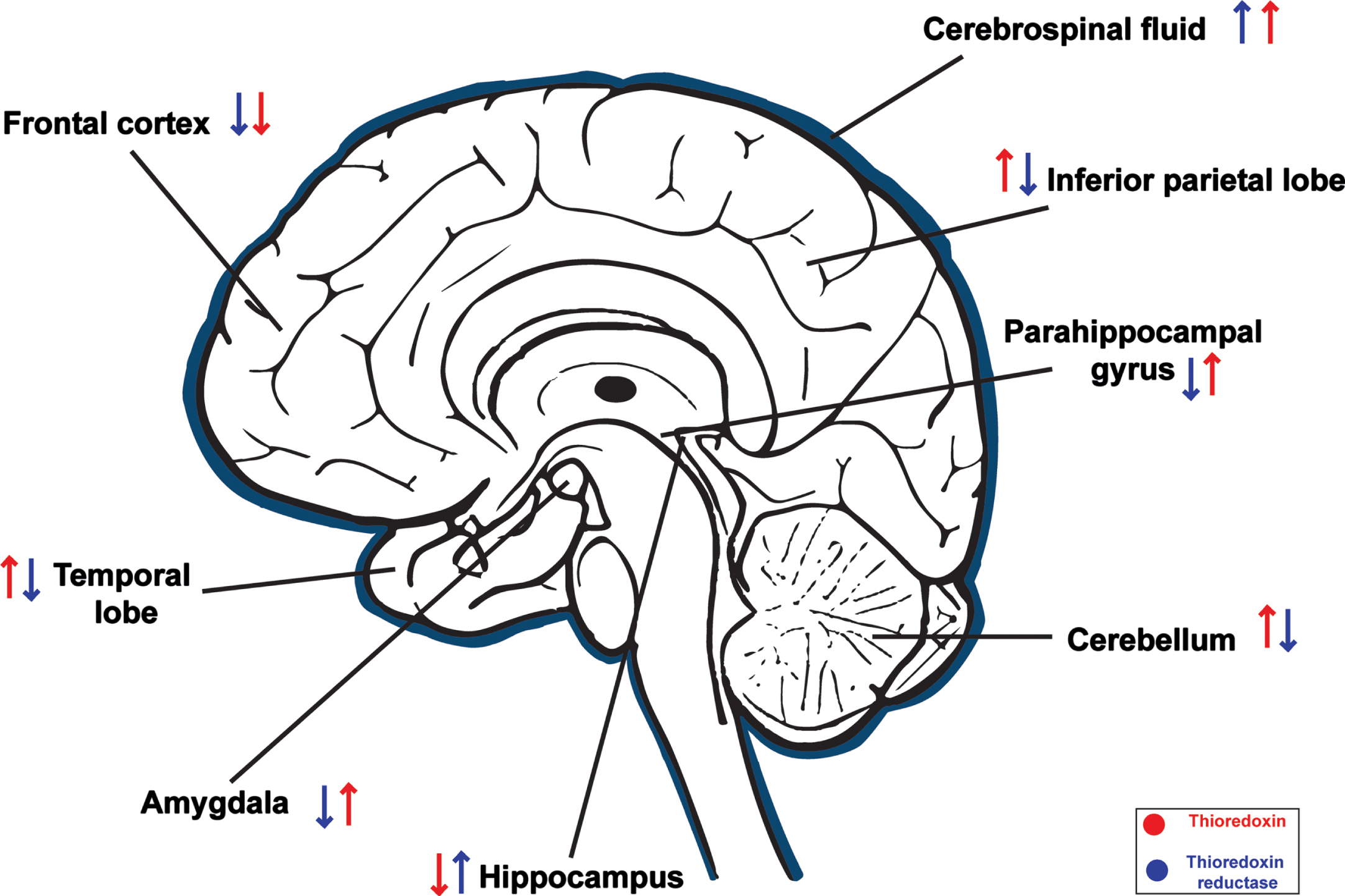
Differential levels (increased ↑ or decreased ↓) of Thioredoxin (red) and Thioredoxin reductase (blue) in different regions of brain in AD patients.
Furthermore, Di Domenico et al. [83] reported that Trx-1 levels in amnestic mild cognitive impairment precede AD. In the amyloid precursor protein/presenilin-1 (APP/PSI) double transgenic mice model of AD, a decrease in the synaptic Trx levels was observed [84]. The effects of Trx on spatial learning and memory in chronic intermittent hypoxia were observed exposed rats [85]. The results demonstrate that the chronic intermittent hypoxia-rats have decreased Trx mRNA and protein in the hippocampus, impairing spatial learning and memory. However, the increased level of Trx mRNA and protein in the hippocampus is correlated with better scores in the Morris water test, and improving spatial learning and memory [85]. In cellular model of AD, SH-SY5Y human neuroblastoma cell with mitochondrial impairment, also demonstrated decreased Trx mRNA and protein levels [86]. ApoE4 targeted replacement mice model also showed decreased levels of Trx-1 [87]. Similarly, Duan and Si [88] reported that hippocampal neurons from Aβ1 - 42-treated mice exhibited decreased levels of Trx and transcription factor, nuclear factor-E2-related factor 2 (Nrf2). Nrf2 is an endogenous modulator that reduces OS. Nrf2 moves from the cytoplasm to the nucleus in response to OS and activates antioxidant or anti-inflammatory gene expression [79, 89]. Despite OS in the hippocampal neurons of AD patients, Nrf2 levels were lower in AD patients [90], reducing Nrf2 nuclear translocation (Fig. 3). The association of dysregulated Trx expression and cognition were also observed in the 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP)-induced Trx-1 transgenic mice model of Parkinson’s disease [91]. Therefore, downregulation of Trx expression markedly increases the vulnerability of neurons to OS [86].
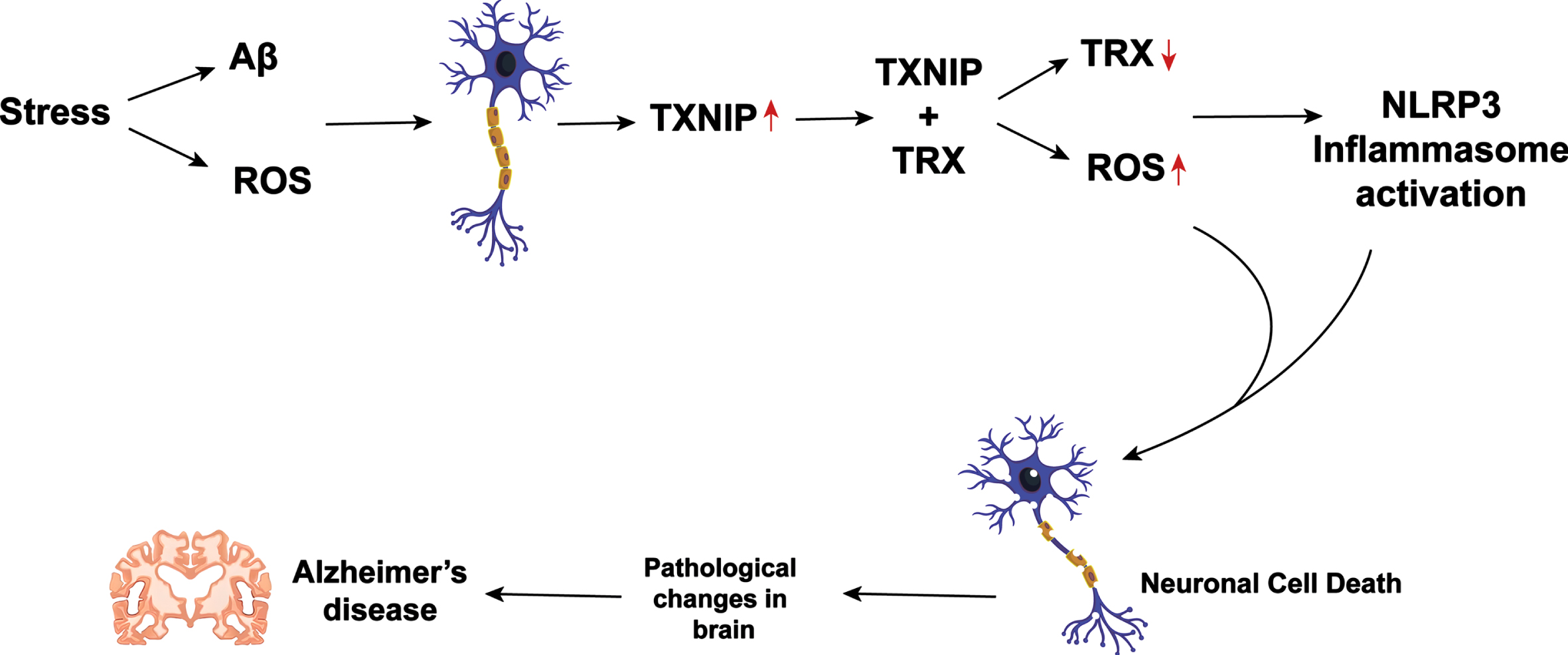
Proposed pathway of Thioredoxin (Trx) and Thioredoxin interacting protein (TXNIP) involvement in AD pathogenesis. Oxidative stress induced by Aβ and ROS increases TXNIP, which interacts with Trx leading to decreased Trx levels. Trx and TXNIP activate NLRP3, contributing to neuronal degeneration in AD.
Thioredoxin-80 (Trx80) concentrations were significantly lower in CSF and brain of AD patients. Trx-80, a product of α-secretase cleavage, is an inhibitor of Aβ’s aggregation and a truncated form of Trx-1 [92]. In addition, increased levels of Trx-80 in serum and decreased levels in the brain of AD patients with APOE4 genotype [93]. Trx80 could be a novel neuron-derived signaling mechanism that regulates microglial function under OS [94]. Aβ1 - 42 aggregation is prevented by Trx80, preventing neurotoxicity. Consequently, a decrease in Trx80 synthesis would result in higher levels of Aβ polymerization and an increase in neuronal susceptibility [92]. The loss in lifespan and locomotor deficits observed in Aβ1 - 42 expressing Drosophila melanogaster models were reversed by Trx80 expression, which also prevents Aβ1 - 42 buildup in the brain. Furthermore, Aβ1 - 42 Trx80 increases the development of autophagosomes and reverses the suppression of the Atg4b-Atg8a/b autophagosome [95]. Therefore, Trx-80 regulates microglial function and prevents neurotoxicity caused by Aβ aggregation. The findings suggest that Trx-80 could be a potential therapeutic target for AD.
A study by Ju et al. [96] reported that exogenous administration of purified recombinant Trx protein exerts significant neuroprotective effects in primary cultures of fetal rat cortical neurons. By analogy, Djukic et al. [21] demonstrated that administration of the antioxidant enzyme glutathione reductase boosted the glutathione cycle and thus enhanced AOD against OS-mediated Pd-induced neurotoxicity in Wistar rats. The former authors also demonstrated that S-nitrosoglutathione exerts neuroprotective efficacy by increasing Trx levels in Aβ-induced toxicity [96]. The activation of the receptor for advanced glycation end products, which is involved in Aβ toxicity and AD pathogenesis, causes an increase in TXNIP expression [97]. In the cortex of postmortem AD human brain samples, TXNIP and its mRNA levels upregulate, as well as caspase 1 and interleukin-1β (IL-1β). Moreover, TXNIP was co-localized near Aβ plaques and p-tau with IL-1β. Therefore, TXNIP overexpression could be associated with AD pathogenesis [98] (Fig. 3). Furthermore, Aβ increases TXNIP, inhibiting Trx and reducing ability but not affecting levels [99]. These findings suggest a complex interplay between Trx and TXNIP in AD pathogenesis, and more research is needed to fully understand their roles.
One potential clinical application is the development of a diagnostic test for early detection of AD. Currently AD diagnosis relies on clinical evaluation and imaging techniques, which may not be sensitive or specific enough to detect early-stage disease. Trx levels in the CSF could serve as a reliable biomarker for early-stage AD, allowing for earlier intervention and treatment, thus it can be used along with TrxR as potential biomarkers for the early detection of AD (Fig. 3).
CONCLUSION
The cellular redox homeostasis is imbalanced in AD. The data from humans, animals, and cellular models of AD confirms the neuroprotective roles of the TS. More studies are required to elucidate the function of Trx-TrxR reductase and the mechanism of regulation in different brain regions. This will help to treat or slow the progression of AD. The TS is a promising avenue for the development of targeted therapy to improve cognitive functions in AD. The AD-affected brain has increased levels of OS markers and decreased AOD. The redox enzymes, including TrxR, protect the cellular environment from OS. The Trx exerts neuroprotective effects against the Aβ induced cytotoxicity. The upregulation of Trx contributes cellular protection against OS-induced cell damage and vice versa. However, the dysregulation of Trx may increase the susceptibility of neurons to death. Further research is needed to fully understand the mechanisms by which Trx exerts its neuroprotective effects and to identify the specific molecular pathways involved in Trx deregulation in AD. It would be useful for future investigations to focus on Trx upregulation on cognitive function and brain pathology in AD animal models or to explore the potential of Trx-targeted therapies, and identifying of novel compounds or interventions that modulate Trx expression or activity, with the goal of restoring cellular redox balance in the brain of AD patients.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.