
Abstract
Introduction:
Tandem pore domain halothane-inhibited K+ channel 1 (THIK-1, coded by KCNK13) provides an upstream regulation of the activation of the NOD-like receptor pyrin domain containing 3 (NLRP3) inflammasome, which has been suggested as one of the key mechanisms of the pathological process in neurodegeneration mainly from in vitro and in vivo model systems studies. However, unequivocal evidence from neurodegenerative disorders has been lacking.
Objective:
To investigate the involvement of the THIK-1/NLRP3 pathway in the pathological process of Alzheimer’s disease (AD) and Parkinson’s disease (PD).
Methods:
This study investigated gene expression of markers in the THIK-1/NLRP3 pathway in an animal model representing AD as well as in human postmortem brains of AD and PD by quantitative real-time PCR. THIK-1 protein expression was determined using automated capillary electrophoresis immunoblotting. Furthermore, DNA methylation of KCNK13 was analysed in AD cohort by pyrosequencing.
Results:
A substantial upregulation of KCNK13, glial activation markers, NLRP3 inflammasome components, and IL1B was observed in the animal study. Increased expression of KCNK13 support an inflammatory glial cell activation in both advanced AD and PD. The increase in KCNK13 expression was also supported by downregulation in DNA methylation of KCNK13 in AD.
Conclusions:
The association between THIK-1 K+ channels expression and pathology changes indicates a THIK-1-induced activation of this glial subtype in AD and PD. Therefore, specific blocks of the microglial THIK-1 K+ channels at the early stage of AD and PD may be beneficial for the patients.
Keywords
INTRODUCTION
Neurodegenerative diseases are the leading cause of morbidity and disability for the aging population worldwide. Among the neurodegenerative diseases, Alzheimer’s disease (AD) and Parkinson’s disease (PD) represent the most common two proteinopathies. Specific proteins, amyloid-β (Aβ) and tau in AD and alpha-synuclein (α-Syn) in PD, along with their misfolding, oligomerization and aggregation, underlie the pathogenic processes of these proteinopathies that eventually leads to neurodegeneration [1]. However, the mechanisms leading to neurodegeneration are multifactorial, caused by endogenous (genetic and aging-related) and exogenous (environmental) factors and their interplay, and remain poorly defined [2]. Several hypotheses have been postulated to explain the pathogenesis of AD and PD, one of which is the involvement of neuroinflammation emerging early in the neurodegenerative process and contributing to its progression [3].
In recent years, the mechanisms of neuroinflammation in the pathological changes of neurodegenerative diseases have been extensively studied. For example, by using animal and cell models, researchers have shown that exposure of glial cells to Aβ or α-Syn results in the elevated secretion of pro-inflammatory cytokines including tumor necrosis factor alpha, interleukin 6 (IL-6), IL-18, and IL-1β, which are detrimental to neuronal function [4–6]. Data from clinical research also links neuroinflammation to the pathogenesis of neurodegenerative diseases. The dysregulation of cytokines and chemokines has been observed in plasma, cerebrospinal fluid (CSF) and brain of AD and PD patients [7–9]. Indeed, accumulating experimental and clinical evidence support the view that immunological mechanisms are closely involved in the onset and progression of neurodegenerative diseases, although whether they act primarily as the cause of pathology or may solely be a consequence of, for example, Aβ deposition is not fully understood.
One potentially important component of the neuroinflammatory process is activation of the NOD-like receptor pyrin domain containing 3 (NLRP3) inflammasome pathway. The NLRP3 inflammasome is composed of three proteins: a cytosolic sensor molecule NLRP3, an apoptosis-associated speck-like protein containing a caspase-activating recruitment domain (ASC, coded by PYCARD gene), and an effector molecule, cysteine protease pro-caspase-1 (CASP1) [10]. This mediator of inflammation in the brain controls microglial activation and IL-1β secretion, with evidence indicating its central role in neurodegenerative disease processes [11]. This mechanism of modulation of innate immunity is in turn controlled by two-pore potassium (K2P) channels; potassium efflux mediated by K2P in glial cells is an upstream signal activating the NLRP3 inflammasome [12, 13]. Tandem pore domain halothane-inhibited K+ channel (THIK-1, coded by KCNK13 gene) is a key K2P channel in this respect, owing to its close association with glial cells [14] and the main potassium channel expressed in microglia [15, 16].
THIK-1 exhibits a distinct function of regulating microglial ramification, surveillance and IL-1β release [14]. Moreover, inhibiting the function of THIK-1 through pharmacological action or gene knockout can depolarize microglia, jeopardize the monitoring function and reduce IL-1β secretion as a result of affecting NLRP3 inflammasome activation [13]. We recently reported that pharmacological inhibition of THIK-1 inhibited caspase-1 activation and IL-1β release from mouse macrophages, mixed glia, and microglia in response to NLRP3 agonists. Similarly, macrophages and microglia from THIK-1 knockout mice had reduced NLRP3-dependent IL-1β release [17].
This evidence suggests that manipulation of THIK-1 may serve as a novel therapeutic approach in neurodegeneration, despite the current absence of direct evidence linking THIK-1 to AD and PD. Therefore, we aimed to determine whether the evidence for a role for THIK-1 activation in cell and animal models also extends to equivalent effects in the human brain. To achieve this, we first determined gene expression of THIK-1 potassium channels and the NLRP3 inflammasome pathway in rats after intracerebral administration of a presumed pathogenic factor in AD, amyloid-β oligomers (Aβo). We subsequently determined expression of KCNK13 and THIK-1 protein in postmortem brain tissue of AD and PD patients, as well as control subjects. Given the role of epigenetic mechanisms in the regulation of inflammasome expression [18], a further investigation into DNA methylation of KCNK13 was undertaken to investigate a potential epigenetic mechanism for the observed upregulation of this potassium channel in AD.
METHODS
Animal husbandry and research ethics committee approval
20 adult female Lister hooded rats, aged 8 weeks, weighing 222±19 g (mean±SD), were obtained from Charles River and housed in groups of five in individually ventilated double decker GR1800 cages (Tecniplast, Italy) on a 12-h light-dark cycle switching at 07 : 00 and 19 : 00 respectively. Environmental conditions were controlled at a temperature of 21±2°C and humidity at 55±5%. Animals had continuous ad libitum access to food and water (Special Diet Services, UK). Animal welfare was assessed daily. Animals were weighed weekly until the day of surgery, after which they were weighed at day 1, 3, and 7.
All surgical procedures and behavioral tests were conducted in strict accordance with the UK Animals (Scientific Procedures) Act of 1986 and the Animal Welfare and Ethical Review Board (AWERB) of the University of Manchester. The protocol was reviewed and approved by the University of Manchester Research Ethical Committee.
Surgical procedure
Rats were randomly assigned to either vehicle or Aβo group (n = 10 per group). Aβo were prepared as described by Rushworth et al [19]. Briefly, synthetic biotin-labelled-Aβ1 - 42 (Anaspec) was dissolved in Hexafluoroisopropanol (HFIP) to disaggregate for 1 h, then aliquoted and HFIP was evaporated off using vacuum centrifugation. Peptide films were dissolved in dimethyl sulfoxide (DMSO) to bring to a 1 mM concentration and then diluted in Ham’s F-12 (Lonza) to a final monomer concentration of 100μM. It was then allowed 16-18 h to aggregate at room temperature. The aggregates tubes were centrifuged at 14,000×g for 20 min to pellet out any fibrillary material and the supernatant was retained as the Aβo. Aβo in the F-12 media was characterized using dot blot before being administered to the animals.
The surgical procedure was carried out as described in earlier publications [20, 21]. Briefly, rats were anaesthetized with 4% (v/v) isoflurane (Piramal) in oxygen (maintained at 2-3% isoflurane) and settled firmly into a stereotaxic frame. Animals then received a sub-cutaneous administration of 0.1 mg/kg of analgesic (Vetergesic® containing 0.3 mg/mL buprenorphine (Orion Pharma)), followed by the bilateral intrahippocampal injections (AP - 3.5 mm, ML 2 mm, DV - 4 mm) [22] of 10μL of either vehicle (DMSO and Ham’s F-12) or 10 nmol Aβo on each side on Day 0. Seven days after surgery (Day 7), animals underwent novel object recognition (NOR) behavioral testing.
Source of human brain tissue and research ethics committee approval
Neuropathologically confirmed postmortem AD brain tissue samples (Table 1) were obtained from the Manchester Brain Bank at Salford Royal NHS Foundation Trust and PD brain tissue samples (Table 2) were obtained from the Multiple Sclerosis and Parkinson’s Tissue Bank at Imperial College London. Matched control samples were also supplied by the two brain banks for each cohort respectively. The Manchester Brain Bank has been approved by the Newcastle & North Tyneside 1 Research Ethics Committee (REC reference 09/H0906/52 + 5). The MS and Parkinson’s Tissue Bank has been approved as a Research Tissue Bank by the Wales Research Ethics Committee (REC reference 08/MRE09/31 + 5).
Demographic parameters for the postmortem AD human brain samples
Demographic parameters for the postmortem PD human brain samples
The brain regions that have been examined in this project were temporal cortex for the AD cohort, substantia nigra (SN) and frontal cortex (FC) for the PD cohort, as these brain regions were indicated as relatively more severely affected by underlying molecular pathologies in the two diseases [23, 24].
Standard procedures were carried out including postmortem brain sample storage, handling and examination according to UK research ethics. Unfixed samples were stored at –80°C immediately after dissection until tissue homogenization. Research was conducted in compliance with the principles of the Declaration of Helsinki.
Clinical data was provided by the brain banks. For the AD cohorts, only demographic parameters and clinical diagnosis were available, while extra clinical data was available for the PD cohort, including age of disease onset, duration of disease, family medical history, clinical history, complications, autonomic and psychiatric symptoms, medication, and clinical cause of death.
Quantitative real-time PCR
RNA expression was quantified using reverse transcriptase semi-quantitative real-time PCR (RT-qPCR) as previously described [25]. All the procedures were performed according to the MIQE guidelines [26].
Briefly, total RNA from postmortem brain tissue of human and rat was extracted using TRIzol™ Reagent (Thermo Fisher, UK) followed by RNA clean up using Monarch® RNA Cleanup Kit (New England Biolabs, UK). RNA purity and concentrations were assessed using a NanoDrop 2100 (Thermo Fisher, UK). Single-stranded cDNA was synthesized from 1 ng of total RNA using M-MLV Reverse Transcriptase (ThermoFisher, UK). cDNA concentrations were measured on a NanoDrop 2100 and diluted to 500 ng/μL in RNase-free water.
Reference genes (RGs) were selected based on previous studies. The stability of RGs were ranked by RefFinder [27], and the top two stable RGs were chosen for delta Ct analysis. Primer pairs were designed by using Primer-BLAST (NCBI, http://www.ncbi.nlm.nih.gov/tools/primer-blast/, accessed on 11 February 2019), exon junctions were included wherever possible to avoid amplification of genomic DNA (Supplementary Table 1). Primers were synthesized by ThermoFisher. Primer specificity was analyzed by dissociation curve. RT-qPCR reactions were carried out using Power SYBR™ Green PCR Master Mix (ThermoFisher, UK) on a 7900HT Real-Time PCR System with 384-well format (Applied Biosystems, UK). The final volume for each reaction was 10μL with 1μL (0.25μM) of corresponding primers, 1μL (500 ng/μL) of total cDNA, 3μL RNase-free water, and 5μL SYBR green. All samples were run in triplicate. The thermal cycler parameters were as follows: UDG activation at 50°C for 2 min, and DNA polymerase activation at 95°C for 10 min, followed by amplification of cDNA for 40 cycles with denaturation at 95°C for 15 s and annealing/extension at respective annealing temperature for each gene for 1 min. Dissociation curve analyses were carried out at the end of each run for PCR product verification. The intra-assay variation was controlled as standard deviation of triplicates < 0.2 and was analyzed by using raw Ct value of RGs and there were no significant variations between plates.
Automated capillary electrophoresis immunoblotting
Protein expression of THIK-1 was quantified using automated capillary electrophoresis immunoblotting as previously described [25]. Briefly, 40–60 mg human postmortem brain tissue was homogenized by 10-fold volume (mg:μL) of lysis buffer (pH 7.4, 10 mM Trizma base, 2 mM EDTA, 320μM sucrose (Sigma-Aldrich, UK)). cOmplete™ Protease Inhibitor Cocktail (Sigma-Aldrich, UK), phenylmethylsulfonyl fluoride (PMSF), and sodium orthovanadate were added to the lysis buffer immediately before use at 4%, 1%, 1% (v/v) concentrations respectively. The differential centrifugation method was used for protein purification [28]. In brief, the tissue homogenate was centrifuged for 15 min (800 x g, 4°C). The pellet containing nuclei was discarded and the supernatant was centrifuged for 20 min (12,000 x g, 4 °C). The supernatant containing soluble protein was stored at -20°C for protein detection. All the procedures were performed on ice wherever possible.
Automated capillary electrophoresis immuno-quantification was conducted on a WesTM instrument for target protein analyses, according to the manufacturer’s protocol (ProteinSimple) [29]. In brief, soluble protein concentration was firstly assessed using Bradford assay, then optimization of sample concentration and primary antibodies was carried out. Sample was diluted to the optimized concentration at 0.4 mg/mL. After addition of a master mix containing dithiothreitol and fluorescent molecular weight marker, samples were heated at 95°C for 5 min for denaturation. Then, prepared samples, blocking reagent (WesTM antibody diluent), target protein primary antibodies, horseradish peroxidase (HRP)-conjugated secondary antibodies, and chemiluminescent substrate were loaded into the allocated wells of a Wes plate, pre-loaded with sample and stacking matrices. Instrument default settings were used. The size-based separation electrophoresis, immobilization, and immunodetection were automatically processed in the capillary array system and the chemiluminescence was detected at multiple exposure times and quantified by the ProteinSimple v3.1 software. Primary antibody used was rabbit anti-THIK-1 (1 : 200, NBP2-41132, Novus Biologicals). The total protein value detected by Total Protein assay (ProteinSimple) of each sample was used for normalization.
DNA extraction, bisulphite conversion, and pyrosequencing
Genomic DNA from temporal cortex of human postmortem tissue of AD and matched controls was purified using QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s instructions. EpiTec Fast DNA Bisulfite Kit (Qiagen) was used to convert unmethylated cytosine to uracil in the CpG sites with a calculated mean conversion of 99%. For analysis, a sequence containing 5 CpGs (TGCGCCCGTTCCCCGGGACCTTCAGCGTTAAGGAGAACTCCGAGG) in the promoter region of KCNK13 (coding gene for THIK-1) (GRCh38.p13, Chromosome 14, 90,061,889 –90,061,933. GenBank accession number BC012779.2, positions 125–169) that contained potential transcription factor binding sites was identified and amplified by PCR using primers, including a biotinylated reverse primer, as follows: 5′-AGGGAGTAGGGGTGGAAATTG-3′ (forward) and 5′-[btn]CTCCACTCCCCCTACTCT-3′ (reverse) (Eurofins MWG Operon). Then, methylation status of the sequence was determined with a PyroMark Q48 Autoprep (Qiagen UK) using 10μl PCR product and employing a sequencing primer, 5′- AAGTAGAAGTGAATTT-3′ (Eurofins MWG Operon). PCR reactions, amplification conditions and the methylation profile were carried out according to our previous studies [30, 31].
Statistical analysis
For the clinical sample, demographic and clinical data were analyzed using descriptive statistics. mRNA analysis was carried out using the 2(-Delta Delta Ct) Method [32]. Raw Ct values were normalized using geometric mean of two RGs as an endogenous internal standard. Data are displayed as mean±SEM. Gaussian distribution was evaluated using the Shapiro-Wilk normality test. Homogeneity of variance was evaluated using Levene’s test. Statistical analyses were conducted in IBM SPSS Statistics 23 and GraphPad Prism 7 software using non-paired parametric Student’s t-test, Welch’s t-test for unequal variances, one-way ANOVA followed with a post hoc test, and Pearson correlation or Spearman rank correlation. Statistical significance was set at p-value≤0.05 (*≤0.05, **≤0.01, ***≤0.001).
RESULTS
Glial cells and NLRP3 inflammasome were activated 7 days after Aβo administration
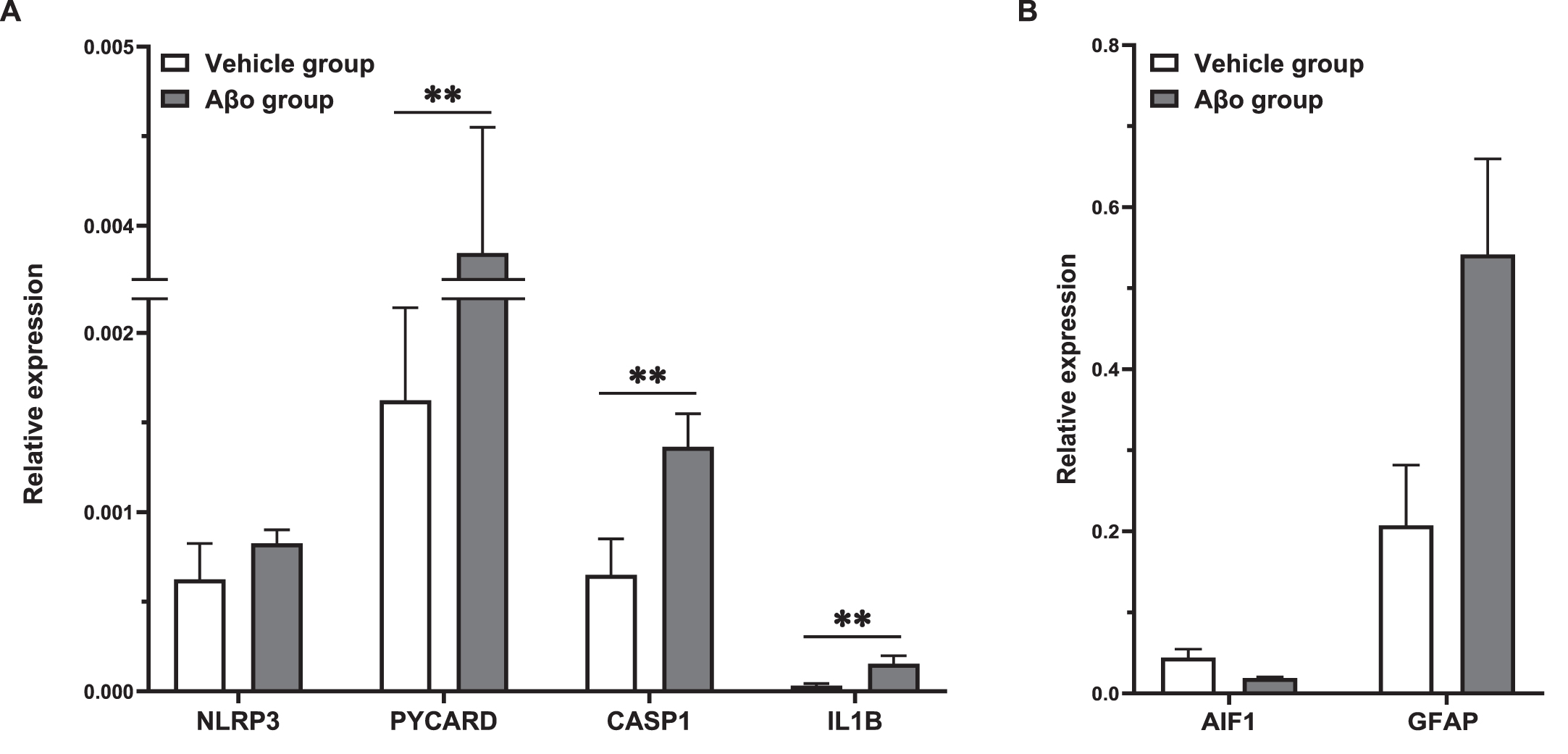
Relative gene expression of inflammatory markers of the NLRP3 pathway determined by qPCR in the left dorsal hippocampus were compared between vehicle and Aβo injected animals 7 days after surgery, when the inflammation is considered to be chronic [33]. As illustrated in Fig. 1 above, gene expression of PYCARD, CASP1, downstream cytokine IL1B as well as the glial markers were significantly elevated in Aβo group, with the highest expression of GFAP (glial fibrillary acidic protein) (Fig. 1).
Gene expression of THIK-1 potassium channels were elevated 7 days after Aβo administration
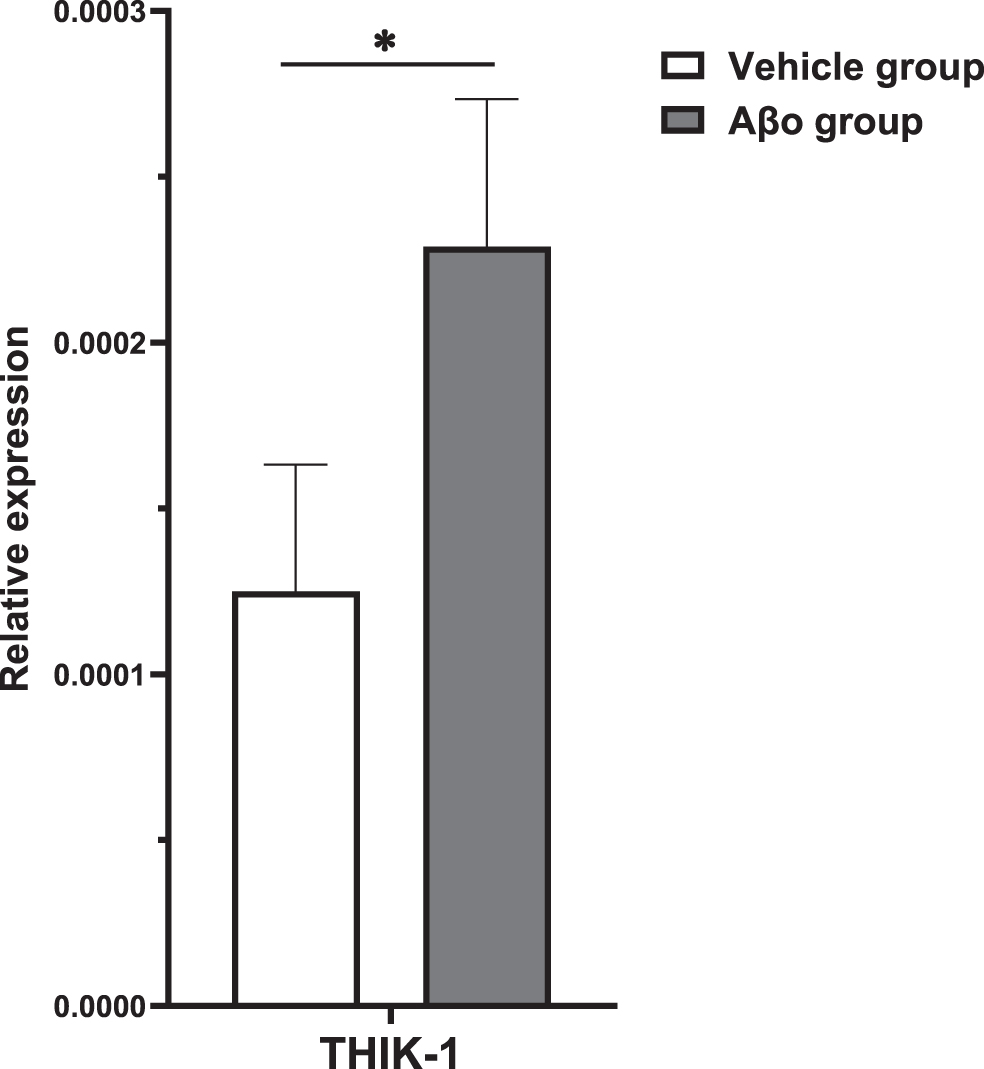
Similar to NLRP3 activation markers, on Day 7, the mRNA expression of KCNK13 was significantly higher in rats following Aβo administration (Fig. 2).
Further analysis of the correlation KCNK13 and NLRP3 inflammasome activation, found gene expression of KCNK13 was positively correlated with gene expression of NLRP3, PYCARD, CASP1, and IL1B as well as GFAP (Table 3).
Correlation between gene expression of THIK-1 and functional indicators of NLRP3 inflammasome
**Spearman’s correlation was conducted. n = 20.
RNA expression of THIK-1 potassium channel was increased in advanced Alzheimer’s disease
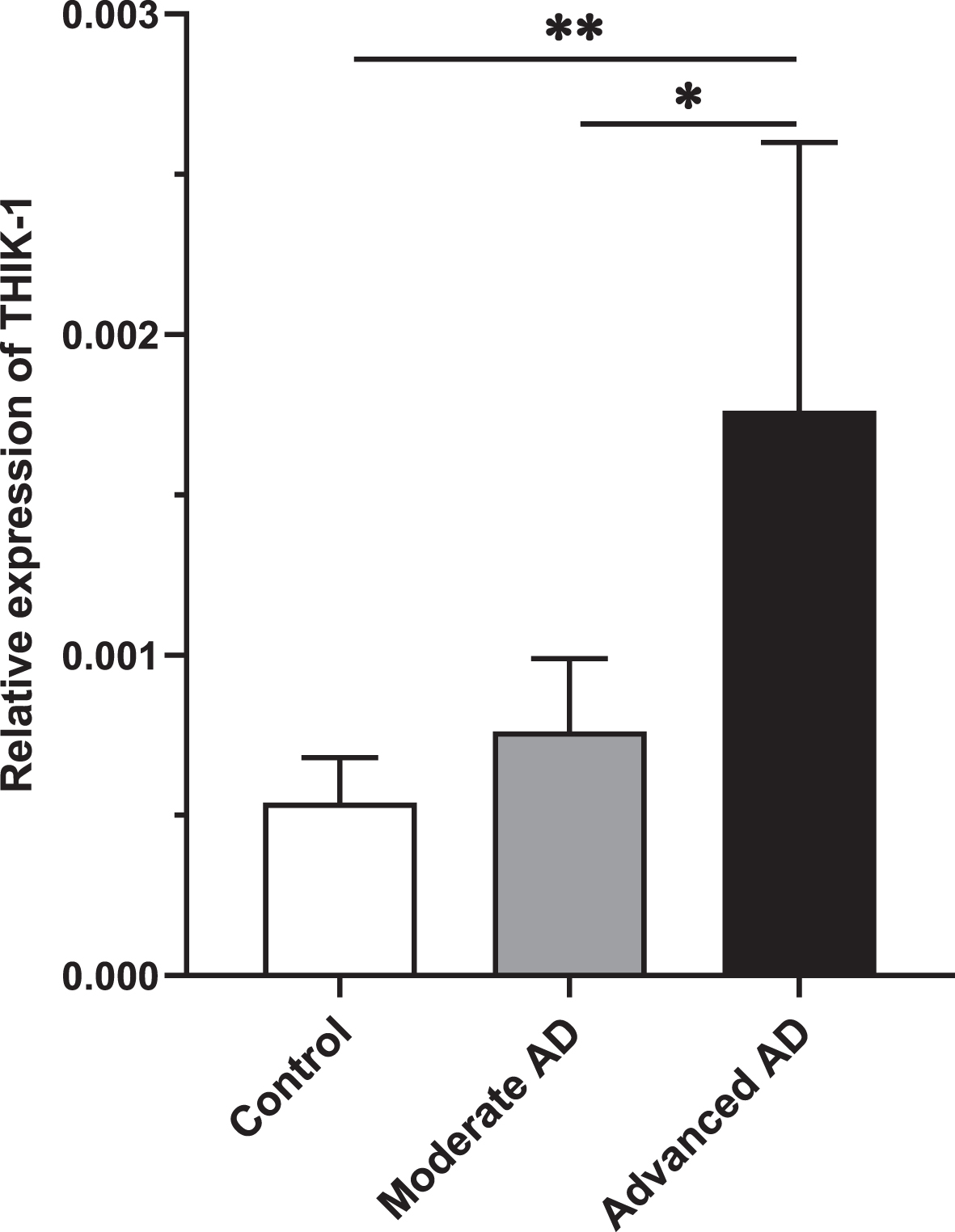
Age, postmortem interval (PMI), or sex did not correlate with any of the quantification value of mRNA or protein expression assessed in this study. Relative gene expression of THIK-1 potassium channel in the temporal cortex of the AD cohort was determined by qPCR. A significant association was seen between KCNK13 expression and AD, with a higher expression in AD (t (112)=2.163, p = 0.033). Further analysis demonstrated an association with the severity of tau pathology of AD showing that KCNK13 was significantly elevated in advanced AD (p = 0.008, 95% CI=(-2.19, -0.29)) compared with controls as well as compared with moderate AD (p = 0.048) but not significantly so in moderate AD (p = 0.581, 95% CI=(-1.34, 0.58)) (Fig. 3).
Protein expression of THIK-1 was not significantly changed in Alzheimer’s disease
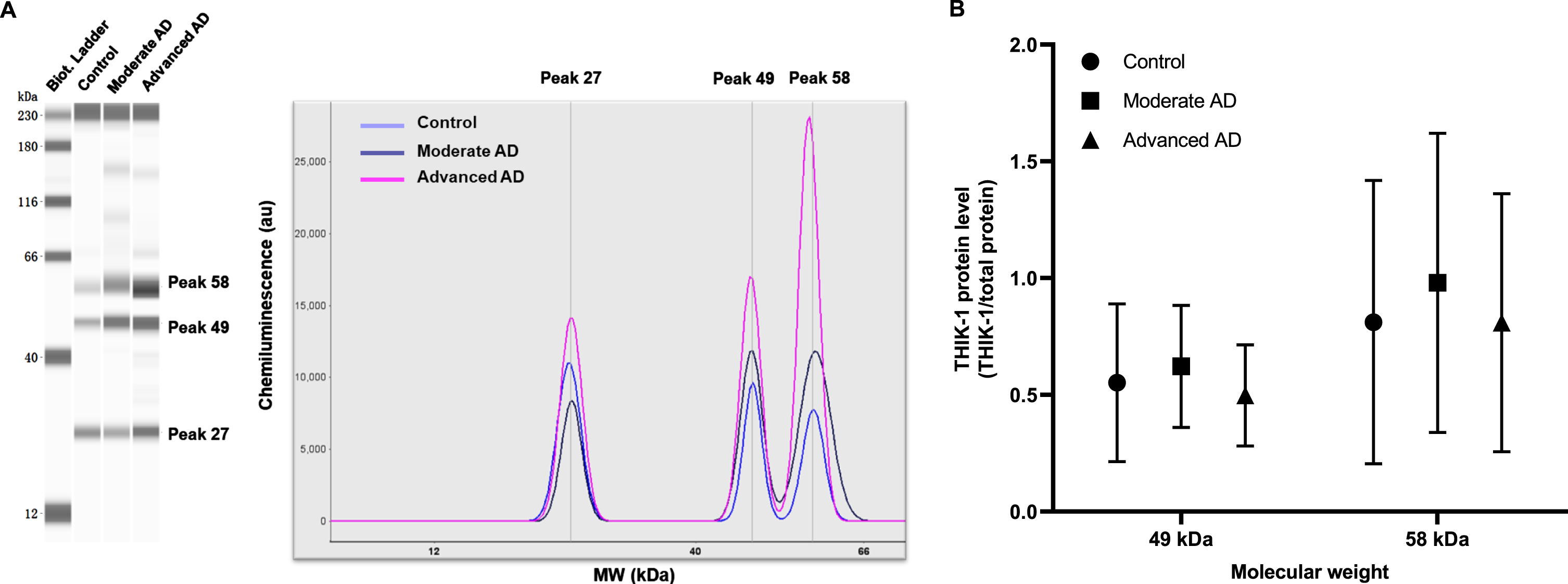
Protein expression of THIK-1 was studied by automated capillary electrophoresis immunoblotting. Three peaks/bands (equivalent to molecular weight (MW): 27 kDa, 49 kDa, and 58 kDa) were detected in the protein assay of THIK-1 (Fig. 4A). The theoretical MW of THIK-1 is 45 kDa (GeneCards, https://www.genecards.org/cgi-bin/carddisp.pl?gene=KCNK13. accessed on October 13, 2021); however, the observed MW by western blot indicated for the anti-THIK-1 antibody is 62 kDa according to product literature (NBP2-41132, Novus Biologicals). Due to the antigen being unavailable, we could not confirm the MW of THIK-1 for our samples. Therefore, expression data for 49 kDa and 58 kDa bands were analyzed on the assumption that they corresponded to THIK-1 protein. No significant differences of 49 kDa protein (one-way ANOVA, F (2. 108)=1.852, p = 0.162) or 58 kDa protein (F (2. 108)=0.977, p = 0.380) expression were observed between groups (Fig. 4B).
DNA methylation of KCNK13 was lower in Alzheimer’s disease
DNA methylation of KCNK13 was studied to follow up the result observed showing an increase of KCNK13 expression in AD. DNA methylation (mean±SEM) on CpG4 in the target sequence of THIK-1 in AD was lower (4.87±2.55) compared with control (6.28±3.85) (t (93)=2.127, p = 0.036) (Fig. 5). However, when compared between AD subgroups, the changes did not reach the significance (one-way ANOVA, F(2. 92)=2.248, p = 0.111). Methylation of either the single CpG site or the mean value of the 5 CpGs was not significantly related to age, PMI or sex in the sample.
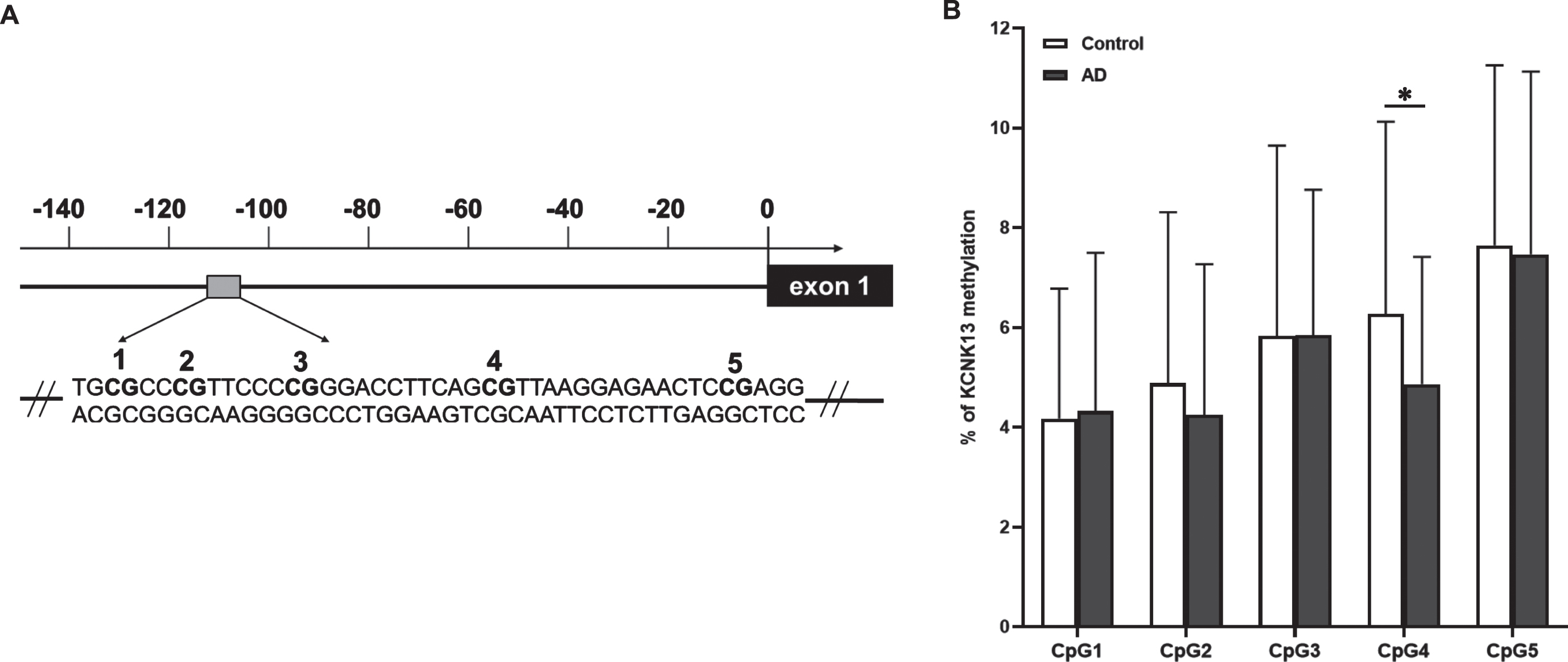
Interestingly, although the methylation status of any CpG site studied, including CpG4, was not significantly correlated with KCNK13 expression, mean percentage methylation, as well as the mean methylation level of each of the five CpGs, were negatively correlated with immunoreactivity of THIK-1 protein (Table 4). This effect was strongest for CpG3-5.
Correlation between % methylation at five CpGs and gene and protein expression of THIK-1 in AD cohort
Analyzed by the Spearman’s correlation test. *p<0.05, **p<0.01.
RNA expression of THIK-1 potassium channel was increased in Parkinson’s disease
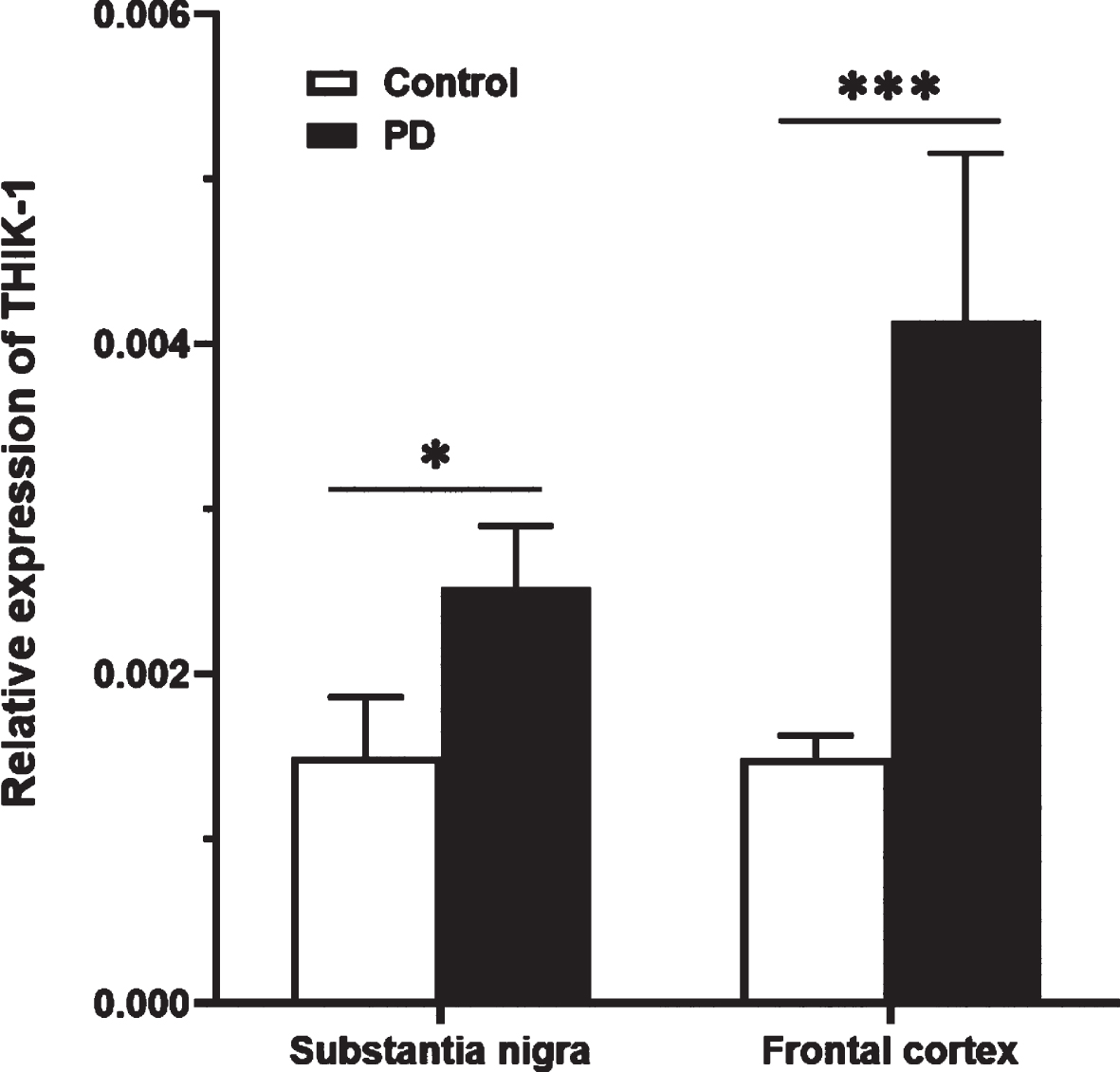
Relative gene expression of KCNK13 was determined in two brain regions of the PD cohort. As shown in Fig. 6, in SN KCNK13 expression was significantly higher in PD compared with control (t (28)=-2.74, p = 0.011). In FC, an elevation of KCNK13 was also seen in the PD group (t (28)=-4.054, p < 0.001).
In addition to the pathology, the brain bank has also provided clinical information including cognitive function in the PD cohort. Given that cognition is more closely associated with FC than SN, and KCNK13 expression was substantially increased in AD in the present study, further analyzing the KCNK13 expression in the symptom-based subgroups of PD (PD with dementia and PD without dementia) was carried out. In the PD with dementia subgroup, the increase of KCNK13 was only found in the FC (p = 0.001, 95% CI=(-2.62, -0.72)) but not in SN (p = 0.248, 95% CI=(-1.90, 0.41)) (Fig. 7). Re-analysis of the FC data after removing one outlier made essentially no difference to the results, with higher expression of KCNK13 in the FC of PD with dementia (one-way ANOVA, F (2, 24)=8.690, p = 0.001). In PD without dementia subgroup, KCNK13 expression was significantly higher in SN (p = 0.05, 95% CI=(-2.02, 0.01)) and FC (p = 0.014, 95% CI=(-1.87, -0.20)) (Fig. 7).

DISCUSSION
We have shown here the involvement of the THIK-1 potassium channel in the activated NLRP3 pathway in an acute animal model of AD. Data at Day 7, representing a sub-chronic inflammatory response to Aβo injection in the brain [33], are consistent with the hypothesis that Aβo can promote NLRP3 activation, potentially via the activation of THIK-1, resulting in an increase in the secretion of pro-inflammatory cytokines. In addition, an elevated expression of KCNK13 gene occurs in human postmortem brains of subjects with AD and with PD with dementia. This highlights the potential role of THIK-1 potassium channel dysfunction in the process of NLRP3 inflammasome activation in dementia-related pathologies. Based on these findings, we further demonstrate a reduction in DNA methylation of the KCNK13 gene promoter sequence in the AD postmortem brains.
The observed increase in expression of CASP1 which mediates the production of IL1B, would be expected to be reflected by an increase of this pro-inflammatory cytokine. The increase in ASC (coded by PYCARD) gene expression, in the absence of a significant elevation of NLRP3, could still indicate an increase in inflammasome-like activity [34]. It was reported that overexpressed ASC can rapidly polymerize and aggregates in the absence of the sensor domain of the inflammasome, such as NLRP3. The ASC speck is functional in regulating caspase-1 activity via recruiting pro-caspase-1 or nucleating CARD domain of caspase-1 filaments [35]. Interestingly, the strong indications of an increase in inflammasome activity leading to a functional pro-inflammatory state in this study occur in the context of a decrease in the expression of AIF-1. While this reduction appears to be inconsistent with NLRP3 activation; however, other such inconsistent results of AIF-1 expression in AD have been reported in previous studies [36]. The results with this microglial marker could reflect a change in cellular form and function which is associated with an inflammatory response. Meanwhile, a substantial elevation in GFAP, indicative of astrocyte activation and proliferation, is also apparent following Aβo injection, which is in accordance with the results of the postmortem study we previously reported [25].
Even though potassium efflux has been known to be an essential upstream event in the process of NLRP3 activation [14, 37], the main potassium channel responsible for AD-related NLRP3 activation is undefined. In the present animal model study, THIK-1 potassium channels showed an increased expression profile in the Aβo group. Importantly, KCNK13 gene expression was positively correlated with NLRP3, PYCARD, CASP1, and IL1B as well as GFAP. The involvement of THIK-1 in AD was further supported by the human postmortem study, with an increased expression of KCNK13 in both advanced AD and PD. THIK-1 function has been little studied in the pathologies of neurodegenerative diseases, the increased expression of the KCNK13 is consistent with another postmortem study [38]. The increase in PD is greatest in the cortex of those patients with dementia compared with other subgroups, although this is not a significant effect, perhaps reflecting the small sample sizes in the PD subgroups. The association between KCNK13 expression and pathological changes, as well as its role in regulating microglial function highlighted in other research discussed above, indicate a THIK-1-induced activation of this glial subtype in AD and PD. Therefore, targeting of microglial THIK-1 channels at the early stage of AD and PD may be beneficial for the patients.
Of note, data of the THIK-1 protein expression was not associated with AD. The THIK-1 protein level in postmortem brain might be low and it can be easily affected by the postmortem process of the brain sample, such as PMI and brain PH [39]. Moreover, agonal factors, including fever, infection, and unconsciousness can contribute to brain gene and protein expression changes [40]. Therefore, our findings for the protein expression of THIK-1 need to be further verified.
DNA methylation, through its effects in modulating gene expression, is found to be involved in cell differentiation, synapse formation, synaptic plasticity, learning and memory behavior in the healthy brain [41]. Thus, it is conceivable that alteration of DNA methylation could have a drastic impact on the ethology and pathology of brain diseases, such as neurological and psychiatric disorders. One potential underlying mechanism is DNA methylation-induced alteration of glial functions in these diseases. It is reported that DNA methylation promotes heightened microglial activation in aged mouse brain [42]. Changes in DNA methylation have also been observed in glial cells in the postmortem AD brain [43]. As THIK-1 is strongly associated with microglia function and NLRP3 inflammasome activation, the reduction in methylation of DNA from a KCNK13 promoter sequence in AD might be expected to enhance transcription and so is consistent with the gene expression findings. The reduction in DNA methylation in the AD group is found to be significant at CpG4, which is a binding site for multiple transcription factors (TFs) (Fig. 8). CpG methylation can negatively, or occasionally positively, influence the binding of TFs to DNA and vice versa [44]. The interactions between TFs and methylated DNA can further affect gene expression, splicing regulation, and chromatin remodeling [45]. Of the TFs with a binding site at CpG4, the forkhead box protein 3 (FOXP3) is particularly involved in immune response.

FOXP3 is mainly expressed in a subset of T cells that play a suppressive role in the immune system [46, 47]. FOXP3 acts also as a TF that suppresses or activates transcription of many genes including cytotoxic T-Lymphocyte antigen4, CD25 and glucocorticoid-induced TNF receptor family gene [46]. Abnormal methylation at this site would suggest a dysregulation of the transcriptional effects of this response, although whether reduced methylation might be a cause or consequence of increased FOXP3 function is unclear. Interestingly, this TF is reportedly increased both in AD brain and in an animal model of AD [48].
The methylation data at CpG4 does not significantly correlate with the expression of mRNA. A mismatch between DNA methylation and transcription is not a rare observation, potentially due to differences in time course as well as other regulatory mechanisms that also contribute to gene expression [49]. Nevertheless, KCNK13 methylation at CpG4 does show a highly significant negative correlation with THIK-1 protein, as does methylation at most of the 5 CpG sites studied (Table 4). This suggests that methylation in the promoter sequence studied might have a functional effect in regulating the amount, and perhaps the activity, of the resultant THIK-1 channels, although the findings across gene and protein expression are not fully consistent in this study.
In summary, the inflammatory outcome in the animal model of relevance to AD is apparent and supports the hypothesis of an involvement of THIK-1/NLRP3 mediated neuroinflammation as a consequence of one pathological feature of AD. The findings suggest a chronic inflammatory change in response to Aβo injection which is consistent with the hypothesis and with human studies. However, in the absence of cognitive behavior changes, assessed by the NOR test (data shown in the Supplementary Material), it is difficult to draw a conclusion whether the inflammatory response at this stage is neuroprotective or neurotoxic. Animal studies do not, of course, suffer from the many limitations of postmortem human investigations, particularly the problem that tissue taken after death is primarily limited to subjects at the very end stage of the disease process. Thus in an animal model, the progression of the pathological process can be monitored at different time-points. In the context of the current study, it would be possible, for example, to determine the temporal relationships between amyloid deposition and the appearance of inflammatory markers. Future studies with a longer time course with different timepoints to assess the THIK-1/NLRP3 induced inflammatory effect on behavioral changes are essential. In addition, THIK-1 knockout models can provide more direct information on its role in regulating NLRP3 activation, which need to be further studied.
While study of human postmortem samples has proved enormously useful in understanding neurobiological disease, it has limitations, as discussed in our previous study [25]. For example, the effects of agonal state can severely reduce the integrity of RNA; while this can to some extent be controlled for with housekeeping genes, it introduces another limitation in interpreting results. Nevertheless, the use of samples from well-matched non-AD subjects provides some control for many of these confounding effects. In addition, even accurate determination of mRNA is only representative of gene expression at a single time point. Moreover, our results in AD are limited to temporal cortex. It would be important in future studies to determine whether elevated KCNK13 is found in other brain regions affected by AD, particularly the hippocampus. While we have a relatively substantial sample size in comparison to many such human brain studies, the sample remains underpowered to identify anything other than relative large effects. Furthermore, changes in the cellular expression of markers may not always be reflected by results in tissue due to recruitment or depletion of the relevant cell [50]. Future research with higher cell specificity and closer examination of protein function in this pathway in human cohorts with clinical information, especially cognitive data, is urgently needed.
Footnotes
ACKNOWLEDGMENTS
We are indebted to patients and their families, who donated these precious samples that made this study possible. We thank contributors who collected samples used in this study. We are grateful for the collaboration with Professor Nigel M. Hooper’s group for the preparation of amyloid-β oligomers.
FUNDING
Opening Foundation of Clinical Medical Centre of Neurology of First People’s Hospital of Yunnan Province. Grant Number: 2021LCZXXF-SJ03, 2022LCZXKF-SJ02.
Hao Tang was funded by China Scholarship Council - The University of Manchester Joint Scholarship Programme, which supported her PhD study. The work presented in this manuscript was conducted as part of her PhD work and reported in her thesis.
Tissue samples were supplied by The Manchester Brain Bank, which is part of the Brains for Dementia Research program, jointly funded by Alzheimer’s Research UK and Alzheimer’s Society.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
All data generated or analyzed during this study are included in this published article and its supplementary information files. Raw CT value of qPCR study are available from the corresponding author on reasonable request.