
Abstract
Apolipoprotein E4 (APOE4), although yet-to-be fully understood, increases the risk and lowers the age of onset of Alzheimer’s disease (AD), which is the major cause of dementia among elderly individuals. The endosome-lysosome and autophagy pathways, which are necessary for homeostasis in both neurons and glia, are dysregulated even in early AD. Nonetheless, the contributory roles of these pathways to developing AD-related pathologies in APOE4 individuals and models are unclear. Therefore, this review summarizes the dysregulations in the endosome-lysosome and autophagy pathways in APOE4 individuals and non-human models, and how these anomalies contribute to developing AD-relevant pathologies. The available literature suggests that APOE4 causes endosomal enlargement, increases endosomal acidification, impairs endosomal recycling, and downregulates exosome production. APOE4 impairs autophagy initiation and inhibits basal autophagy and autophagy flux. APOE4 promotes lysosome formation and trafficking and causes ApoE to accumulate in lysosomes. APOE4-mediated changes in the endosome, autophagosome and lysosome could promote AD-related features including Aβ accumulation, tau hyperphosphorylation, glial dysfunction, lipid dyshomeostasis, and synaptic defects. ApoE4 protein could mediate APOE4-mediated endosome-lysosome-autophagy changes. ApoE4 impairs vesicle recycling and endosome trafficking, impairs the synthesis of autophagy genes, resists being dissociated from its receptors and degradation, and forms a stable folding intermediate that could disrupt lysosome structure. Drugs such as molecular correctors that target ApoE4 molecular structure and enhance autophagy may ameliorate the endosome-lysosome-autophagy-mediated increase in AD risk in APOE4 individuals.
INTRODUCTION
Alzheimer’s disease (AD) is an irreversible neurodegenerative disease and the most common cause of dementia among elderly individuals, accounting for an estimated 60% to 80% of cases. AD currently affects 35 million people worldwide; this number is projected to nearly triple by 2050 [1]. Aside from classifying AD as early-onset AD (EOAD) and late-onset AD (LOAD) based on the age of onset, AD is classified as sporadic AD (sAD) and familial AD (fAD) based on inheritance. fAD, also known as autosomal dominant Alzheimer’s disease (ADAD), accounts for up to 2% of AD cases and is caused by pathogenic mutations in the amyloid precursor protein (APP) gene and presenilin (PSEN) genes: PSEN1 and PSEN2. However, sAD accounts for up to 98% of AD cases with multifactorial etiology. Although the strongest risk factor for developing sAD is ageing, the pathogenesis of sAD is modulated by epigenetic mechanisms and several putative susceptibility genes [2, 3], of which the APOE polymorphism is considered the greatest.
The APOE gene is located on the long arm of chromosome 19 in sub-band 32 of region 13 (19q 13.32) and exists in three allelic forms: ɛ2, ɛ3, and ɛ4 based on T/C nucleotide substitution at two single nucleotide polymorphisms (SNPs), rs7412 and rs429358, in the coding region of exon 4 (Fig. 1A). Despite the variations among populations, the APOE ɛ3, ɛ4, and ɛ2 alleles have a prevalence of 79%, 14%, and ∼7%, respectively [2]. The inheritance of the APOE ɛ3 allele seems not to influence AD risk and bearing even one copy of APOE ɛ2 allele appears to reduce the risk of developing AD by up to 40%. APOE4 carriers, on the other hand, make up a significant proportion of AD cases in most clinical studies [4–7], highlighting the importance of APOE4 in AD pathogenesis. APOE4 promotes the development of AD-relevant features (Table 1); APOE4 dose-dependently increases an individual’s risk and lowers the age of onset of AD. Having one copy of APOE ɛ4 allele (ɛ3/ɛ4) can increase your risk by 2 to 3 times while two copies (ɛ4/ɛ4) can increase the risk by 12 times [8, 9]. Nonetheless, the mechanisms via which APOE4 increases AD risk remain unclear.
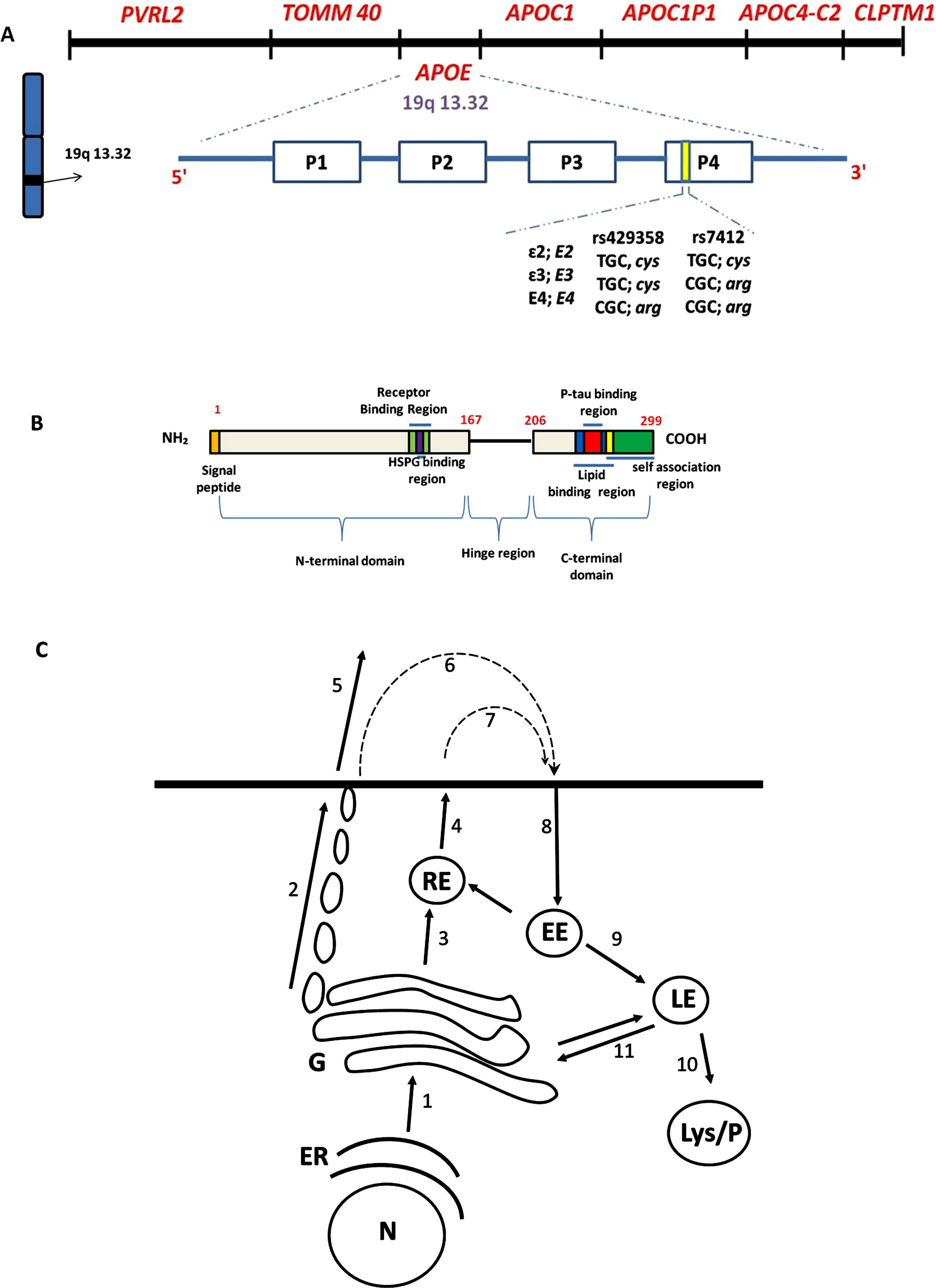
APOE locus, functional domain of ApoE protein and ApoE secretion via the classical constitutive pathway (A). The APOE gene is located on the long arm of chromosome 19 in sub-band 32 of region 13 (19q 13.32). The poliovirus receptor-related 2 (PVRL2) and the translocase of mitochondrial membrane 40 (TOMM40) genes lie upstream of the APOE locus whiles the apolipoprotein (APO) C1 [APOC1], APOC1 pseudogene 1 (APOC1P1), APOC4-C2, and the cleft lip and palate transmembrane protein 1 (CLPTM1) lie downstream of APOE [40]. The SNPs in the fourth exon distinguish the three APOE alleles. The fourth exon bears two thymine/cytosine (T/C)-single nucleotide polymorphisms (SNPs), rs429358 and rs7412; these SNPs define the three APOE allelic forms: ɛ2 (which is scarce), ɛ3 (which is most common), and ɛ4 [41]. (B) Functional segments on ApoE. N-terminal domain (1–167) has receptor binding region and HSPG binding region, hinge region (168–205) and C-terminal domain (206–299) has lipid binding domain, p-tau binding region, and self-association region. (C) represents the biology of ApoE. Newly synthesized ApoE follows the classical constitutive secretion pathway. ApoE is synthesized as a 38,500 Mr protein designated as pre-apoE, containing an NH2-terminal 18-AA extension (B) that acts as a signal peptide and mediates entry of pre-ApoE into the ER. After cleavage of the signal peptide in the ER, ApoE travels to the Golgi and trans-Golgi network (Arrow 1) for post-translational modification: ApoE is O-glycosylated on threonine 194 and extensively sialylated. ApoE is then trafficked to the plasma membrane in tubulo-vesicular carriers (Arrow 2) [42, 43]. Secreted ApoE (which may occur in glycosylated, sialylated, or partially desialylated form) is released as phospholipid discs, varying in their density and association with lipids (Arrow 5).
The effect of APOE4 on AD-relevant features
++ indicates studies that covered endosome, lysosome, and/or autophagy; Glia dysfunction result in AD features including Aβ accumulation, synaptic dysfunction, and lipid dyshomeostasis.
The hallmark pathologies of AD include the extracellular accumulation of amyloid plaques, intracellular accumulation of neurofibrillary tangles whose major component is phosphorylated tau protein, as well as extensive neuronal and synaptic losses. In AD, one of the brain pathologies that occur decades before amyloid plaque deposition and cognitive decline involves the cellular organelles of the endocytic and autophagy pathways: endosome, autophagosome, and lysosome [10–12]. The endosome, autophagosome and lysosome play an integral role in cell functions, including the trafficking and degradation of proteins [13]. Also, amyloidogenic processing of transmembrane protein AβPP (amyloid-β protein precursor), that generates amyloid-β (Aβ) mostly begins at these intracellular compartments, highlighting a potential role of anomalies of these organelles to promote AD features. Independent studies have reported on changes in the endosome, autophagosome and lysosome in APOE4 human postmortem brains, as well as in vitro and in vivo models [12, 14–18]. Yet, the contributions of the endosome-autophagosome-lysosome-associated changes to developing AD-like pathologies are a little clearer now with direct evidence from AD patients [12]. Therefore, in this review, we summarize how APOE4-mediated endosome-autophagy-lysosome dysfunction promotes AD-associated pathologies, namely Aβ accumulation, tau hyperphosphorylation, lipid dyshomeostasis, glial dysfunction, and synaptic defects. Also, we have highlighted the ApoE4 protein as an instigator of endosome-autophagy-lysosome dysfunction. Additionally, we have suggested various therapies that target the ApoE4 protein, endosomes, lysosomes, and autophagy to ameliorate AD-associated pathologies. Furthermore, we have proposed other variables that could enhance the understanding of the mechanisms underlying APOE4-mediated endosome-lysosome-autophagy changes. Finally, we have highlighted the potential involvement of the endosome-lysosome pathway and autophagy in other AD-related pathogenesis.
APOE4
ALTERS THE BIOLOGY OF ENDOSOMES
Endosomes are membrane-bound cytosolic vesicles whose main function is to internalize materials (hereafter referred to as cargoes) into the cytoplasm [19]. Endosomes start as early endosomes and transform into either recycling endosomes or late endosomes via the cargo sorting system proteins Retromer and ESCRT (endosomal sorting complex required for transport), respectively. The early endosomes are Rab5+ vesicles, recycling endosomes are Rab11+ and Rab4+ vesicles whereas late endosomes are Rab7+ vesicles [13]. During the maturation step into late endosomes, there occurs a decrease in the intraluminal pH and the early endosomes move in space from the cell periphery towards the microtubule organizing centre [20]. Also, vesicles bud from the membrane of early endosomes and accumulate, thereby forming a multivesicular body [13, 20]. The multivesicular body could fuse with the plasma membrane to release the vesicles to the extracellular compartment as exosomes.
APOE4 affects the endosomal pathway in AD and pre-clinical AD brains (Table 1). For example, in the neocortex of pre-clinical AD brains endocytic uptake and recycling were activated in pyramidal neurons. In the neurons, Rab5+ endosomes were enlarged, and the enlargement was associated with translocation of Rab5 to endosomes and elevated levels of Rab4 (indicative of increased recycling). APOE4, however, accentuated the early endosome enlargement in the preclinical AD brain [11, 21]. These changes were corroborated by findings in ApoE4 mice, a non-AD model, in which there occurred an age-dependent increase in the number and size of early endosomes [14]. In another study, APOE4 was found to alter the recycling machinery, resulting in the reductions of cell membrane receptors including LRP1 (LDL receptor-related protein 1) [15], Apoer2/Lrp8 receptor and glutamate receptors (AMPA and NMDA) [16]. APOE4 astrocytes secrete more poorly-lipidated ApoE [22, 23], and the trapping of ABCA1 (cholesterol transporter) in endosomes results in low abundance of the receptor in the plasma membrane and lesser secretion of cholesterol onto nascent ApoE to form HDL-like particles [22]. This example highlights the inhibitory potential on pathways activated by these receptors. Also, the delayed trafficking of the endosomes to the plasma membrane could promote the amyloidogenic processing of AβPP, resulting in the generation of Aβ. Furthermore, the trapping of receptors in the endosomes could be a common pathology that occurs in APOE4 individuals and models. Regarding the mechanisms underlying endosomal anomalies, transcriptomic analysis of the entorhinal cortex from ApoE4 mice showed that the differentially expressed entorhinal cortex genes were enriched in genes related to endosomes and extracellular exosomes. These genes included Rab GTPases (Rab 5, Rab 7, Rab 9, Rab 14, Rab 15, Rab 28, Rab gef1, Rab if, Rab I2) [14]. The findings suggest that the APOE4 neuron may have an active endosomal pathway characterized by endocytosis and the formation of early endosomes (Rab 5 and Rab gef1), formation of late endosomes (Rab 7), traffickings of endosomes (Rab 14) and late endosomes (Rab 9) to trans-Golgi network, routing of late endosomes to the lysosomes (Rab 28) and exocytosis (Rab if).
APOE4 could cause endosomal acidification and this observation may be associated with altered expressions of proton pumps. Transcriptomic analysis of the entorhinal cortex from ApoE4 mice revealed an upregulation of 9 vacuolar H+-ATPases (V-ATPases), namely Atp6ap1, Atp6ap2, Snx3, Snx15, Vps4a, Vps24, Vps29, Vta1, and Mp6r [14]. Also, aberrant endosomal acidification was increased in APOE4 astrocytes relative to APOE3 astrocytes. The abnormal endosomal acidification was attributed to a downregulation of the Na+/H+ exchanger (NHE6) [15]. This phenotype was inducible in normal fibroblasts by overexpressing APP, βCTF, γ-secretase inhibitors, or Rab5, which supports its dependence on APP-βCTF, but not Aβ [24].
Additionally, APOE4 compromised brain exosome production by downregulating exosome biosynthesis and release. In the brains of ApoE4 mice, the expression levels of the exosome pathway regulators TSG101 (tumour susceptibility 101) and RAB35 were reduced at both mRNA and protein levels [17]. TSG101 is a component of the ESCRT machinery. Rab35 is required for the fusion of intraluminal vesicle-containing multivesicular bodies with the cell plasma membrane before exosome secretion. The compromised exosome production could diminish a cell’s ability to eliminate materials from the endosome-lysosome compartment which may result in proteasomal stress. Aside from the alteration in the exosome secretion, the extracellular vesicle membranes in APOE4 carriers contained higher levels of cholesterol and ceramide than those in non-carriers [18], indicative of lipid abnormalities. In all, the hypothetical APOE4 cell, aside from being ladened with endosomes, also may have limited capability to produce exosomes.
APOE4
ALTERS AUTOPHAGY
Autophagy is one of the principal cellular degradatory pathways. In mammalian cells, three primary types of autophagy exist, namely, microautophagy, chaperone-mediated autophagy, and macroautophagy [25], of which macroautophagy is the major type. During macroautophagy (hereafter referred to as autophagy), double-membrane vesicles called autophagosomes are synthesized de novo from the endoplasmic reticulum to sequester cargo. The sequestered cargo is transported to the lysosome [25] for digestion of the cargoes and autophagosomes. The degradation of the autophagosomes by the lysosomes is measured as autophagic flux. The autophagosome ultimately fuses with the lysosome to form an autolysosome. The activation and completion of autophagy are often assessed by quantifying the levels of the proteins p62 and LC3B (microtubule-associated proteins 1A/1B light chain 3B) (which exist as LC3B-II and I). Autophagy activation decreases p62 levels and increases the conjugation of the lipid phosphatidyl ethanolamine to LC3B-I to form LC3B-II. When autophagosomes are degraded by lysosomes, proteins such as p62 that colocalize with autophagosomes are also downgraded.
APOE4 alters the autophagy machinery in both non-dementia individuals and AD patients [12]. Different studies have explored how APOE4 impacts autophagy dynamics. For example, studies with mouse ApoE4 astrocytes and brains of APOE TR mice showed that APOE4 impaired basal autophagy, autophagy induction and autophagic flux [26, 27] which were evidenced by an increase in p62 levels and reduced LC3-II/LC3-I ratio compared to their APOE3 counterparts. In AD patients, relative to APOE3 AD patients, homozygous APOE4 AD patients exhibited lower brain mRNA levels of the autophagy genes LC3 and p62, and the lysosomal glycoprotein LAMP2, and the lower expression of autophagy genes was shown to involve the transcription factor EB (TFEB) [12]. Overall, the APOE4 model could be prone to proteasomal stress. Additionally, the APOE4 neuron may be inefficient in clearing intracellular Aβ and tau via autophagy, resulting in intracellular accumulation of tau and Aβ.
Another APOE4-associated autophagy dysfunction is related to the phosphorylation of a transcription factor called forkhead box O-3a (FOXO3A). FOXO3a mediates a variety of cellular processes including apoptosis, proliferation, cell cycle progression, DNA damage and tumorigenesis. FOXO3 is regulated by the phosphoinositide 3-kinase class I/protein kinase B (PI3K-AKT) signalling; active AKT translocates into the nucleus, where it phosphorylates FOXO3a at three conserved residues (Thr 32, Ser 253, and Ser 315) [28]. FOXO3a directly regulates an autophagy network to functionally regulate proteostasis [29]. APOE4 attenuated autophagy via FoxO3a repression in the brain [30], which was evidenced by increased phosphorylation of FoxO3a at Ser253 and reduced expressions of Atg12, Beclin-1, BNIP3 in APOE4 carriers, likely leading to dysfunction of autophagy in APOE4 carriers [30]. The impaired autophagy in APOE4 models could also be due to the kinase mTOR (mammalian target of rapamycin) which is activated by PI3K-AKT signalling. The activation of canonical autophagy is preceded by the downregulation of MTOR and/or the phosphorylation of the mTOR at residues such as Ser2448 yet the activation state of mTOR in APOE4 cells and models is not reported. mTOR overactivation could underpin the impaired autophagy in APOE4 individuals; mTOR overactivation was implicated in impaired autophagy observed in human ALS SOD1 G93A astrocytes [31].
APOE4
ALTERS BIOLOGY OF LYSOSOMES
Lysosomes are membrane-bound vesicles that are involved in the degradation and recycling of extracellular and intracellular materials [13]. The lysosomes serve as a converging point for cellular structures including the endosomes and autophagosomes [25]. Lysosomes, like endosomes, have an acidic lumen (pH 4.5–6.5) compared to the cytoplasm. The acidic pH is due to the activity of the membrane ATP-dependent proton pumps, including vacuolar ATPase (v-ATPase) and proton H+-ATPase, which pump protons into the lysosomal lumen. The acidic pH is optimal for the hydrolases and other enzymes present [32].
APOE4 affects lysosomes in both AD and non-AD individuals and models. APOE4 iPSC-derived astrocytes [33, 34] and microglia [34] exhibited cholesterol accumulation, and these have been attributable to the subcellular localization of cholesterol in lysosomes [33]. The impact of APOE4 on lysosomes is corroborated by the fact that in cells that stably expressed the APOE isoforms, APOE4 increased lysosomal trafficking, caused ApoE protein to accumulate in enlarged lysosomes, and altered the proteomic contents of lysosomes following internalization [35]. Additionally, APOE4 could promote lysosome formation in AD-like conditions; neurons of human-APOE4 transgenic mice treated with thiorphan (an inhibitor of the Aβ-degrading enzyme neprilysin) displayed an increased number of lysosomes compared with those of ApoE3 mice [36].
Available literature suggests that at least multiple gene dysregulations underpins APOE4-associated lysosomal anomalies. For example, transcriptomic analysis of the entorhinal cortex from APOE4-targeted mice showed that differentially expressed entorhinal cortex genes were enriched in lysosomal membranes [14], and ApoE4 mice showed elevated levels of the cathepsin D, a lysosomal protease, in the hippocampal region of the mouse brain [37]. Another study that focused on cells using an ingenuity pathway analysis uncovered that iPSC-derived microglia exhibited downregulation in differentially expressed lysosome-associated genes, including LAMP1/2 (lysosomal associated membrane protein 1/2), NPC1/2 (Niemann–Pick type C disease 1/2), SMPD1 (sphingomyelin phosphodiesterase 1), and LIPA (lipase A, lysosomal acid type) [34]. LAMP1 and LAMP2 are the dominant proteins in the lysosomal membranes. Studies have shown that LAMP2 is needed for chaperone-mediated autophagy. LAMP1 and LAMP2 bind and inhibit the channel activity of TMEM175 to facilitate lysosomal acidification for optimal hydrolase activity. Additionally, LAMP1 and LAMP2 bind tightly to NPC1 and NPC2 proteins that export cholesterol from lysosomes [38]. In the cell, endocytosed LDL cholesteryl esters first reach the early endosomes where they are hydrolysed to free cholesterol. The free cholesterol is routed to the late endosomes and then to lysosomes. Thereafter, the free cholesterol is incorporated into the plasma membranes and membranes of organelles by the coordinated actions of Niemann-Pick type C (NPC) 1 and NPC2 proteins. Thus, a downregulation of LAMP1 and LAMP2 in the microglia could result in alkalinisation of lysosomal pH, accumulation of cholesterol in lysosomes and impaired autophagy and other digestive activities. Also, a downregulation in at least NPC1/2 could cause glial-associated cholesterol accumulation. Moreover, in another study, a significant correlation between APOE polymorphisms and the lysosomal disease Niemann–Pick disease type C1 was identified in human subjects: APOE4 and APOE2 were associated with increased and decreased disease severity, respectively [39]. In all, the APOE4 cell may exhibit enlarged lysosomes with upregulated cathepsin D and downregulated LAMP1/2 and NPC1/2. These findings suggest that APOE4 could alter lysosomal biology.
APOE4
PROTEIN IMPAIRS THE BIOLOGY OF ENDOSOMES, LYSOSOMES AND AUTOPHAGOSOMES
Apolipoprotein E (ApoE), the translated product of APOE, is a 34 kDa glycoprotein of 299 amino acids that is involved in cholesterol transport and the catabolism of triglyceride-rich lipoproteins. ApoE associates with lipid particles; ApoE is a component of both very low-density lipoprotein (VLDL) and high-density lipoprotein (HDL). Structurally, ApoE protein consists of an N-terminal domain (NT) [residues 1–167], C-terminal (CT) domain [residues 206–299], and a hinge (168–205) [44] (Fig. 1B). There are three isoforms of ApoE (ApoE2, ApoE3, and ApoE4) which are the translated products of the APOE ɛ2, ɛ3, and ɛ4 alleles, respectively (Fig. 1A). The three ApoE isoforms differ at the NT domain by the amino acids cysteine (Cys) or arginine (Arg) at positions 112 and 158. ApoE2 has Cys at both positions 112 and 158: ApoE4 has Arg at both positions 112 and 158; ApoE3 has Cys and Arg at positions 112 and 158, respectively [ApoE2 (Cys 112/Cys 158), ApoE3 (Cys 112/Arg 158), ApoE4 (Arg 112/Arg 158)]. The Cys/Arg substitutions at residues 112 and 158 confer different properties on the ApoE isoforms, with ApoE4 either gaining toxic functions or losing protective functions.
Available literature suggests that ApoE secretion follows classical constitutive secretory pathway; following synthesis, ApoE trafficks from the endoplasmic reticulum to the Golgi body and then through tubulovesicles to plasma membrane for secretion by exocytosis (Fig. 1C). Extracellular ApoE is internalized into early endosomes, then trafficked to either the Golgi body, recycling endosomes or late endosomes to the lysosomes for degradation. ApoE interacts with the endosomes [45, 46] lysosome [35], intraluminal vesicles [47] and autophagosomes [35, 48], highlighting the potential of ApoE4-gain-of-toxic functions to alter the biology of the cellular structures. Regarding endosomes, studies in neurons [49] and haematoma cells [50] uncovered that the ApoE4 protein impaired vesicle recycling, resulting in the sequestration of receptors in intracellular compartments and reduction in plasma surface expression of the receptors, including ApoER2 [49], and insulin receptor [51], highlighting inhibitory potential of ApoE4 on receptor-mediated pathways.
Additionally, ApoE4 dysregulated endosomal trafficking and caused an age-dependent increase in early endosome number and size [14] similar to APOE4-associated endosomal changes. Also, ApoE protein isoforms impact lysosomal activities as well as Aβ42-induced lysosomal changes. Compared to ApoE3, recombinant ApoE4 caused a disruption of lysosomal integrity and a shift in the localization of cathepsin D in both human primary cortical neurons and neuroblastoma cells [37]. Additionally, exogenous ApoE4 exacerbated Aβ42 and Fe2 +-induced lysosomal changes in the APOE4 models [52]. Moreover, in the lysosomes of cultured neuro-2a cells, ApoE4 disrupted lysosomal membranes to potentiate Aβ42-induced lysosomal leakage; and this anomaly occurred following internalization of ApoE and Aβ42. Furthermore, in smooth muscle cells treated with ferrous ions than ApoE3. ApoE accumulated in lysosomes in the form of monomers, dimers, apoE-containing complexes, and ApoE fragments. However, ApoE4 and apoE4-containing complexes persisted in smooth muscle cells longer than apoE3 and its complexes [53]. Similarly, in immortalized neuron-like and hepatic cells, and mouse brain tissue, ApoE4 accumulated in enlarged lysosomes, altered autophagic flux, and changed the proteomic contents of lysosomes following internalization [35].
The biophysical property of ApoE4 has been suggested as a molecular basis of APOE4-associated endosome-lysosome-autophagy related anomalies. The properties of ApoE that are affected by the cys/arg substitution include the type of lipids the ApoE isoforms associate with, the lipid-binding capacity, domain-domain interactions, and stability [54]. The greater lipid-binding ability of ApoE4 coupled with the large area of very-low-density lipoproteins (VLDL) being covered with phospholipids [55] promotes the preferential binding of ApoE4 to VLDL instead of HDL [56]. In addition, ApoE4 has a greater tendency to exist as monomers (which are the functional lipid-binding form) in solutions instead of a tetramer [57]. Like ApoE3 and unlike ApoE2, the ApoE4 protein unfolds to form a stable intermediate. However, compared to the unfolded ApoE3, the unfolded ApoE4 more readily forms a stable intermediate that is similar to a molten globule, which has relevance in both health and disease [58]. These altered biophysical properties have impact on the endosome, lysosome and autophagosome.
Ligand/receptor dissociation is necessary for ApoE-mediated maturation of the early endosomes to allow subsequent cargo delivery and for the endosomal content to rapidly recycle to the cell surface. The release of ApoE from its receptor in the early endosome is pH-dependent [59–61]. The early endosomal pH, which triggers ligand-receptor dissociation, closely matches the isoelectric point of ApoE4 [16] which resulted in the loss of net-surface charge. The loss of net surface charge at the isoelectric point is accompanied by reduced solubility in an aqueous environment, leading to impaired dissociation of ApoE4 from its receptors [16] and a greater propensity of ApoE4 to form a molten globule configuration under acidic conditions [58] which could antagonize the structure and functions of the endosomes. In the lysosmes, the pathological behavior of ApoE4 may arise from altered molecular properties of this protein at the acidic pH of the lysosome. Generally, at low pH, rate constants for association and dissociation are 2–10 times faster than those at pH 7.4. Aggregation beyond the tetrameric form is also more evident at lower pH values. Stability is found to be considerably greater than that at neutral pH and to be isoform-dependent. Also, the amino-terminal domain (which forms the basis of ApoE polymorphism) of ApoE4 is less susceptible to chemical and thermal denaturation than the apoE3 and apoE2 domains [58]. At acidic pH, ApoE4 forms a stable folding intermediate folding that has the characteristics of a molten globule [58]. This stable folding intermediate could be the cause of ApoE4-mediated endosome-lysosomal changes [58]. Alteration of the lysosomal pH may serve as therapeutic potential. Studies by Ji et al. (2006) showed that neutralizing lysosomal pH with bafilomycin or NH4Cl abolished the apoE4 potentiation of Aβ-induced lysosomal leakage and apoptosis in neuro-2a cells. Using an in vitro model, it was found that at acidic pH, apoE4 bound more avidly to phospholipid vesicles and disrupted them to a greater extent than at pH 7.4, and that aggregation is essential for apoE4 to potentiate Aβ-induced lysosomal leakage [52]. This finding suggests that ApoE4 disrupts lysosomal membranes to cause lysosomal leakage.
APOE4
PROTEIN COULD INSTIGATE
APOE4
-ASSOCIATED ENDOSOME-LYSOSOME-AUTOPHAGY CHANGES TO PROMOTE AD FEATURES
The ApoE4 protein could instigate APOE4-mediated endosome-lysosome-autophagy changes to promote the hallmark AD features, namely Aβ accumulation and tau phosphorylation. The ApoE4/AD studies have focussed mostly on amyloid pathology and tau hyperphosphorylation, highlighting a possible information gap regarding ApoE4’s contribution to other AD pathologies. The involvement of ApoE4 on endosome/lysosome/autophagy-mediated APOE4-mediated AD features is subsequently discussed.
Aβ ACCUMULATION
APOE4 promotes Aβ accumulation and several mechanisms have been reported (Table 1). Findings from independent studies suggest that ApoE4 protein could be pivotal in Aβ accumulation and could involve the endosome, lysosome and/or autophagy.
There occurs decreased sialylation of ApoE4 that increases the interaction between ApoE and Aβ, and promoting Aβ fibrillization [62]. Also, ApoE4 protein was found to mediate APOE4-associated impaired clearance of Aβ from the neuropil. One study reported that there occurs increased reflux of Aβ-ApoE4 complex into the brain interstitium due to reduced affinity of the Aβ-ApoE4 complex to laminin and inefficient perivascular clearance of ApoE4-Aβ complex [63]. Also, ApoE binds the receptors RAGE (receptor for advanced glycation end products) and LRP1 (LDL receptor-related protein 1) to mediate the clearance of Aβ across the blood-brain barrier (BBB) [64]. Using tracer studies, it was uncovered that Aβ binding to 125I-labeled ApoE4 redirected the rapid clearance of free Aβ40/42, in the form of Aβ-ApoE4 complex, from the fast-internalizing LRP1 receptor to the slow-internalizing VLDL receptor (VLDLR). In contrast,125I-labeled-ApoE2 and 125I-labeled-ApoE3, as well as 125I-labeled-Aβ-ApoE2 and 125I-labeled-Aβ-ApoE3 complexes, are cleared at the BBB via both VLDLR and LRP1 at a substantially faster rate than Aβ-ApoE4 complexes [65]. Aside from the role of the cys/arg-112 substitution-conferred different physical property on ApoE4, the altered internalization of the Aβ-ApoE4 complex could be due to altered cellular expression of the LRP1 receptor at the plasma membrane. Astrocytes are an important component of the BBB. Though not investigated in the study, it is reported that the aberrant endosomal acidification in APOE4 astrocytes could trap the LRP1 receptor within intracellular compartments, leading to loss of surface expression and impaired Aβ clearance [15]. Furthermore, studies with human neuroblastoma cell lines (OAN) showed that exogenous ApoE3 and ApoE4, but not ApoE2 enhanced the internalization of Aβ42. However, relative to ApoE3, the endocytosed Aβ42 was hardly catabolized at all in the presence of ApoE4 [66]. Enhanced uptake of ApoE4-mediated Aβ uptake could be corroborated by a finding that there is increased LDL receptor (LDLR) and LRP1-mediated uptake of Aβ-ApoE4 complex into synaptosomes within presynaptic terminals [67]. A recent publication proposes that ApoE influences Aβ metabolism not through direct binding to Aβ in solution but through its actions with other interacting receptors/transporters and cell surfaces. It was found that ApoE competes with soluble Aβ for the low-density lipoprotein receptor-related protein 1 (LRP1)–dependent cellular uptake pathway in astrocytes [68]. In all, these findings corroborate the role of cys/arg-112-substitution-altered ApoE4 property in Aβ accumulation.
Once within the cell, Aβ traffics from the endosomes to lysosomes for degradation. This process serves as a major intracellular Aβ clearance pathway and is differentially regulated by the ApoE isoforms. Majority of endocytosed Aβ in neurons traffics through early and late endosomes to the lysosomes for degradation. Also, a portion of endocytosed Aβ traffics through recycling endosomes. In neuro2a neuroblastoma cells, recombinant ApoE4 less efficiently mediated Aβ42 uptake, Aβ42 trafficking through early endosomes and late endosomes to the lysosomes, Aβ42 recycling and Aβ42 degradation [69], thereby promoting intraneuronal accumulation. The potential of ApoE4 to impair Aβ degradation in lysosomes was corroborated in APOE knocked-out mice-derived primary microglia that was treated with apoE4-containing HDL particles [70]. More so, it was found that that Aβ degradation in lysosomes is facilitated by the lipidation status of ApoE. Lipidated ApoE is derived by activating liver X receptors; the ApoE4 was poorly lipidated [70]. Furthermore, ApoE4 protein could alter autophagy to inhibit Aβ degradation in lysosomes. Autophagy is activated in the glia to degrade Aβ intracellularly. However, in APOE4 glia, there occur autophagy defects such as impaired lysosomal clearance, thereby contributing to Aβ accumulation [12]. Further tests uncovered a direct involvement of ApoE4 in dysregulated autophagy. Computational modelling using brain samples of AD patients predicted an avid specific binding of ApoE4 protein to CLEAR motifs. In vitro binding assays suggested that there existed a competition between ApoE4 and the transcription factor EB (TFEB) at CLEAR sites. Thus, ApoE4-CLEAR interactions may account for defective autophagy in APOE4 carriers [12]. Furthermore, considering the effect of ApoE4 on dysregulating phospholipid membranes, it has been opined that ApoE4 could disrupt the membranes of autophagosomes [71].
ApoE4 protein could promote Aβ production. The binding of lipidated ApoE to ApoER2/Lrp8 receptor triggered the endocytosis of AβPP, β-secretase and ApoER2 in neuroblastoma cells, leading to the production of Aβ. This mechanism was mediated by the cytoplasmic adaptor protein X11α or X11β (X11α/β) whose PTB (phosphotyrosine-binding) domain binds to AβPP and the cytosolic domain of ApoER2 [72]. ApoE4-ApoER2 binding inhibited AβPP endocytosis, but increased cell surface AβPP levels and AβPP association with lipid rafts to promote amyloidogenic processing [73]. Thus, this effect of ApoE4 could be gain of toxic functions. On the other hand, poorly lipidated apoE4, through the low-density lipoprotein receptor-related protein (LRP) pathway, increased Aβ production in rat neuroblastoma B103 cells stably transfected with human wild-type APP695 to a greater extent than poorly lipidated apoE3 (60% versus 30%) due to more pronounced stimulation of AβPP recycling by apoE4 than apoE3 [74]. Another study suggested how the endolysosomal enzyme δ-secretase, which is elevated in aged mice and human AD brains, contributes to the ApoE4-associated Aβ accumulation. δ-secretase cleaves AβPP at both N373 and N585 residues in the ectodomain and facilitates Aβ production by decreasing the steric hindrance for β-secretase (BACE1) [75]. ApoE4 and 27-hydroxycholesterol (27–OHC) co-activate CEBPβ/δ-secretase signalling in neurons, by mediating lysosomal δ-secretase leakage, activation, secretion and endocytosis [76]. The δ-secretase-derived truncated AβPP C586-695 fragment directly binds to CCAAT/enhancer-binding protein beta (CEBPβ) and elicits its nuclear translocation to augment the transcriptional activities on APP, MAPT (microtubule-associated protein tau), δ-secretase and inflammatory cytokine mRNA expression [75, 77]. Thus, brain Aβ accumulation could be enhanced via a positive feedback mechanism that involves at least ApoE4, Aβ, andδ-secretase.
Another mechanism via which the ApoE4 protein could cause the accumulation of amyloidogenic proteins may involve cell-specific activities that convert toxic oligomeric forms of amyloidogenic proteins to less toxic fibrillar forms. In retinal pigment cells, ApoE functions in the ESCRT-independent sorting mechanism of premelanosome (PMEL) protein onto intraluminal vesicles and regulates the endosomal formation of PMEL-derived amyloid fibrils in vitro and in vivo. However, compared to the APOE3 and APOE2 counterparts, potentially toxic premelanosome (PMEL)-derived amyloid peptides accumulated in the endosomes of retinal pigment epithelium from APOE4-TR mice. The authors found that the ApoE4 exhibited decreased neutralizing potential of the toxic amyloidogenic fragments of PMEL [47].
Meanwhile, more studies are required to understand the role of the endosome-autophagy-lysosome compartment in APOE4-mediated Aβ accumulation. Recently, an “advanced” in vivo study used a neuron-specific transgenic mRFP-eGFP-LC3 probe of autophagy and pH, multiplex confocal imaging and correlative light electron microscopy to demonstrate autophagy-lysosomal dynamics in five AD mouse models [78]. The study showed that autolysosome acidification declined in neurons well before extracellular amyloid deposition, associated with markedly lowered vATPase activity and build-up of Aβ/AβPP-βCTF selectively within enlarged de-acidified autolysosomes. In more compromised yet still intact neurons, profuse Aβ-positive autophagic vacuoles (AVs) packed into large membrane blebs and formed flower-like perikaryal rosettes, a pattern termed PANTHOS [poisonous anthos (flower)] that is also present in the AD brain. Additional AVs coalesce into peri-nuclear networks of membrane tubules where fibrillar Aβ accumulates intraluminally. This study, when replicated in APOE models, could reveal dynamics in the endosome-autophagy-lysosome compartment [78].
TAU HYPERPHOSPHORYLATION
APOE4 enhances and shortens the age of onset of AD-like site-specific tau hyperphosphorylation even in the absence of AD [79–83]. ApoE4 protein has been implicated in tau hyperphosphorylation. ApoE is reported to regulate tau phosphorylation by decreasing the phosphorylation of tau kinases (CDK5/p25, GSK 3β, and p35/MAPK) [84] and inhibiting tau phosphorylation at Thr171 and Ser202/Thr205 epitopes in neurons [85]. However, exogenous ApoE4 decreased the activation of GSK-3β and p35, relative to ApoE3 [84], suggesting a loss of function for ApoE4.
The endosome-lysosome compartment is involved in APOE4-associated tau hyperphosphorylation, and the involvement of the ApoE4 protein has been uncovered. Thus far, the endo-lysosome enzyme δ-secretase has been implicated, not only in Aβ generation, but also in tau hyperphosphorylation. In the noradrenergic locus coeruleus where the earliest detectable pre-tangle tau occurs, the inhibition of noradrenaline transport by ApoE4 can induce the aggregation of tau protein [86]. ApoE4 selectively binds to the vesicular monoamine transporter 2 (Vmat2) and inhibits neurotransmitter uptake. The exclusion of norepinephrine (NE) from synaptic vesicles leads to its oxidation into the toxic metabolite 3,4-dihydroxy phenyl glycolaldehyde (DOPEGAL), which subsequently activates asparagine endopeptidase (AEP)/δ-secretase-mediated cleavage of tau at N368 and the subsequent triggering of LC neurodegeneration [87].
There are other potential mechanisms via which the endosome-lysosome compartment and autophagy could alter tau biology. Similar studies can focus on ApoE4 to understand how ApoE4 promotes AD-associated tau pathology. Firstly, the inheritance of APOE4 can alter the phosphoinositol biphosphate (PIP2) homeostasis of the endosome-lysosome-autophagy compartment to cause tau hyperphosphorylation, and this has been attributed to elevated levels of synaptojanin 1 (synj1), a lipid phosphatase and P1P2 (phosphoinositol bisphosphate) degrading enzyme [88, 89]. The loss of Synj1 increases the number and sizes of early endosomes, impairs recycling but not trafficking to lysosomes, does not affect late endosomes, and slightly alters lysosome morphology [90]. Synj1 deficiency promotes autophagosome formation [91], highlighting its role in the biology of endosome, lysosome and autophagosome. Thus, the upregulation in SYNJ1 could be an adaptation to rectify APOE4-associated changes in the endosome/lysosome/autophagosome compartment. The upregulation of SYNJ1 has been attributed to the downregulation of miR-195. Overexpression of miR-195 in ApoE4 mice reduced the expression of synj1 and ameliorated tau hyperphosphorylation alongside cognitive deficits and amyloid plaque burden [92]. Moreover, overexpression of miR-195 in iPSCs of APOE4 AD patients rescued AD-related lysosomal defects [92]. A possible association between miR-195 and ApoE4 is yet to be reported though both molecules associates with the endosome, lysosome and autophagosome.
In addition, the components of the endosome-lysosome compartment namely charged multivesicular body protein 6 (CHMP6), tumor susceptibility 101 (TSG101), other components of the ESCRT complex, and Rab GTPase regulate vesicle cargoes that are routed from the endosome to the lysosome to affect tau trafficking, degradation, or secretion [93]. Also, exosomes and larger vesicles have been found to deliver aggregated tau and other proteins to neurons both in vitro and in vivo. In cultured cells, the internalized exosomes containing aggregated tau ended up inside lysosomes. Rather than digesting this material, the lysosomes ruptured and spilt their contents, sparking the aggregation of cytosolic tau. The authors hypothesized that exosomes resist digestion, forcing lysosomes to become more acidic in trying to clear them. Eventually, this acidity ruptures the exosomes, but also the lysosomal membrane. Alkalizing the lysosomes arrested this pathology. The addition of ammonium chloride to the cell culture cut tau aggregation by two-thirds. Another illustration of the involvement of the endosomal compartment in tau hyperphosphorylation has to do with RAB7, which is needed for the fusion of endosomes with lysosomes. Suppressing RAB7 attenuated tau aggregation by half, whereas overexpressing RAB7 boosted tau aggregation [95]. Recently, the ApoE receptor LRP1 has been identified as an endocytic receptor for tau [96]. LRP1 rapidly internalized 125I-labelled tau, which is then efficiently degraded in lysosomal compartments. LRP1 binds weakly and less efficiently internalizes phosphorylated forms of recombinant tau. Moreover, ApoE inhibited LRP1-mediated uptake of tau, and the ApoE4 isoform was the most potent inhibitor, likely because of its higher affinity for LRP1 [96].The endosome-lysosome compartment and autophagy may be involved in the formation of GVB. The core and membrane of GVBs contain prototypical markers of the autophagic and endocytic pathways such as the endosomal protein CHMP2B (charged multivesicular body protein 2b). Recently, it is reported that seeding-induced tau aggregation leads to a specific type of lysosomal structures, i.e., granulovacuolar bodies, in neurons [97]. It would be of interest to investigate the influence of ApoE status on the formation of such structures.
PERSPECTIVES
Potential involvement of endosome-lysosome pathway and autophagy in other AD-related pathogenesis
AD pathogenesis does not only include Aβ accumulation and tau hyperphosphorylation, but also synaptic defects, glial dysfunction, lipid dyshomeostasis, mitochondrial anomalies and ApoE fragmentation, all of which are altered in APOE4 humans and models. Meanwhile, glial dysfunction partly contributes to synaptic dysfunction and lipid dyshomeostasis. The involvement of endosome/lysosome and autophagy dysfunction in the AD features, namely glial dysfunction, synaptic defects, and lipid dyshomeostasis, is a bit clear. However, the contribution of the ApoE4 to the afore-mentioned AD-features via endosome/lysosome and autophagy dysfunction are not clearly reported. The possible association between the endosome-lysosome-autophagy and glial dysfunction, mitochondrial anomalies and ApoE fragmentation are subsequently highlighted.
Glial dysfunction. The neuroglia (comprising astrocytes, microglia, and oligodendrocytes) support neurons in diverse ways. However, the APOE4 neuroglia are “dysfunctional” relative to their APOE3 counterparts [83, 98] (Table 1), resulting in reduced neuronal support. The endosome-lysosome-and autophagy dysfunction has been implicated in APOE4-associated neuroglia dysfunction.
Alterations in the endolysosomal compartment have been implicated in glia-associated synaptic changes in APOE4 individuals and models. In an astrocyte-neuron co-culture, the iPSC-derived APOE4 astrocytes weakly promoted neuronal survival and synaptogenesis compared to the iPSC-derived APOE3 astrocytes [23]. Also, astrocytes in the hippocampus of APOE4 knock-in mice exhibit reduced synaptic pruning relative to astrocytes from APOE2 and APOE3 knock-in mice [99], highlighting APOE4 astrocyte-associated synaptic impairment. The involvement of the lysosomes in APOE4 glia-associated synaptic changes has been suggested, and could involve the phagocytic system; astrocytes actively phagocytose and eliminate synapses in the CNS [100]. Phagocytic pathways end with phagosomes fusing with lysosomes for subsequent hydrolysis. Even though the lysosomes in APOE4 models have some defects, it has not been established whether the fusion of the phagosome and lysosome is impaired. Additionally, the endosome marker Rab5, which is elevated in APOE4, can also regulate phagocytosis [101]. Therefore, APOE4-associated Rab5 pathology could contribute to impaired phagocytosis and synaptic pruning. Recently, iPSC-derived oligodendrocytes and oligodendrocytes from the brains of humans and targeted replacement mice exhibited cholesterol accumulation, resulting in poor myelination [102], highlighting a possible link between cholesterol abnormality and synaptic impairment. However, the study did not investigate the role of the endosome- lysosome compartment and autophagy in this cytopathology.
Alterations in the endolysosomal compartment have been implicated in glial-mediated enhancement of neuronal circuit and glia-associated lipid anomalies in APOE4 individuals and models. Astrocytes are the chief secretors of ApoE and the main backbone of neural circuits; Ca2 + signalling is the basis of astrocytes’ excitability. In APOE4 astrocytes, retention of the cholesterol transporter ABCA1 in the endosomes leading to ApoE hypolipidation has been reported. However, the role of ApoE4 protein in this anomaly was not investigated in the astrocytes. Nonetheless, it has been found that enhanced expression of ADP-ribosylation factor 6 (ARF6) [22] causes the trapping of ABCA1 in late endosomes and impairs the recycling of ABCA1 to the plasma membrane [103]. Ca2 + hyperactivity of APOE4 astrocytes has been attributed to dysregulation of Ca2 + handling in lysosomal-enriched acidic stores [104]. Furthermore, using human iPSC-derived astrocytes, the overexpression of PICALM (phosphatidylinositol binding clathrin assembly protein) is suggested to reverse endocytic disruptions [105].
Mitochondrial anomalies. The inheritance of APOE4 is associated with mitochondrial abnormalities [106] (Table 1). A recent report about the interactions among the endosomes, lysosomes and mitochondria [107] suggest that dysfunctions in the endosome-lysosome compartment and autophagy could alter mitochondrial activities. The endosome-lysosome compartment and autophagy are involved in the clearance of defunct mitochondria through sequestration into endosomes, and mitophagy, and is sometimes exported for degradation to other cells through extracellular vesicles [107]. Moreover, cholesterol and iron which are respectively needed by the mitochondrion for steroid hormone synthesis and iron-sulphur cluster enter the cell via receptor-mediated endocytosis, ending up in the endosomal system [107]. Through physical interactions, also described as transient ‘kiss and run’ interactions, the transferrin-containing endosomes directly transfer iron to the mitochondrion depending on endosomal iron concentration [108, 109]. Also, the lysosomes physically interact and deliver cholesterol to the mitochondrion, in a process that requires the late endosome protein MLN64 (metastatic lymph node protein 64). The endolysosomal system is claimed to regulate mitochondrial dynamics, including fission and fusion. Mitochondrial fission is regulated by Rab7 [110] and Vps35, a core component of the retromer [111]. Vps35 decreases mitochondrial fusion by promoting the degradation of the mitofusin 2 (Mfn2) [112] and stimulates mitochondrial fission by causing the removal of inactive DRP1 and stimulating the activity of active DRP1 molecules present in the mitochondria [113, 114]. The Vps35-dependent regulation of mitochondrial dynamics also requires the endocytic fission protein and ATPase EHD1 [115]. Furthermore, there is increasing attention for a role of early-life stress being at the origin of mitochondrial dysfunction in AD [116]. Given that stress could influence ApoE expression, perhaps there is a connection with ApoE as well, that is early life stress-ApoE-mitochondrial dysfunction in AD.
Fragmentation of ApoE. ApoE undergoes proteolysis to yield C-terminal (CT)-truncated and CT ApoE fragments [117]. The ApoE fragments, particularly the CT-truncated ApoE, enhance AD-like features in an isoform-dependent manner (ApoE4 > ApoE3 > ApoE2). ApoE fragments, especially the CT fragments, could cause tau hyperphosphorylation [118–120], inflammation, and oxidative stress. The roles of ApoE fragments in AD pathogenesis have been reviewed [121, 122]. Two groups of proteases have been suggested to mediate ApoE fragmentation, an aspartic protease and multiple serine proteases. Cathepsin D, the only aspartic protease, may mediate intraneuronal fragmentation of ApoE [123]. ApoE4 (∼87%) has been found to associate more with the cathepsin D-positive late endosomes/lysosomes compared to ApoE3 (∼9%) [124]. The serine proteases [chymotrypsin-like serine proteases [119, 125], high-temperature requirement serine peptidase A1 (HtrA1) [126] and thrombin [127] may mediate extracellular fragmentation of the protein. Exosomes could mediate the transport of the chymotrypsin-like protease into the extracellular milieu. The endosome-lysosome compartment degrades ApoE fragments; the abundance of ApoE4 fragments in the cytoplasm of APOE4 neurons has been attributed to the peptide’s potential to escape the endosome-lysosome compartment.
Targeting endosome-lysosome pathway and autophagy to ameliorate APOE4-mediated increase in AD risk
Over the years, different studies have investigated endosome-lysosome and autophagy-related mechanisms to mitigate APOE4-associated increased AD risk. Some approaches targeted the autophagy pathway and have observed the therapeutic potential of the autophagy inducer rapamycin. In one study, treating ApoE4 astrocytes with rapamycin, enhanced Aβ plaque degradation and rescued vascular, metabolic and learning deficits in APOE4 transgenic mice with pre-symptomatic AD [128]. Another approach sought to mitigate the gain of toxic functions by ApoE4. The arg112cys substitution in ApoE4 introduces domain-domain interaction in the 3D structure of ApoE4, and this interaction underlies the toxic gain of function by ApoE4. In this approach, inducing proper 3D structure restored protein function and ameliorated APOE4-conferred increased AD risks. To that effect, APOE4 models have been treated with small-molecule correctors, the phthalazinones [CB9032258, PH-001, PH-002, PH-003, PH-004, PH-005, PH-006, PH-007, PH-008] [129, 130]. The phthalazinones represent a chemical series of highly active ApoE4 structure correctors that block domain interaction in apoE4-expressing cells without affecting apoE3-expressing cells. Besides that, some studies have targeted the lysosomes in APOE4 models. Ji et al. (2006) showed that neutralizing lysosomal pH with bafilomycin or NH4Cl abolished the apoE4 potentiation of Aβ-induced lysosomal leakage and apoptosis in neuro-2a cells. Nonetheless, validation of this therapeutic intervention is necessary as most evidence suggests that increased lysosomal pH is detrimental and AD models benefit from lysosomal acidification. Another study in a mouse model of tauopathy showed that activating lysosomes pharmacologically using a farnesyltransferase inhibitor reduced tau pathology. The authors found that lonafarnib (a farnesyltransferase inhibitor) induces lysosomal-mediated tau degradation and prevent pathology in a tau mouse model via a mechanism that involves targeting the GTPase protein Rhes [131]. The therapeutic potential of farnesyltransferase inhibitors in APOE4 models can be assessed. In all, drugs such as molecular correctors that target ApoE4 molecular structure and enhance autophagy may ameliorate the endosome-lysosome-autophagy-mediated increase in Alzheimer’s disease risk in APOE4 individuals.
Understanding the mechanisms underlying APOE4-mediated endosome-lysosome-autophagy changes
The possible role of the mammalian target of rapamycin (mTOR). APOE/AD research for long has not extensively investigated the kinase mTOR-dependent pathways in APOE4 models. mTOR, in a complex of mTORC1, serves as a convergent protein for two major cell survival pathways: phosphoinositide 3-kinase class I/protein kinase B /mammalian target of rapamycin (PI3K-I/Akt/mTOR) and the mitogen-activated protein kinase / extracellular signal-regulated kinase 1/2 (MAPK/Erk1/2/ mTOR). APOE expression is activated by the PPAR-γ (peroxisome proliferator-activated receptor-gamma) and LXR (liver X receptor), all of which, have mTOR as a major activator [132]. The kinase mTOR controls several activities including autophagy, lipid metabolism, and cell proliferation [133]. Though not reported in the APOE4 brains and its cells, previous studies in other models have established links between the mTOR and major APOE4-associated dysregulated pathways: the proinflammatory cyclophilin A (CypA) in CypA–nuclear factor-kB (NFκB) – matrix-metalloproteinase-9 (MMP-9) pathway in pericytes [134], VEGF in the vascular endothelial growth factor (VEGF)-dependent pathway [135], calcineurin in the calcineurin/nuclear factor of activated T cells (NFAT) signalling pathway [136], and cPLA2 (cytosolic phospholipase A2) in cPLA2-induced inflammation and oxidative stress [137, 138]. Not only is mTOR associated with late endosomes and lysosomes but the activation thereof. Nonetheless, the effect of the endosome-lysosome compartment on mTOR activities in APOE4 models has not been reported. Additionally, a somewhat controversial literature was recently published. The authors reported that replicative senescent cells accumulate ApoE and the accumulated ApoE mediates senescence by destabilizing heterochromatin [139]. In that study, the authors uncovered ApoE-LC3 interaction in the senescent cells and found that the inhibition of the autophagy degradation pathway with bafilomycin A1 (BFA1) [an indication of autophagic flux inhibition] restored the protein levels of KAP1 and Emerin that decreased in APOE-overexpressing human MPCs. In this study, the authors did not report on the levels of mTOR in the replicative senescent cells. Aside from mTOR being involved in cellular senescence [140–142], the activation of mTOR could inhibit autophagic flux [143, 144] and can cause the accumulation of LC3 [145, 146] as was observed in the senescent cells. Additionally, this study also suggests that autophagy flux inhibition could be a therapeutic option in senescent cells.
Cytokine involvement. The glia are the principal source of cytokines such as interleukin 1β (IL-1β), interleukin 6 (IL-6), and tumour necrosis factor alpha (TNF-α) in AD and other neurodegenerative diseases, and even more in APOE4 individuals. These cytokines directly or indirectly interact with the endosome-lysosome-autophagy compartments. For example, IL-6 type cytokines signal from endomembranes in particular the endosome, and situations have been reported in which endocytosis of receptor complexes is a prerequisite of intracellular signalling [147]. Uncleaved TNF-α is endocytosed in macrophages [148]. IL-1β induces nuclear to cytoplasmic translocation of NEDD8 resulting in ubiquitination, which is a necessary step in tagging unwanted or spent entities for lysosomal autophagy [149]. The secretion of IL-1β is even enhanced upon autophagy activation. Meanwhile, the cytokines affect cellular signalling pathways such as MAPK-p38 and mTOR which have the potential to affect the activities of the endosomes, lysosomes, and/or autophagy. For example, elevated levels of IL-1β have been linked to a variety of neuropathologies, including phosphorylation of tau, and elevation of acetylcholinesterase and α-synuclein due to its control of kinases, in this case, MAPK-p38 [150, 151]. It will be helpful to know how the cytokines independently contribute to the changes in the endosome, lysosomes, and autophagy in APOE4 models.
Beyond the 112/158 Cys/Arg ApoE polymorphism factor
Over a decade since it became known that APOE4 increases AD risk, several studies have been conducted in vivo and in vitro to unravel how APOE4 increases AD risk. A major yet-to-be-unravelled issue regarding APOE4-conferred AD risk is why only some carriers of APOE4 develop AD. Inter- and intra- ethnic variabilities could influence APOE4-associated AD risk; individuals with African ancestry are claimed to have a lower risk than those with European or Asian ancestry, and even among people with African ancestry, there are differences in APOE4-associated effects [152, 153].
One of the mechanisms involves epigenetic activities at APOE CpG island [CpGI]; methylation of the APOE CpGI in the AD brain is reduced [154, 155], and the glia is proposed to be the major contributor [155]. The SNPs that define the three common alleles of APOE ɛ2, ɛ3, and ɛ4 reside in the coding region of exon 4, which overlaps with a well-defined CpGI. The two SNPs of APOE 2/3/4 polymorphism, rs429358 and rs7412, directly affect the CpG content by either contributing to or disrupting a single CpG site in the APOE CGI [156]. It has been hypothesized that the presence of APOE4 changes the DNA methylation landscape of the APOE CpGI and that such epigenetic alteration contributes to AD susceptibility [154, 155].
Another mechanism may involve single amino acid substitutions in ApoE. Recently, in a 54-year-old Korean man with sporadic early-onset AD, a whole exome sequencing revealed a novel mutation, APOE Leu159Pro which is in the LDLR region of APOE [157]. Also, an APOE variant called the APOE Christchurch mutation (APOE3Ch) has been identified. The APOE Christchurch mutation is an arginine-to-serine substitution at amino acid 136 (R136S) that lies between amino acids Cys/Arg 112 and Cys/Arg 158 [158]. The APOE Christchurch mutation halts the downstream effect of elevated Aβ levels [159]. The arginine-serine 136 substitution decreases the ApoE’s affinity for heparan sulfate proteoglycans (HSPGs), which are thought to promote Aβ accumulation and neuronal uptake of tau [159].
Additionally, the role of APOE haplotypes, instead of a single risk allele, in the differential susceptibility of individuals to develop AD is gaining attention [34]. Genetic studies of the SNPs within the APOE4 locus have revealed that there exist APOE haplotypes and local variants across populations, which may explain why not all APOE4 bearers develop AD. Most APOE-related studies have focussed on the two coding haplotypes, rs429358 and rs7412, which are used to classify the APOE ɛ2/ɛ3/ɛ4 alleles. Additionally, there exist at least 3 haplotypes, namely rs440446, rs769449 and rs769450 [160], which are intronic, and rs449647, rs769446, andrs405509 APOE promoter polymorphism [161–163], which are non-coding variants. Recently, using the 1000 genome haplotype approach, a transcriptomic study of SNPs flanking APOE (APOE±50kbp) in iPSC cell lines defined 23 common and 11 rare haplotypes, with APOE4 occurring exclusively on 7 unique haplotypes and APOE3 occurring on 12 different haplotypes. Studies with the iPSC models demonstrated that APOE4 local haplotype, rather than a single risk allele, contributed to risk [34]. Global transcriptomic analyses revealed human-specific, APOE4-driven lipid metabolic dysregulation in astrocytes and microglia [34]. Mechanistic studies on the contribution of the different haplotypes to the APOE4-conferred AD risk are therefore eminent to unravel how individual and ethnic variabilities influence APOE4-conferred AD risk.
CONCLUSION
APOE4, through yet-to-understood mechanisms, increases the risk of developing AD, which is thus far, a progressive but incurable neurodegenerative disease. Available literature suggests that APOE4 increases endosomal acidification, impairs endosomal recycling and downregulates exosome production (Fig. 2). APOE4 impairs autophagy initiation and inhibits basal autophagy and autophagic flux. APOE4 promotes lysosome formation and trafficking and causes ApoE to accumulate in lysosomes. In the absence of AD, APOE4-mediated changes in the endosome, autophagosome, and lysosome could promote AD-related features including Aβ accumulation, tau hyperphosphorylation, glial dysfunction, lipid dyshomeostasis, and synaptic defects (Fig. 2). ApoE4 protein could be the instigator of APOE4-mediated endosome-lysosome-autophagy changes. ApoE4 impairs vesicle recycling and endosome trafficking, impairs the synthesis of autophagy genes, resists being dissociated from its receptors and degradation, and forms a stable folding intermediate that could disrupt lysosome structure. Drugs such as molecular correctors that target ApoE4 molecular structure or enhance autophagy may ameliorate APOE4-mediated increase in Alzheimer’s disease risk.

The effects of APOE4 on the endosome-lysosome pathway and autophagy. Features of the endosome-lysosome compartment and autophagy that are altered by APOE4 (purple letters) and how defects in the endosome-lysosome compartment enhance AD-relevant pathologies (red letters).
Footnotes
ACKNOWLEDGMENTS
The authors are grateful for the financial support of the following organizations: Natural Science Foundation of China, Natural Science Foundation of Hebei Province, China Scholarship Council, Hebei University Science and technology research project, and Excellent Overseas researcher Program in Hebei Provincial Department of Human Resources and Social Security.
FUNDING
This work was supported by grants from the following organizations: Natural Science Foundation of China (Grant No. 81801278); Natural Science Foundation of Hebei Province (Grant No. H2019206637); China Scholarship Council (Grant No. 201608130015); Hebei University Science and technology research project (Grant No. ZD2019049); Excellent Overseas researcher Program in Hebei Provincial Department of Human Resources and Social Security (Grant No. C20190509); Key Natural Science Foundation of Hebei Province (Grant No. H2020206557); and Natural Science Foundation of Hebei Province (Grant No. H2023206266).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.