
Abstract
Background:
Cognitive decline is a common consequence of COVID-19, and studies suggest a link between COVID-19 and Alzheimer’s disease (AD). However, the molecular mechanisms underlying this association remain unclear.
Objective:
To understand the potential molecular mechanisms underlying the association between COVID-19 and AD development, and identify the potential genetic targets for pharmaceutical approaches to reduce the risk or delay the development of COVID-19-related neurological pathologies.
Methods:
We analyzed transcriptome datasets of 638 brain samples using a novel Robust Rank Aggregation method, followed by functional enrichment, protein-protein, hub genes, gene-miRNA, and gene-transcription factor (TF) interaction analyses to identify molecular markers altered in AD and COVID-19 infected brains.
Results:
Our analyses of frontal cortex from COVID-19 and AD patients identified commonly altered genes, miRNAs and TFs. Functional enrichment and hub gene analysis of these molecular changes revealed commonly altered pathways, including downregulation of the cyclic adenosine monophosphate (cAMP) signaling and taurine and hypotaurine metabolism, alongside upregulation of neuroinflammatory pathways. Furthermore, gene-miRNA and gene-TF network analyses provided potential up- and downstream regulators of identified pathways.
Conclusion:
We found that downregulation of cAMP signaling pathway, taurine metabolisms, and upregulation of neuroinflammatory related pathways are commonly altered in AD and COVID-19 pathogenesis, and may make COVID-19 patients more susceptible to cognitive decline and AD. We also identified genetic targets, regulating these pathways that can be targeted pharmaceutically to reduce the risk or delay the development of COVID-19-related neurological pathologies and AD.
Keywords
INTRODUCTION
The coronavirus disease 2019 (COVID-19) first appeared in early December 2019 in Wuhan, China, and in January 2020, the World Health Organization (WHO) announced a Public Health Emergency of International Concern. As of April 2023, COVID-19 had infected more than 762 million cases and took over 6.8 million lives worldwide [1]. Although extensive research has been performed in the last 3 years to reveal mechanisms underlying COVID-19 pathogenesis, it remains poorly understood. Mounting evidences suggest adverse effects of COVID-19 infection on different human organs raising concerns about the long-term sequelae of this disease and its potential role in inducing other diseases [2]. Therefore, recently, investigating the association of COVID-19 with other diseases has gained attention among researchers leading to increased reports on the role of COVID-19 in neurodegenerative diseases, diabetes, and cardiovascular diseases [2, 3]. A recent comparison of frontal cortex transcriptome data from COVID-19 patients and the elderly population showed an enrichment of aging-like molecular signatures in the brain of COVID-19 patients [4], which suggests a potential role of COVID-19 in accelerated aging and age-related diseases. In this regard, epidemiological studies have also indicated an increased risk of developing Alzheimer’s disease (AD) following infection with COVID-19 (HR: 1.69, 95% CI: 1.53–1.72) [5]. Moreover, molecular studies have shown the presence of main AD pathological hallmarks, including amyloid-β (Aβ) and phosphorylated tau protein (p-tau) deposition in the brain of COVID-19 infected patients [6]. AD is the most common form of dementia and a leading cause of death globally [7]. Currently, over 50 million people have dementia, and AD contributes to almost 75% of the cases, which is estimated to hit 150 million in 2050 [8]. Given the recent reports suggesting COVID-19 as the risk factor for developing AD, a higher number of AD cases are expected than estimated before COVID-19 pandemic hit the world. AD is a complex disease with different risk factors, typically characterized by initial memory and learning impairment followed by cognitive dysfunction, which remains incurable with only two recently approved drugs against Aβ [9, 10]. So far, a limited number of studies have investigated potential molecular mechanisms underlying the causative role of COVID-19 infection in AD development and reported possible causative mechanisms; however, molecular changes underpinning this association are still unclear [6, 11].
High-throughput technologies have proved efficient and reliable tools for comprehensively analyzing biological changes at different molecular levels [12]. Transcriptome and proteome analyses, among the most popular high-throughput approaches, significantly contributed to our understanding of diseases and are now a pre-requisite in biological studies. Currently, high-throughput approaches generate biological data at much higher pace than could be interpreted. Therefore, the challenge is to extract new knowledge from these vast existing data. Meta-analysis of existing results from multiple high throughput studies are a popular approach to summarize and extract the most reliable data by taking advantage of the increased statistical power provided by larger combined sample sizes [8]. Herein, we used a novel Robust Rank Aggregation (RRA) method [13, 14], which reduces errors or biases between multiple datasets to perform the meta-analysis on transcriptomic datasets from AD to identify and prioritize gene lists, and find commonly overlapping differentially expressed genes (DEGs). A comparative analysis between AD and COVID-19 infection regulated gene signatures in the brain identified common DEGs between the two conditions. We further investigated protein-protein interaction and identified the hub genes within the constructed network. Subsequently, hub gene-miRNA and hub gene-transcription factor interaction network analyses have been carried out to find potential molecular targets altered commonly in both diseases.
Our analyses show a downregulation of cyclic adenosine monophosphate (cAMP) signaling and taurine metabolism pathways, and an upregulation of neuroinflammatory related pathways such as Neutrophil extracellular trap (NET) formation pathway are a common features in both AD and COVID-19 pathogenesis. These data suggest that COVID-19 patients may be more susceptible to cognitive decline and AD. This information will provide a foundation for future animal and clinical studies and will lead to a better understanding of the molecular mechanisms underpinning the observed association between COVID-19 and AD. Studies using large cohorts of COVID-19 and AD patients are thus needed to assess the potential therapeutic targets related to these pathways and determine the diagnostic and therapeutic potential of identified miRNA and TFs.
MATERIALS AND METHODS
Dataset selection and processing
The raw count files of transcriptome datasets from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) were included in our study if they met the following inclusion criteria: 1) the dataset was original; 2) reported gene expression in the same brain region of AD patients or COVID-19 infected patients; 3) both cases and controls were included. A list of differentially expressed genes (DEGs) between AD/COVID-19 cases and healthy controls were analyzed using the limma or DESeq2 R packages; Genes with p-value<0.05 were considered upregulated and downregulated if log2 fold change > 0.263 and log2 fold change< -0.263 respectively.
Identification of robust DEGs in AD and COVID-19 datasets
Herein, we employed the RRA method by using the “RobustRankAggreg” R package (Version: 1.2.1) [13], to identify robust DEGs from each dataset. To do this, the identified lists of down and upregulated genes from each dataset were separately ranked based on their fold changes. These lists were combined into a single file, which was then subjected to the RRA method. Unlike the Venn diagram analysis, which identifies shared genes, RRA identifies genes that exhibit significant fold changes across datasets, even if they are not present in all of them [13, 15]. Robust DEGs with a Bonferroni-corrected p-value less than 0.05 were considered statistically significant. Then, the list of AD robust DEGs was compared with the list of DEGs from COVID-19 patients using the “upset plot” to obtain common down- and upregulated DEGs between AD and COVID-19 datasets.
Network analysis
Protein-protein interaction (PPI) networks were analyzed using the Cytoscape-String App plugin with a confidence score > 0.05, as previously described [8]. Briefly, robust DEGs shared between AD and COVID-19 were uploaded into Cytoscape. Next, the Homo sapiens database in the StringDB was selected to reveal the protein interaction between differentially expressed proteins. Finally, to identify the hub genes within the protein network, CytoHubba; a plugin within Cytoscape, was utilized, and hub genes were selected based on the Maximal Clique Centrality (MCC) algorithm [16].
Tissue-specific expression of hub genes
Genotype-Tissue Expression (GTEx) Project data, including RNA-seq data from 53 human tissue samples, was used to analyze the tissue-specific expression of the identified hub genes [17]. GTEx was accessed through the Expression Atlas database. The tissue expression levels were measured in transcripts per million (TPM), and Z-score normalization was applied to the expression levels for data visualization with a heatmap.
Functional enrichment analysis
For functional enrichment analysis of common DEGs between AD and COVID-19, we utilized ShinyGo, a web-based tool for comprehensive gene set enrichment analysis (http://bioinformatics.sdstate.edu/go/, ShinyGO 0.77) [18]. KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway, GO Biological process (BP), and GO Molecular function (MF) were then used to find the enriched terms from the submitted list of DEGs. Enriched terms with an adjusted p-value less than 0.05 were considered statistically significant for down- and upregulated DEGs.
Gene–miRNA and gene-transcription factors interaction analysis
The gene–miRNA interaction analysis was carried out in the NetworkAnalyst tool [19], which uses collected data of validated miRNA-gene interaction from TarBase (which showed the complete list of miRNA for most of all DEGs) [20]. The miRNAs-DEGs network was then visualized using Cytoscape. The list of the top 5 miRNA based, on network topology measurements, including degree and betweenness centrality, was reported.
Similarly, for the gene-transcription factor (TF) interaction analysis, we employed the NetworkAnalyst tool. Official gene symbols were submitted, and related TFs were explored from ChIP-seq data, ChIP Enrichment Analysis (ChEA) (which provided the most comprehensive list of TF for all DEGs) [21]. The gene-TF interaction network was also visualized using Cytoscape.
Literature search for the validation of the identified miRNAs and TFs via text mining
To search miRNAs and TFs from our analysis and their relation with AD or COVID-19, based on the published literature, the “batch_pubmed_dowload” function from the easyPubMed R package [22] was employed. The articles, including our identified targets, were thoroughly discussed in the discussion.
RESULTS
Analysis of transcriptome data from 638 brain samples reveals data heterogeneity between AD datasets and profound transcriptomic changes in AD and COVID-19 cohorts
Three transcriptome datasets, obtained from the frontal cortex of postmortem brains of 377 AD patients and 223 healthy controls (GSE118553, GSE48350, and GSE33000), and a dataset for COVID-19 (GSE188847) with 17 cases and 21 controls were included in our study (16-18, 4) (Table 1). DEGs in each brain region were extracted using the limma and DESeq2 R packages (p-value<0.05, and log2|FC|>0.263) [23, 24]. The numbers of down- and upregulated DEGs from each AD dataset were varying from 664 (downregulated: 268 and upregulated: 396, GSE118553) to 1672 (downregulated:1009 and upregulated: 663, GSE48350) for the AD datasets (Table 1). Interestingly, analyses of the COVID-19 dataset revealed 2,471 down- and 3,134 upregulated genes (Table 1), indicating COVID-19 infection causes more profound transcriptomic changes in comparison to the AD patient cohort (Fig. 1, Supplementary File 1). Furthermore, a comparison of DEGs between the three AD datasets showed only 1 down- and 7 upregulated genes, shared among them (Supplementary File 1, Supplementary Figure 1). This data indicates variations between existing AD datasets, which could be related to subtype heterogeneity across AD patient cohorts [25]. It is also possible that the differences in post-mortem sample collection times between studies might cause variation in the transcriptome dataset, as was shown previously [26]. Among the common genes, rabphilin 3a (RPH3A) is a small G protein that acts in the late stages of neurotransmitter exocytosis that was found to be downregulated between all three AD datasets. Downregulation of RPH3A has been shown to be associated with dementia severity and increased Aβ concentrations [27]. On the other side, common upregulated genes between all three AD datasets are involved in different biological pathways mostly in immune response including, Human leukocyte antigen - DR alpha (HLA-DRA), Fc fragment of IgG receptor IIa (FCGR2A) and Cluster of differentiation 74 (CD74). HLA-DRA gene encodes a protein subunit of the Major histocompatibility complex class II (MHC II) molecule. FCGR2A gene encodes a receptor that binds to the fragment crystallizable region (Fc region) of IgG antibodies [28]. The FCGR2A receptor is expressed on immune cells such as macrophages, neutrophils, and natural killer (NK) cells; when the receptor binds to IgG antibodies, it triggers immune effector functions such as phagocytosis and antibody-dependent cell-mediated cytotoxicity (ADCC) [29]. CD74 gene encodes a protein that plays a role in antigen processing and presentation, and it is also involved in regulation of cell proliferation and survival [30] (Supplementary File 1).
Characteristics of the selected datasets analyzed in this study

Differentially expressed genes from each dataset. Volcano plots demonstrate the thresholds for differentially expressed genes in GSE33000 (A), GSE 48350 (B), GSE118553, and GSE188847. Each data point represents a single gene. The x-axis represents the log2 fold change in expression (AD or COVID-19 versus Control), and the –log (p-value) is plotted on the y-axis. Blue and red points indicate significantly (p-value<0.05) down- and upregulated genes with fold change lower than 0.83 and over 1.2. AD, Alzheimer’s disease; CTRL, Control; FC, Fold change.
Integrated transcriptome analysis revealed robust AD genes in COVID-19 infected brains
The RRA analysis using the integrated list of down- and upregulated genes, including 1,628 down- and 1,401 upregulated genes across AD datasets, sorted based on their fold changes yielded 44 down- and 42 upregulated AD robust DEGs (Fig. 2, Supplementary File 1). The comparison between the AD robust DEGs and COVID-19 DEGs showed 23 up- and 26 downregulated common genes (Fig. 2). Neuronal differentiation 6 (NEUROD6), Corticotropin releasing hormone (CRH), and Glutamic acid decarboxylase 2 (GAD2) are the top three deregulated DEGs shared between the COVID-19 dataset and robust AD genes. NEUROD6 is a basic helix-loop-helix transcription factor involved in neuronal development and differentiation, and its downregulation has been suggested as a potential biomarker for AD [34]. CHR is a gene involved in neuroendocrine responses to stress, and its abundance is decreased in AD patients [35]. GAD2 encodes an enzyme that catalyzes the production of gamma-aminobutyric acid from L-glutamic acid [36]. Downregulation of GAD2 and high levels of L-glutamic acid have been reported in AD patients leading to neuronal death, a phenomenon generally termed excitotoxicity [37]. While the top three shared upregulated DEGs, including Cluster of differentiation 74 (CD74), C-X-C chemokine receptor type 4 (CXCR-4), also known as CD184, and Complement component 5a receptor 1 (C5AR1) or known as CD88, are mainly involved in immune response [38]. Additionally, one DEG, Tachykinin precursor 1 (TAC1), involved in cell-cell signaling and inflammatory response [39], was upregulated in AD but downregulated in COVID-19.
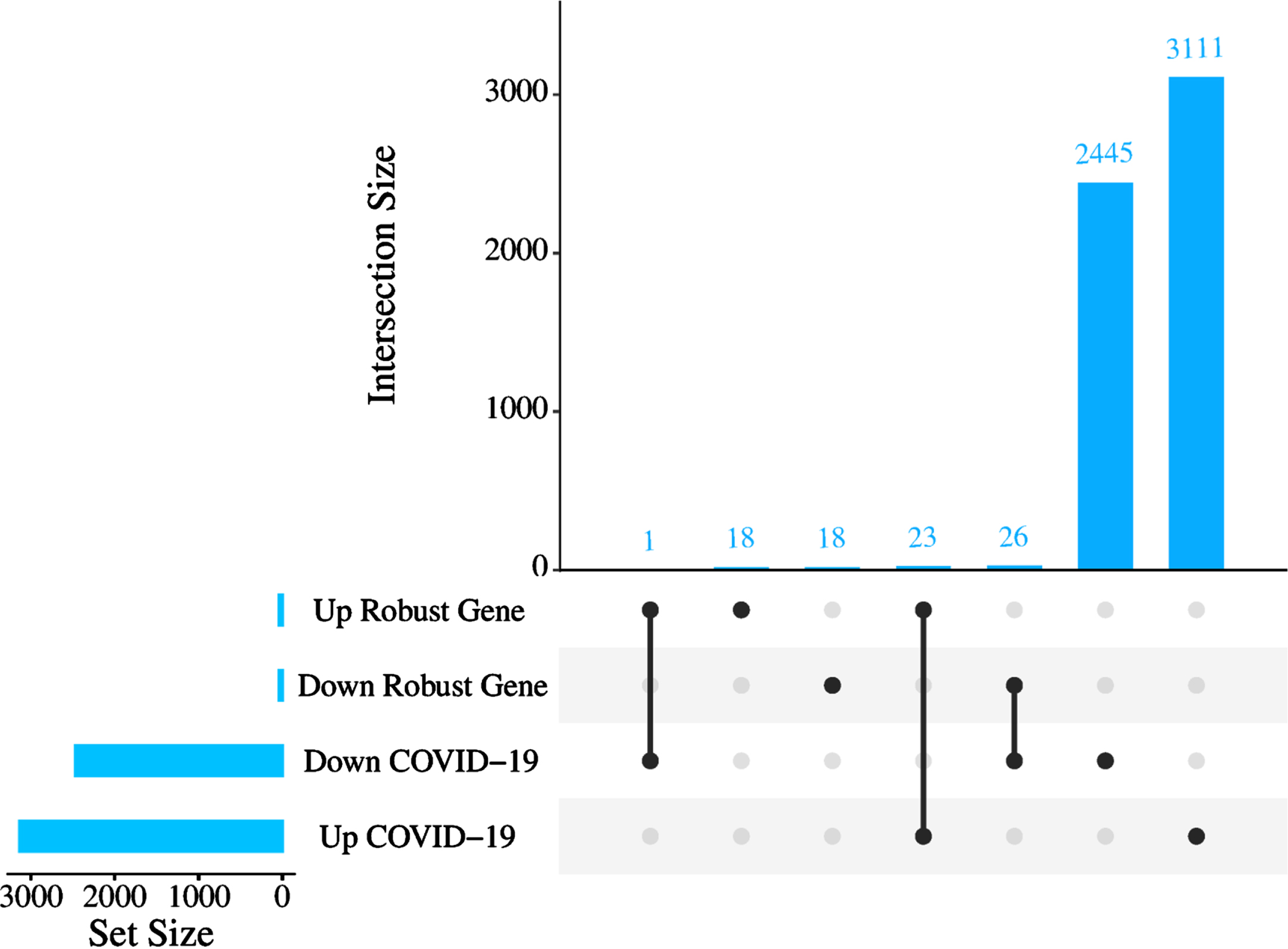
The number of commonly altered genes between AD and COVID-19. Upset plot indicating the overlap of COVID-19 DEGs with either increased or decreased expression in robust AD DEGs.
PPI network analysis of robust DEGs identified common hub genes associated with both diseases
To understand interactions between common DEGs between AD and COVID-19 and find the hub genes within their network, we performed PPI interaction and hub gene analysis. Our analyses revealed Brain-derived neurotrophic factor (BDNF), Somatostatin (SST), Glutamic acid decarboxylase 1/2 (GAD1/2), CRH, Vasoactive intestinal peptide (VIP), Glial fibrillary acidic protein (GFAP), Adenylate cyclase activating polypeptide 1 (ADCYAP1), CXCR-4 and NEUROD6, as the hub genes within the PPI network of common DEGs between AD and COVID-19 (Fig. 3). In addition, our tissue-specific expression analysis further validated that these hub genes are mainly expressed in the brain, except CXCR4, a transmembrane protein of CXC chemokine receptor that showed higher expression in the blood and spleen cells (Fig. 4, Supplementary File 1).
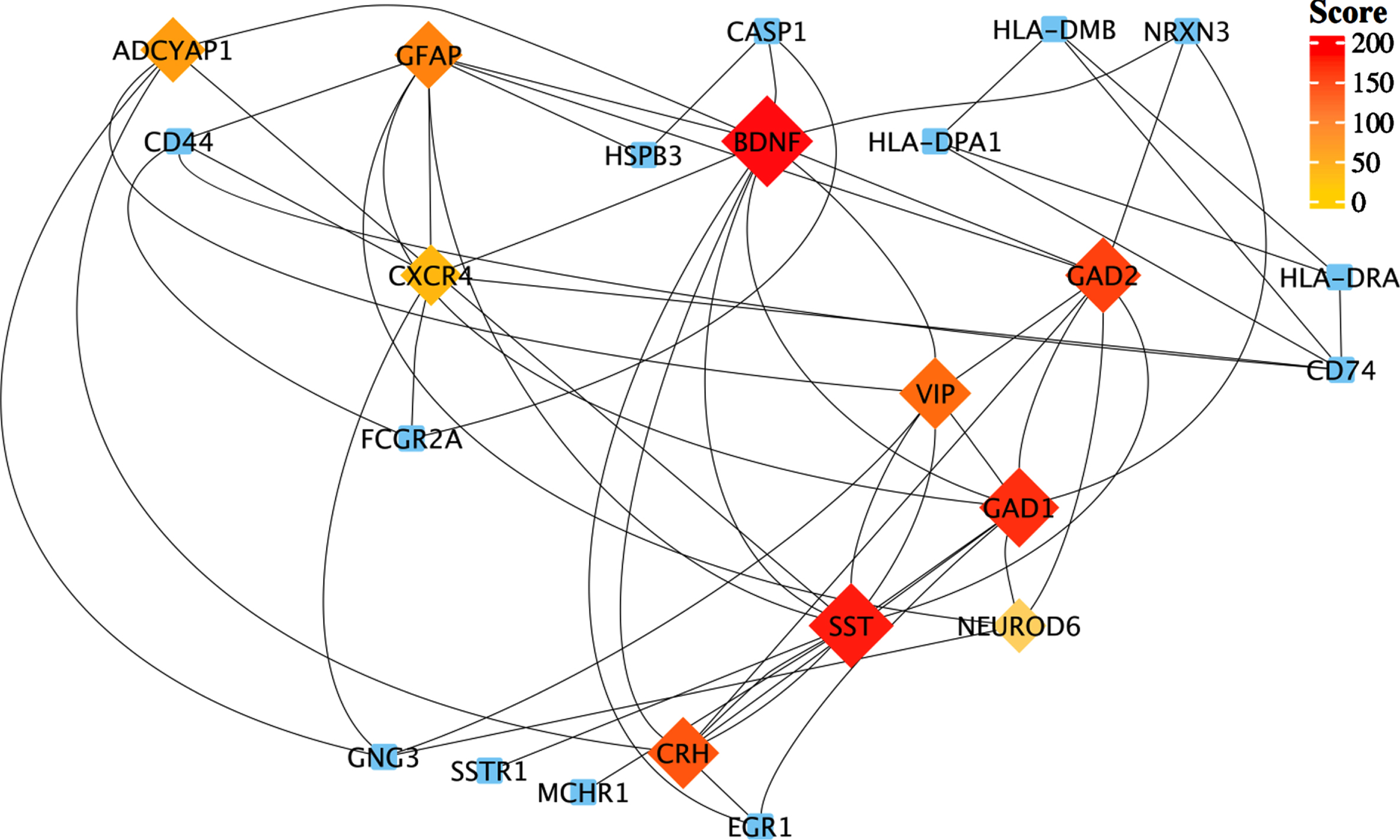
Protein-protein interaction network of those shared DEGs between robust AD and COVID-19 genes. Top-ranked hub genes are shown in rhombi shape and highlighted based on their score from the Maximal Clique Centrality (MCC) algorithm in the cytoHubba plugin of Cytoscape.

Tissue-specific expression of hub genes. The heatmap shows the expression levels of the hub genes in different human tissues based on the GTEx database. Expression levels are normalized using the Z-score normalization method.
Functional enrichment analysis revealed key pathways shared between COVID-19 and AD
We performed functional enrichment analysis to identify the common pathways altered in both AD and COVID-19. The cAMP signaling was the most significant downregulated pathway in both diseases (Enrichment FDR = 2.24E-07) (Fig. 5). The other significantly downregulated enriched pathways included taurine and hypotaurine metabolism and GABAergic synapse pathway (Fig. 5, Supplementary File 1). On the other hand, commonly upregulated genes were enriched in pathways involved in inflammatory responses and cell death, such as NETs, structures that are formed as a defense mechanism by neutrophils to trap and neutralize invading pathogens. However, these structures can also contribute to the development of various pathophysiological conditions, including sterile inflammation and autoimmunity [40] (Fig. 5, Supplementary File 1). Intriguingly, pathway analysis of identified hub genes within the PPI network (Fig. 3) also returned the cAMP signaling pathway as the most commonly altered pathway (Fig. 6), indicating a key role of this pathway in linking COVID-19 and AD. The cAMP signaling pathway plays an important role in a wide range of biological processes including metabolism, gene expression, ion transport, cell growth and differentiation, and neurotransmitter release [41]. In addition, it plays a role in the regulation of cyclic AMP response element-binding protein, a transcription factor that plays a critical role in learning and memory, as well as in neuronal development and plasticity [42].
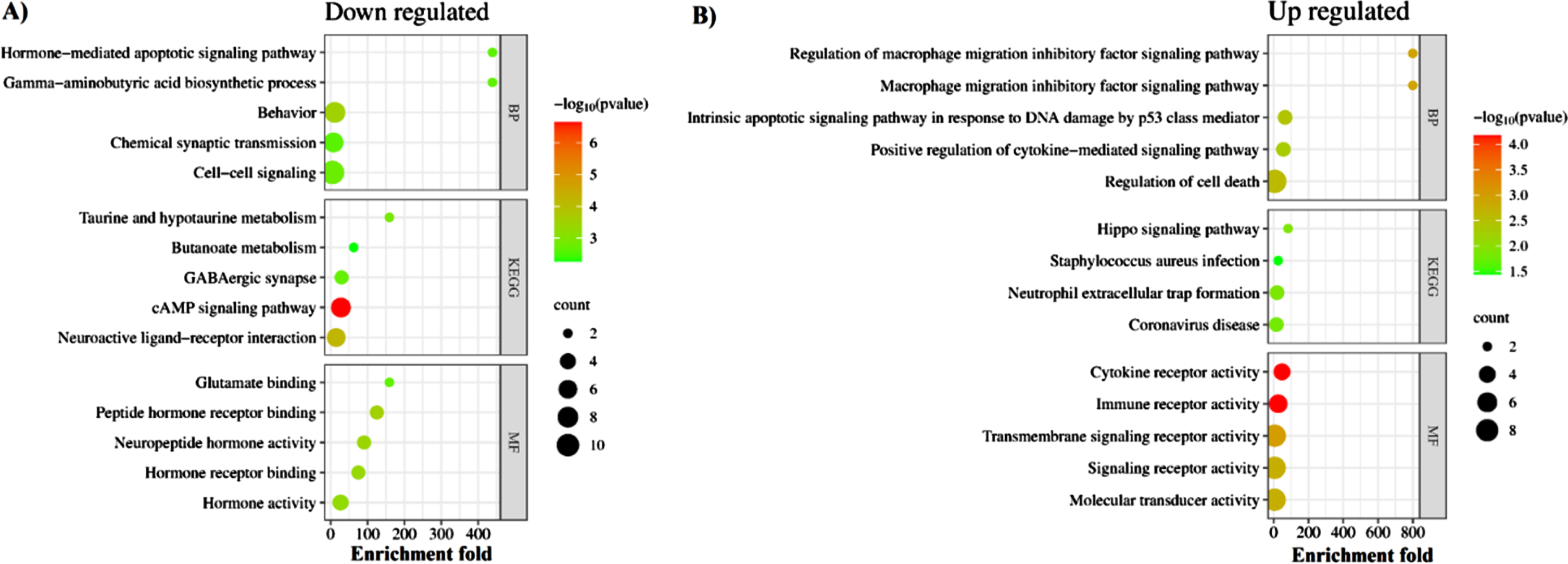
Molecular commonalities of AD and COVID-19 based on the gene expression signatures. Top 5 most significantly down- (A) and up- (B) regulated KEGG pathways, biological processes, and molecular functions enriched among the shared DEGs between robust AD and COVID-19 DEGs. The complete list of enriched terms is provided in Supplementary File 1.

KEGG pathways enriched by identified hub genes. The left panel shows the identified hub genes connected with their corresponding KEGG pathway. The cAMP signaling is the main altered pathway; however, other pathways are enriched almost by two or three genes. GAD1/GAD2 showed to be involved in most of the identified pathways. The right panel shows the gene ratio for each enriched pathway. Dot size and color indicate gene count and log10 p-values for each enriched pathway.
Gene–miRNA and gene-TF interaction analysis revealed regulatory networks of miRNAs and TFs interacting with common hub genes
We also performed a gene-miRNA interaction analysis to identify miRNAs interacting with identified hub genes. This analysis yielded a list of 100 known and unknown miRNAs regarding their association with AD and COVID-19 (Fig. 7). Among them, has-mir-16-5p, has-mir-27a-3p, has-mir-130a-3p, has-mir-107, and has-mir-182-5p were identified as the top five interacting miRNAs based on their degree and betweenness within the network. Moreover, as one of the identified hub genes, BDNF was characterized by the highest number of interacting miRNAs with 35 connections. However, our analyses yielded no interacting miRNA for NEUROD6 and CRH (Fig. 7).
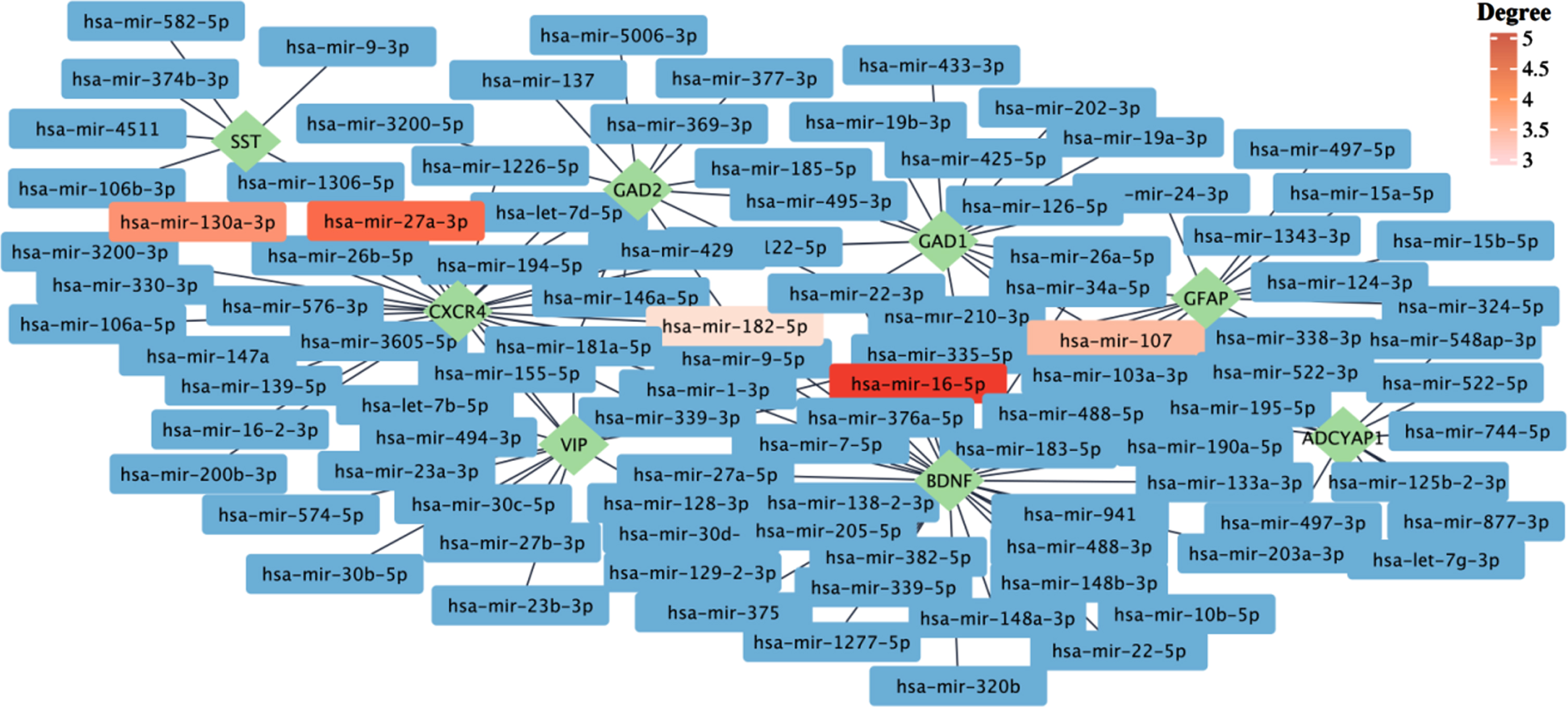
The gene–miRNA interactions network. The gene–miRNA interactions network is generated by using the TarBase database. The green rhombi show the hub genes, and the rectangles show the miRNA interacting with hub genes. The top five miRNAs based on their degrees are highlighted. The complete list of miRNAs interacting with hub genes can be found in Supplementary File 1.
To comprehensively understand the gene regulatory network of common hub genes of COVID-19 and AD, we next performed gene-TF interaction analysis. Our analysis identified 104 known and unknown TFs related to their association with AD and COVID-19 (Fig. 8). The top five TFs (based on network degree) include; Suppressor of zeste 12 protein homolog (SUZ12), Switch-independent 3 family transcription repressor B (SIN3B), RE1-silencing transcription factor (REST), Signal transducer and activator of transcription 3 (STAT3) and B-cell-specific moloney murine leukemia virus integration site 1 (BMI1) (Fig. 8).
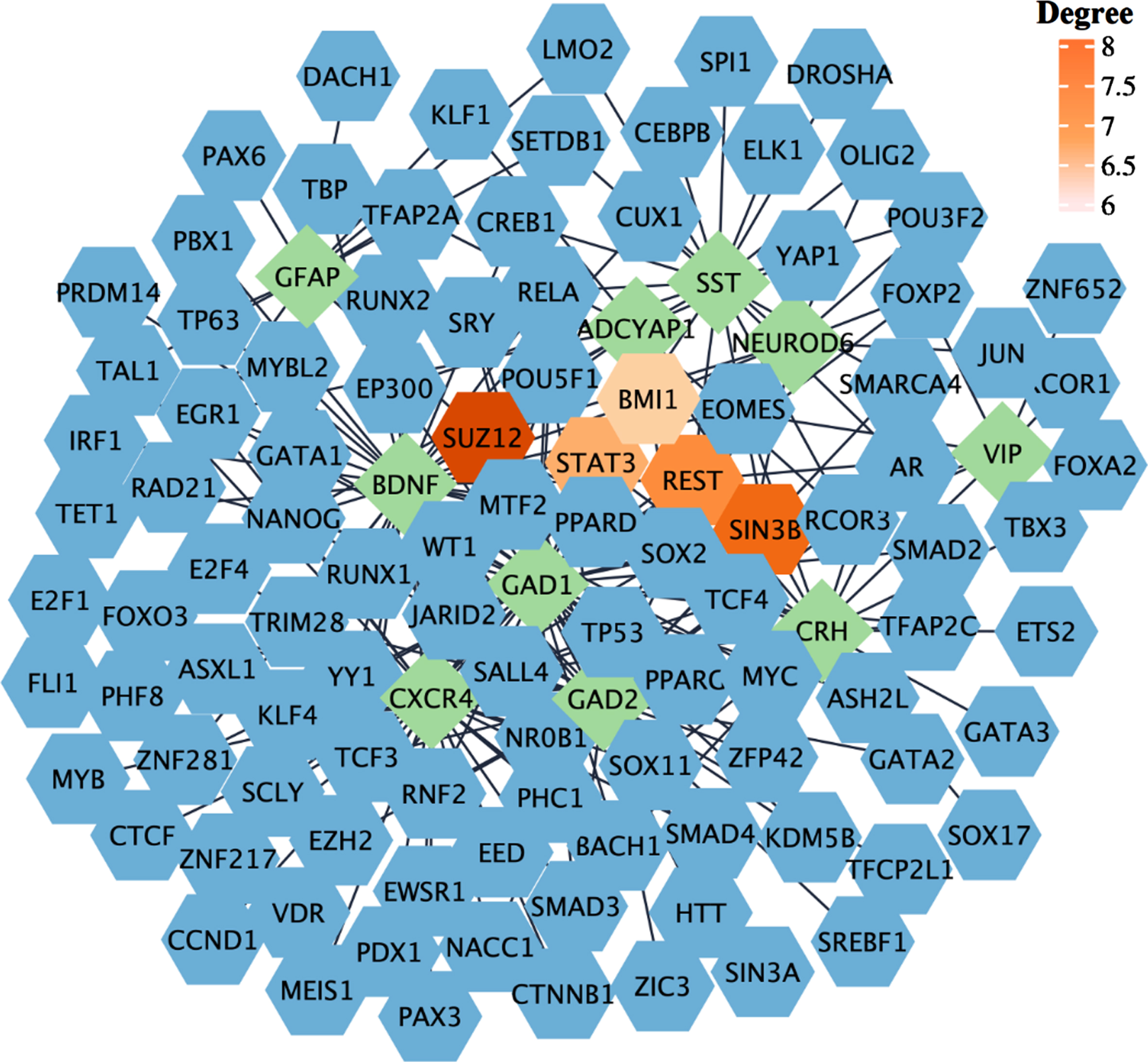
Gene-TF interaction network. The network was generated based on the ChEA database. The green rhombi show the hub genes, and the blue hexagons show the TFs interacting with hub genes, The top five identified TFs based on their degree are highlighted.
Data mining approach for validating association of identified miRNA and TF targets in AD
Data validation using a literature mining approach was performed as a complementary analysis to our bioinformatic study to validate and interpret our results more comprehensively and accurately. Data validation was performed for the top five identified miRNAs and TFs. The results of this analysis further identified published reports on the role of some of these targets in AD and/or COVID-19 and suggested that these identified miRNAs and TFs might be utilized as pharmacological targets. In addition, our analysis also revealed no published reports for some of our identified targets. The number of published papers on the top 5 miRNAs and TFs in each disease has been shown in Table 2 (Details are provided in Supplementary File 1). It is important to note that in this study, we used the names of identified miRNAs or TFs along with disease names to retrieve relevant publications on their association with COVID-19 and AD. However, it is possible that some of the identified publications may not be directly related to our study. For example, our analysis identified one related paper for has-mir-182-5p, but it focused on multiple miRNA changes in prion-infected animals and discussed the role of some of these miRNAs in the AD [43]. While the involvement of has-mir-182-5p in multiple pathological conditions such as cancer, chronic heart failure, and ischemia/reperfusion (I/R) kidney injury has been widely investigated [44–46], its specific role in AD and COVID-19 remains to be investigated. However, other miRNAs provided interesting evidence in line with our results, such as mir-27a-3p. Micro-27a is located on human chromosome 19 and is processed to form miR-27a-3p [47]. miR-27a-3p is implicated in various cancer types and has recently been found to be decreased in the serum and cerebrospinal fluid (CSF) of AD patients [48]. A negative association has been observed between the level of miR-27a-3p and the degree of brain Aβ deposition, indicating its involvement in the progression of the AD [48]. This miRNA has recently been found to specifically interact with one of the six regions on the viral RNAs that are primarily bound by specific miRNAs, and its level has been found to be increased in hospitalized COVID-19 patients as well [49, 50]. Likewise, the top identified TFs, such as STAT3, showed the highest number of published evidence for both diseases. In contrast, we found no information regarding the role of SIN3B in either disease. STAT3 is a TF with multiple key roles in development, and its increased phosphorylation has been found in the brain of both AD animal models and patients [51]. While there is currently no evidence regarding the role of SIN3B in COVID-19 or AD, its expression has been shown to be required for cellular senescence, and its downregulation is associated with tumor progression [52].
Number of published studies on the role of identified miRNAs and TFs in AD and COVID-19
DISCUSSION
In this study, we comprehensively analyzed transcriptome datasets of 638 brain samples, using a novel integrated genomic approach RRA method to identify robust genes altered in AD and COVID-19 infected brains. Our analysis showed that downregulation of cAMP signaling, Taurine and hypotaurine metabolism, and GABAergic synapse pathways, and upregulation of inflammatory pathways, such as neutrophil extracellular trap formation pathways, are common signatures of both disease conditions. The cAMP signaling pathway is crucial for many biological processes. Its deregulation has been associated with aging and age-related diseases, and increases in cAMP levels have been shown to reverse some of the adverse effects of aging [53]. It has also been shown to play a key role in long-term memory formation [54]. Decreased levels of cAMP has been reported in AD animal models, and pharmacological interventions that increase cAMP levels have been shown to be beneficial for neuronal protection, learning, and memory improvement in animal AD models [55, 56]. Also, the elevation of intracellular cAMP proved protective against COVID-19 Immunoglobulin G-induced procoagulant platelets and was suggested as a potential therapeutic target [57]. Intriguingly, taurine deficiency has been suggested as a critical driver of aging and taurine supplementation increased health and life span [58]. In addition, the level of taurine has been reported to be decreased in the blood and CSF of AD patients and was associated with cognitive scores [59]. Similarly, the serum level of taurine is decreased in COVID-19-infected patients [60]. Neuroprotective effects of taurine have also been reported in animal models of AD and Parkinson’s disease, reducing the Aβ aggregation and inhibiting neuroinflammation and microgliosis [59]. Intriguingly, adding taurine to drinking water significantly improved cognitive impairment and memory in mouse models of the AD [61]. In contrast, there are also reports indicating increased plasma levels of taurine in both COVID-19 and AD patients [62, 63]. Increased transportation of taurine across the blood-brain barrier has been reported during oxidative stress conditions, and NEUROD6 has been shown to be a key regulator of taurine transport through the blood-brain barrier [62].
On the other hand, the upregulation of inflammatory pathways, such as the neutrophil extracellular trap formation pathway, has been reported to be involved in AD [64]. The role of microglia, the brain’s resident population of immune cells, in neuroinflammation and AD has been widely investigated. However, it has been recently suggested that infiltrating blood-derived neutrophils into the central nervous system (CNS) can also contribute to AD pathogenesis and cognitive impairment [65]. In addition, neutrophils enter the CNS before the onset of cognitive impairment and are found to be highly abundant when memory loss is first observed. Blocking this process might have therapeutic potential to restore cognitive function [65]. Neutrophils are potent sources of reactive oxygen species, and their activation and associated oxidative stress have been shown to be associated with AD pathology in humans, and neutrophil-related inflammatory factors have been suggested as the potential biomarkers to predict memory and executive function decline in patients with mild AD [66]. In both human and animal models of AD, neutrophils are found to be co-localized with senile plaques and stained for NET markers (27). In addition, neutrophil adhesion in brain capillaries decreased cortical blood flow, leading to memory impairment in the mouse AD model [67]. Furthermore, neutrophils play a key role in pathogen clearance through phagocytosis, NETs, and generating reactive oxygen species [68]. SARS-CoV-2 infected human and animal models showed an increased number of neutrophils and proteins associated with the neutrophil degranulation [69, 70], and neutrophil degranulation has been identified as one of the main enriched pathways in proteomics analysis of COVID-19 patients [71]. Altogether, the results of functional enrichment analysis showed a downregulation of biological functions related to anti-inflammatory responses and, accordingly, upregulation of inflammatory responses may, in part, contribute to post-COVID-19 cognitive impairment and possibly AD development.
The result of hub gene analysis also showed some of the well-known genes involved in signal transduction, memory, and cognition, such as BDNF, a key neuroplasticity regulator. Its downregulation has been reported as one of the primary mediators of AD, multiple sclerosis, and Parkinson’s disease pathogenesis [72]. Interestingly BDNF also protects neurons against hypoxia and inflammation-induced pathogenesis, the key pathological events in COVID-19 [73, 74]. Recent studies reported lower serum levels of BDNF in COVID-19 infected patients and indicated a direct association between BDNF and cognitive decline in COVID-19 patients [75]. SST, another identified hub gene, is a multi-functional neuropeptide in a subpopulation of GABAergic interneurons [76]. SST’s expression level is shown to decrease with age and contributes to the formation of Aβ plaque deposition [76, 77]. While no data is available about Somatostatin in COVID-19 patients, its analogs are suggested as potential drugs for treating respiratory failure in diseases like COVID-19 [78]. Most of the identified hub genes are retrieved from downregulated genes; however, there are two hub genes from upregulated genes, including GFAP and CXCR4. GFAP, an astrocytic cytoskeletal protein, was upregulated in AD patients and cognitively normal older adults at risk of AD and correlated with amyloid-PET positivity and worse outcomes in global cognition [79, 80]. Increased plasma level of GFAP is also suggested as a potential prognostic marker in COVID-19, associated with the mortality risk [81]. CXCR4 is another upregulated hub gene in AD and COVID-19 patients [82, 83]. CXCR4 is a G protein-coupled receptor that binds to CXCL12 and triggers downstream signaling pathways associated with inflammatory pathways [82]. CXCR4 is involved in neuronal guidance and apoptosis via astroglial signaling and microglial activation [84]. Aβ plaques have an attraction effect on microglia, leading to the activation of an inflammatory cascade. This cascade involves the stimulation of CXCR4-dependent signaling by CXCL12 in both microglia and astrocytes, resulting in the release of pro-inflammatory cytokines, including tumor necrosis factor α (TNF-α) [85].
Next, we analyzed the miRNA-gene interaction network to identify miRNAs interacting with hub genes. During the last decade, exploring the involvement of miRNAs in various human diseases has gained much attention, mainly due to their crucial function in gene regulation, through mediating mRNA degradation and regulating transcription and translation via both canonical and non-canonical mechanisms [86]. Our analysis returned 100 miRNAs; among them, has-mir-16-5p was the most significant (Degree: 5; Betweenness: 1132.11) node, interacting with hub genes. Others include miRNA has-mir-27a-3p, has-mir-130a-3p, has-mir-107, and has-mir-182-5p among the top five identified miRNAs. Hitherto, results about the role of mir-16-5p in AD are inconsistent. While some indicated that upregulation of this mir-16-5p by Aβ deposition can lead to neuronal cell apoptosis through targeting BCL-2 [87], others suggested a protective role of this miRNA against Aβ-induced injury by targeting BACE1 [88]. The expression level of hsa-miR-16-5p was shown to be lower in COVID-19 patients than in healthy controls [50]. This miRNA can target many identified DEGs in macrophages (n = 15) and T cells (n = 10) in COVID-19 infected patients [89]. hsa-mir-16-5p was found to affect cell cycle, survival, and differentiation of T cells, and modulate inflammatory signaling and cytokines, including IL-1β, IL-6, and TNF-α, and NF-κB mTOR-related pathways and genes [90]. In addition, a recent study of in silico analysis suggested a regulatory role of has-mir-16-5p and has-mir-27a-3p in the angiotensin-converting enzyme 2 (ACE2) network [90]. The ACE2 receptor, found in several human organs, is the entry point for SARS-CoV-2 and SARS-CoV into host cells [91]. A lower level of mir-27a-3p has been reported in AD patients’ CSF, accompanied by high tau levels and low levels of Aβ [92]. It has been reported that miR-27a-3p can downregulate glycogen synthase kinase 3β (GSK3β) and activate the wnt/β-catenin signaling pathway, which ultimately helps in maintaining the integrity of the blood-brain barrier [93].
Although, in our analysis, we have only performed data mining for the top five identified miRNAs, there is also evidence of the association of several other enriched miRNAs with AD and COVID-19. For example, the downregulation of mir-124-3p, one of the mir-targeting identified hub genes, has been reported in AD patients [94]. Interestingly, mir-124-3p showed to decrease abnormal hyperphosphorylation of tau protein and subsequent cell apoptosis by regulating caveolin-1, phosphoinositide 3-kinase (PI3K), phospho-Akt (Akt-Ser473)/Akt, phospho-GSK3β (GSK3β-Ser9)/GSK3β pathway [95]. Conversely, upregulation of mir-7-5p has been shown in AD patients, contributing to AKT and GSK3β dephosphorylation and insulin resistance in neuronal cells, accelerating the progression of Aβ plaque and neurofibrillary tangles formation via multiple mechanisms [96, 97]. Decreased expression levels of mir-124-3p and mir-7-5p have been observed in COVID-19 patients compared to healthy controls [98]. Intriguingly, mir-7-5p and mir-24-3p were found to directly inhibit S protein expression and SARS-CoV-2 replication [99].
In addition, there are pieces of evidence on some of the identified TFs such as REST, and SUZ12. Consistent with the literature, our analyses revealed that while the expression of REST decreased, SUZ12 expression increased in AD brains in comparison to the control [100, 101]. REST has been shown to protect against AD via downregulating genes that promote cell death and AD pathology and trigger stress response gene expression. In addition, REST also protects neurons against Aβ induced toxicity and oxidative stress, and its deficiency leads to age-related neurodegeneration [100]. The protective role of glial REST against neurodegenerative diseases also has been linked to inhibitory effects on innate immunity and inflammation [100].
STAT3 is another top-identified TF involved in various physiological processes, including immune reactions [102]. Increased phosphorylation of STAT3 and its abnormal activation has been reported in both the hippocampus of AD mouse models and postmortem AD brain, which is critical for the secretion of cytokines involved in neuroinflammation, and correlated with the presence of reactive astrocytes in animal models of AD [103]. Additionally, STAT3 could serve as a transcriptional regulator for BACE1, the principal enzyme involved in the production of Aβ, and STAT3 inhibition was shown to reduce the level of BACE1 and neuroinflammation [51]. The STAT-3 inhibition as a downstream element in the IL-6/JAK/STAT-3 axis is also suggested as a therapeutic strategy to mitigate COVID-19 severity [104]. STAT-3 may play multiple roles during COVID-19 infection, such as instigating pro-inflammatory reactions, initiating the cytokine storm, disrupting the immune response balance, impairing anti-viral immune responses, and intensifying lymphopenia [104, 105]. Overall, our gene regulatory network analysis identified known and unknown genetic components of biological pathways associated with COVID-19 infection and AD development. The identified miRNAs and TFs can be targeted for therapeutic purposes to reduce the risk or delay the development of COVID-19-related neurological pathologies and AD.
In conclusion, our study has unveiled significant insights into the molecular mechanisms linking COVID-19 infection and AD development. Through comprehensive transcriptomic analysis, we have identified shared transcriptional signatures in the frontal cortex of individuals affected by AD and COVID-19, thereby illuminating the common pathways and their genetic modulators involved in the progression of both diseases. Notably, we have observed downregulation of the cAMP signaling pathway and perturbations in taurine metabolisms, alongside upregulation of neuroinflammatory pathways such as NET formation. These findings suggest that the convergence of these molecular components may render COVID-19 patients more vulnerable to cognitive decline and AD. Moreover, our study has pinpointed several promising therapeutic targets for COVID-19 and AD, encompassing genes, miRNAs, and TFs. Restoration of cAMP levels and supplementation of taurine hold potential as neuroprotective interventions.
Modulating inflammatory responses and targeting specific hub genes, miRNAs, and TFs also present prospects for mitigating cognitive impairment in individuals experiencing post-COVID-19 complications and reduce the risk of developing AD. Nevertheless, it is crucial to note that the therapeutic targets identified in our study necessitate experimental validation to ascertain their clinical efficacy and safety. Therefore, further investigation employing animal models and large-scale clinical cohorts is imperative to validate and expand our findings.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This work was supported by NIH/NIA 1K01AG060040 and The Jeffress Trust Foundation Interdisciplinary Research Award to AK.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and/or its supplementary material.