
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by cognitive decline, memory loss, and behavioral impairments. Despite extensive research efforts, effective treatment options for AD remain limited. Recently, gene therapy has emerged as a promising avenue for targeted intervention in the pathogenesis of AD. This review will provide an overview of clinical and preclinical studies where gene therapy techniques have been utilized in the context of AD, highlighting their potential as novel therapeutic strategies. While challenges remain, ongoing research and technological advancement continue to enhance the potential of gene therapy as a targeted and personalized therapeutic approach for AD.
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia among the elderly [1]. There are two forms of AD, a familial form that affects less than 5% of the population and a sporadic form that is the most widespread [2, 3]. The latter is a combination of environmental and genetic risk factors. The major risk factors for sporadic AD are age and the presence of at least one copy of the apolipoprotein (APOE) ɛ4 allele. The presence of this allele is associated with a higher risk of developing AD and facilitates amyloid-β (Aβ) aggregation [2, 5]. In addition, APOEɛ4 inhibits Aβ clearance and degradation from the brain [6]. Other risk factors like cardiovascular disease, diabetes, and hypertension have also been associated with an increased likelihood of developing AD [7, 8]. The familial AD is caused by mutations in three genes, amyloid-β precursor protein (AβPP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2). In this form, the symptoms manifest earlier than in sporadic AD, normally between 30 and 50 years of age [9]. Clinically, the earliest symptoms include memory problems, as difficulty remembering recent events or conversations, changes in mood, anxiety, and alteration in sleep patterns and these changes can appear years before receiving a diagnosis of AD. As the disease progresses, depressive symptoms, apathy, and withdrawal are highly prevalent in preclinical or early stages of AD until the individual becomes disoriented and confused and memory loss is so severe to disrupt daily life [10, 11]. The major neuropathological hallmarks of AD are the accumulation of extracellular amyloid plaques and intracellular neurofibrillary tangles [1, 12], which contribute to synaptic and neuronal loss that leads to a severe atrophy of the brain (Fig. 1) [1, 13]. Amyloid plaques are made of a small, misfolded peptide called Aβ that is formed by the sequential cleavage of the AβPP by the β-secretase (or BACE) and γ-secretase enzymes. There are two main forms of Aβ, Aβ40 or Aβ42, respectively formed by 40 or 42 amino acids. The latter is the most abundant, insoluble with a higher rate of fibrillization [14]. Aβ peptide tends to assemble and form different types of aggregates referred to as oligomers, protofibrils, or mature amyloid fibrils that are the species found in the dense-core plaques [15]. Of all Aβ species, oligomers are considered the most toxic for neurons [16–18]. Neurofibrillary tangles are extracellular aggregates made of the tau protein [19]. Human tau is encoded by the MAPT gene located on chromosome 17q21. The physiological function of tau is to assembling and stabilizing microtubules and its activity is regulated by phosphorylation [19]. However, in AD tau becomes hyperphosphorylated, detaches from microtubules, consequently contributing to synaptic dysfunction [19, 20].
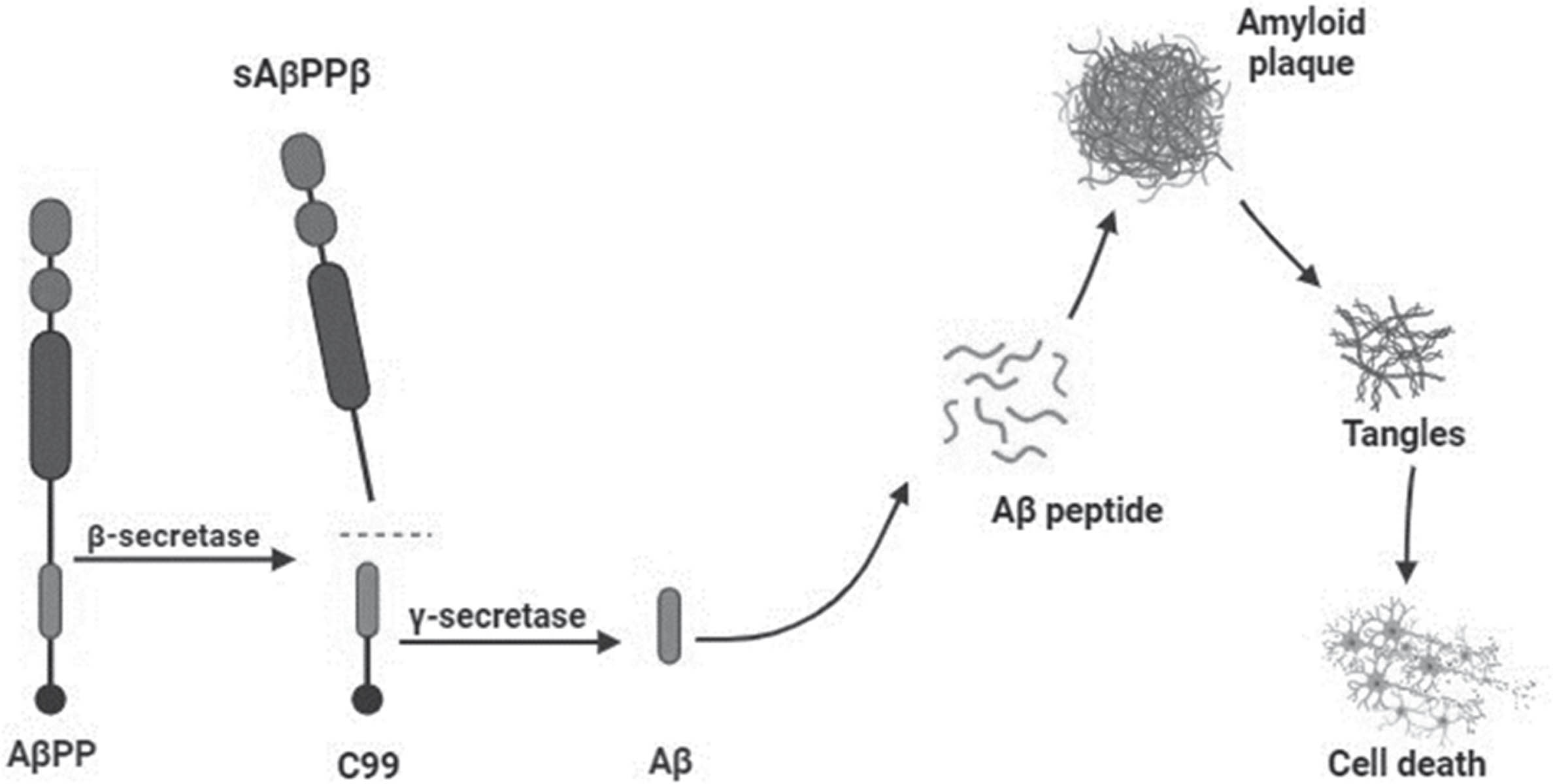
Aβ cascade hypothesis. Formation and involvement of Aβ in neurodegeneration.
Aβ and hyperphosphorylated tau have synergistic effect on impairing synapse function [21]. Emerging research suggests that Aβ and tau pathology can impact neurotrophins availability and signaling, compromising their trophic support and therefore synaptic function. Neurotrophins and synaptic function work in conjunction during the process of synaptic plasticity. Both are required to stabilize new synaptic structures and the loss of either can lead to cognitive impairments [22]. Neurotrophins are a family of proteins that play a crucial role in the development, survival, and function of neurons in the nervous system. Nerve growth factors (NGFs) together with the brain-derived neurotrophic factor (BDNF) form the neurotrophins protein family [23]. NGFs regulate neuronal growth, differentiation, and survival by binding to specific receptors on the surface of nerve cells [24, 25]. Several studies have shown that Aβ1 - 42 binds to the NGFR/p75NTR causing an increase in the extracellular levels of Aβ, which leads to neurotoxicity [26–28]. In AD, there is a progressive loss of neurons, particularly in regions critical for memory and cognitive functions, such as the hippocampus and cortex. This neuronal loss contributes to the characteristic cognitive decline and memory impairment observed in AD patients [29, 30]. NGFs are known to support the survival and function of neurons in these brain regions [31]. BDNF is involved in the survival and maintenance of neurons in the brain, as well as in promoting the growth and branching of dendrites, which are essential for neuronal communication [32]. Studies have shown that BDNF levels are decreased in the brain of individuals with AD. This reduction in BDNF levels is believed to be one of the factors that contribute to the degeneration and dysfunction of neurons in key brain regions involved in memory and cognition [33]. Additionally, among the factors known to promote synaptic plasticity, neurotrophins like BDNF have been proven to be also crucial for learning and memory processes [34]. It seems that one of the causes of impaired synaptic plasticity, observed in AD, is believed to be reduced availability of BDNF [35]. The decline in neurotrophins levels and impaired signaling observed in AD may contribute to the progressive neurodegeneration and cognitive decline seen in the disease [36–38].
It is estimated that of the 35 million people suffering from dementia around 75% are affected from AD [39], and this number is destined to approximately double every 5 years [1] unless a cure is found. The recent approval of three disease-modifying therapy for AD is viewed by some as a breakthrough, but many remain unconvinced by the data underlying this approval [40–42]. The latter lose their efficacy with the progression of the disease and the side effects overcome the benefits. The question to be determined is how much clinical effect we can expect from modifying only one aspect of AD pathology. Pre-symptomatic treatments and combination therapy with multiple disease-associated targets seem likely to be a promising approach to fight a complex disorder such AD. Toward this end, there is an absolute need to develop novel therapeutic strategies to slow down the progression of the disease and improve cognitive decline in AD patients. Gene therapy could be a valid strategy on the road to find a cure for AD due to its potential to address the underlying genetic and molecular factors contributing to the development and progression of the disease. By altering or inducing the expression of specific proteins it could provide neuroprotection and eventually improve the underlying pathogenic mechanisms [43]. In this review, we will present a summary of the recent findings in the field of gene therapy in AD, and we will focus on the use of adeno associated viruses (AAVs) as vectors for delivering therapeutic genes into target cells. In addition, we will explore the advantages and potential risks of this groundbreaking field to gain a deeper understanding of whether gene therapy could be the promising avenue to ultimately discover a cure for AD.
AAV MEDIATED GENE THERAPY
Utilizing AAVs as system to deliver therapeutic genes holds great promises in the field of neurodegenerative disease [43]. AAVs are small and non-pathogenic viruses particularly suited for this purpose, due to their ability to efficiently deliver therapeutic genes to specific target cells in the brain [44]. In doing so, it could be possible to restore or enhance the function of damaged neurons [44]. AAVs can also deliver gene modifying tools such RNA interferences (RNAi) or CRISPR-Cas9 system to target, silence or edit specific genes [44, 45]. This approach holds the potential to not only halt disease progression but also reverse or slow down the debilitating symptoms associated with neurodegenerative disease [46–48]. The mechanism by which AAVs work in gene therapy involves several key steps. AAVs are engineered to carry the desired therapeutic genes, RNAi or CRIPR-Cas9 system (Fig. 2A–C) [49, 50]. These modified AAV vectors are then injected intravenously or directly into the brain. AAVs can infect a wide range of cells, but their tropism, or the cells they can specifically target, can be modified by altering the viral capsid proteins [49, 51]. Once the AAVs reach the target cells, they attach to specific receptors on the cell surface. The virus is then taken up into the cell through endocytosis, the viral capsid proteins are shed, allowing the viral genome to be released and exposed within the cell. The AAV genome, containing the therapeutic gene, is a single-stranded DNA molecule. In the nucleus of the target cell, the viral DNA is converted into a double-stranded molecule and integrates into the host cell’s genome. Once integrated, the therapeutic gene becomes a permanent part of the target cell’s genetic materials. The host cell’s machinery then reads the therapeutic gene and produces the corresponding protein. The specific protein being produced depends on the nature of the therapeutic gene inserted into the AAV vector [49, 52]. As far as RNAi, once inside the cell regulates gene expression by silencing specific target genes. In RNAi-based gene therapy, short interfering RNAs (siRNAs) or small hairpin RNAs (shRNAs) are used to target and degrade specific mRNA molecules, thereby preventing the production of the corresponding protein [50, 53]. CRISPR-Cas9 is a powerful gene editing tool that enables precise modification of DNA sequences. The Cas9 protein, guided by a small RNA molecule called a guide RNA (gRNA), can be programmed to target specific DNA sequences, and introduce genetic changes, such as gene knockout, gene insertion, or gene correction. AAVs can be engineered to carry the Cas9 gene along with the gRNA sequence. Once delivered into the target cells, the AAV vectors can release the Cas9-gRNA complex, which then binds to the target DNA sequence and initiates the gene editing process [50, 55]. For neurodegenerative diseases, the goal is often to produce proteins that can compensate for or correct the underlying genetic defect, restore normal cellular function, or provide protection to the affected neurons [43]. In neurodegenerative diseases such as AD, Parkinson’s disease and Huntington’s disease, where the underlying cause often involves genetic mutations or dysfunctions, gene therapy offers a potential avenue for treatment [47].
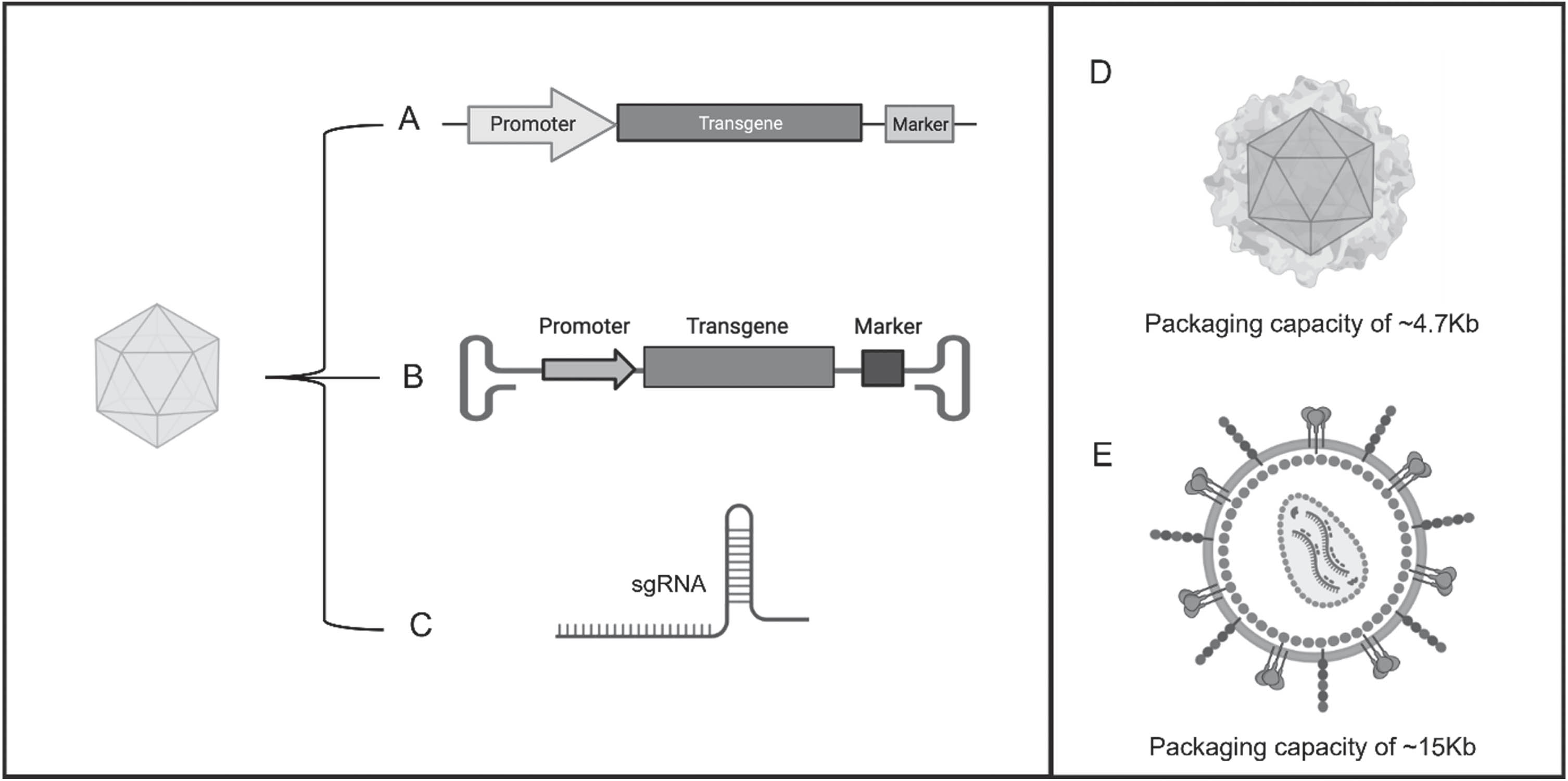
Adeno-associated virus (AAV) systems for gene therapy. AAVs are vectors designed to carry (A) the desired therapeutic genes, (B) RNAi to allow for the transient or stable knockdown of the gene of interest, or (C) single guide RNA (sgRNA) designed to effectively target site-specific cleavages of DNA. (D, E) Difference in packaging capacity between AAVs and lentivirus. AAVs are smaller and can carry up to a maximum of 4.7 Kb of genomic DNA, whereas a lentiviral vector is much bigger and can carry up to 15 Kb of genomic DNA.
Using AAVs as a tool in gene therapy has several advantages and disadvantages. One of the major advantages is the efficiency of gene delivery. AAVs have a high transduction efficiency, meaning they can effectively deliver therapeutic genes to target cells. They can infect both dividing and non-dividing cells, making them suitable for a wide range of tissues and organs, including the brain. Most importantly, growing evidence indicates that AAVs have a low immunogenic response compared to other viral vectors [56]. This means that they are less likely to trigger an immune response in the body, reducing the risk of adverse reactions, and they also have a low pathogenicity and are not associated with causing severe diseases in humans. Another important feature of using AAVs as gene delivery system is the long-lasting gene expression once the therapeutic gene is integrated into the host cell’s genome, which can persist for an extended period, potentially providing sustained therapeutic effects [57–59]. Most importantly, AAVs integrate their genetic material mainly into a specific region of the host genome known as the AAVS1 (AAV integration site 1) locus. AAVS1 is located on human chromosome 19, and it is considered a relatively safe integration site due to its association with minimal disruption of essential genes, therefore minimizing side effects [60, 61].
One of the principal limitations of AAV vectors is represented by the limited cargo capacity for carrying therapeutic genes. Their small size restricts the amount of genetic material that can be packaged into the viral vector. This limitation can be a challenge when delivering larger genes or multiple genes simultaneously. This problem could be solved by utilizing other viral vectors such as lentiviruses that have successfully been used for delivering larger genetic constructs (Fig. 2D, E) [62–64]. However, it needs to be taken into consideration that unlike AAVs, lentiviruses do not have a specific integration site. They integrate into the host genome at random locations, and this can lead to a potential risk of insertional mutagenesis, where the viral integration disrupts or activates nearby host genes, causing possible side effect [65]. Toward this end, while both AAVs and lentiviruses are widely used for gene delivery, their different integration mechanisms can influence the choice of vector based on the specific goals of a given application. Another problem with AAVs is that even though they have a low immunogenicity compared to other viral vectors, they can still trigger an immune response in some individuals. This immune response may limit the effectiveness of the therapy or cause adverse reactions. AAVs have certain tissue tropisms, meaning they prefer infecting specific types of cells. This can be advantageous in targeting specific tissues, but it can also limit their efficacy in other tissues or organs [57–59]. It is important to note that ongoing research and advancements in gene therapy are continuously addressing these limitations and optimizing the use of AAVs as a therapeutic tool.
GENE THERAPY TARGETS IN AD
Gene therapy is considered a valid strategy for AD due to its potential to address the underlying genetic and molecular factors contributing to the development and progression of the disease [66]. Although the traditional view of gene therapy is the modification of a single gene in a monogenic illness, its feasibility for illnesses with multifaceted etiologies, such as neurodegenerative diseases, is sometimes questioned. While there is ongoing research in this area, it is important to note that gene therapy for AD is still in the experimental stages and has not yet been approved as a standard treatment option. However, several potential targets have been identified and investigated [67]. This review will provide an overview of major targets of genetic therapy in AD, the preclinical and clinical trials to date, and examine the key hurdles that remain to be surpassed. A summary of these studies can be found in Tables 1 and 2.
Summary of gene therapy-based preclinical studies for AD discussed in this review
Summary of gene therapy-based clinical trials for AD discussed in this review. Information sourced from ClinicalTrials.gov
Targeting neurotrophins
In the context of AD, neurotrophins have attracted significant attention due to their potential to protect and promote the health of brain cells [68]. Studies examining the role of neurotrophins in AD have predominantly explored the role of NGF and BDNF in the cholinergic basal forebrain (CBF) and the hippocampus. CBF neurons require an ongoing supply of neurotrophins for maintenance and survival through their lifespan [69]. The idea is to introduce the neurotrophins into specific cells of the brain, which would then produce and release NGF or BDNF proteins, potentially leading to neuroprotection and improve cognitive function. Several preclinical studies have explored the use of NGF gene therapy for AD. In 1989, Ernfors and colleagues showed that implanting genetically modified cells that produced NGF in the brain of rats with a cholinergic loss increased the survival of CBF neurons [70]. In other studies, continuous NGF delivery after cell grafting in the CBF led to functional recovery in spatial memory in aged rats and reduction in Aβ deposition in aged monkeys [71, 72]. The development of AAVs as a system for in vivo gene delivery has opened a new venue for the delivery of NGF directly to the target cells, enabling them to produce NGF within their own cellular machinery [73]. This delivery method is also important because NGF is unable to pass through the blood-brain barrier naturally, but the utilization of gene therapy methods enables the delivery of NGF to the basal forebrain [74]. In two independent studies, using young rats, AAV-mediated NGF delivery resulted in enhancement of cognitive functions and neuroprotection. These effects have been observed even months after the initial viral injection, indicating the long-lasting impact of AAV-NGF therapy [73, 75]. Similar results were found in rats with cholinergic deficit induced by intraseptal injection of 192IgG-saporin [76]. Nagahara et al. [77] injected a lentivirus encoding NGF into the CBF region of elderly rhesus monkeys. They did not notice any adverse effects, the NGF expression was maintained for at least 1 year, and the number of cholinergic p75-labeled neurons in aged monkeys recovered to the levels of young monkeys.
To explore the potential benefits of NGF in human subjects Tuszynski et al. implanted into the forebrain of 8 individuals with mild AD autologous fibroblast modified to express human NGF [78]. In this phase I clinical trial (NCT00017940), after a period of 22 months, no long-term negative effects associated with NGF were observed. It was also reported that there was a potential improvement in the rate of cognitive decline and positive changes in brain metabolism [79]. After the discovery of adeno-associated virus serotype 2 (AAV2) vectors, which allow for secure and durable gene transfer to the brains of monkeys and humans, a phase I clinical trial (NCT00087789) was conducted using an AAV2-NGF [80]. Over a period of two years, the administration of AAV2-NGF was found to be safe and well-tolerated by the patients and no signs of accelerated decline were observed. Most interestingly, brain autopsy tissue analysis confirmed the sustained and targeted expression of NGF through gene-mediated mechanisms, validating its long-term bioactivity [80]. These encouraging results led to a multicenter controlled phase II clinical trial (NCT00876863) on 40 AD patients receiving AAV2 vectors expressing human NGF to the nucleus basalis of Meynert. Unfortunately, this trial failed to demonstrate significant improvements in primary clinical outcomes [81]. Due to the limited spread of the injected AAV2-NGF vectors coupled with incorrect stereotactic targeting, this multicenter clinical trial was unable to stimulate the cholinergic pathways needed for the improvement of cognition in AD patients and thus the efficacy of AAV2 therapy with NGF is still unanswered [82].
Another neurotrophins, BDNF, produces its effects through the modulation of neuroinflammatory processes [83]. Several studies have examined the effect of BDNF gene therapy in models of AD. Nagahara et al. proved that lentiviral-mediated BDNF gene transfer to J20 transgenic mice improved synapse loss and memory impairment independent of the effects on Aβ load [84]. These findings were confirmed in Tg2576 mice [85], aged rats and primates [84]. More recently, magnetic resonance imaging (MRI) has been used to guide AAV2-BDNF delivery to the entorhinal cortex of non-human primates to maximize targeted expression. This technique elevated BDNF labelling in the dentate gyrus, but the authors did not report any functional and behavioral changes [86]. In addition to these targeted and selective BDNF manipulations, the indirect enhancement of BDNF expression by gene therapy has also been investigated. Increasing levels of cyclic adenosine monophosphate-response element binding protein (CREB) binding protein (CBP) via lentiviral delivery induced BDNF expression and rescued learning and memory impairment in 3xTg-AD mice [87]. Interestingly, to eliminate virus-related safety issues and invasive procedures, Arora et al. considered brain-targeted lipid-based nanoparticles to deliver plasmid encoding BDNF to brain. These bifunctionalized nanoparticles enhanced BDNF expression and reduced toxic Aβ levels and plaque load in 6- and 9-month-old APP/PS1 mice, whereas synaptic proteins, synaptophysin, and PSD-95 were found to be increased in both age groups [88]. No adverse effects were observed throughout the treatment period, indicating that the strategy employed to address AD pathology was safe and effective [88].
Recently, a new phase I clinical trial using an AAV2-BDNF has been announced (NCT05040217). A total of 12 participants will be included, 6 early AD subjects and 6 mild cognitive impairment (MCI). The gene therapy vector used will be precisely delivered into the brain using stereotactic administration under MRI guidance. The subjects will be monitored for a predetermined period of 24 months and will continue to be followed indefinitely thereafter. The objective of this study is to minimize the loss of neurons and repair synapses in the brains of individuals diagnosed with early AD and MCI and to see if continuously administer BDNF into the brain could improve patients’ memory. At the same time, this trial would test the safety and tolerability of the therapy.
Although some of the results of preclinical and clinical studies have been promising, it has been challenging to precisely target neurotrophins to the appropriate brain regions. AD affects multiple regions of the brain, and delivering neurotrophins to specific areas while avoiding potential side effects in other regions is crucial. Additionally, the safety and long-term effects of neurotrophins gene therapy need further investigation. The disease is unlikely to be “cured” by increasing neurotrophins levels alone. AD is characterized by the accumulation of Aβ and neurofibrillary tangles, loss of synapses and neurons. It is not feasible to expect that a treatment focused solely on cholinergic neurons will effectively interrupt these extensive pathological mechanisms. Additionally, NGF may synergistically enhance the efficacy of anti-Aβ treatments, potentially surpassing the effectiveness of either treatment alone.
Targeting AβPP, Aβ production, and clearance
AβPP is particularly attractive as a target for gene therapy due to its central involvement in the production of Aβ. By modifying the expression or processing of AβPP through gene therapy, it is possible to achieve a persistent reduction in Aβ production. Another strategy could be to enhance its clearance from the brain by targeting enzymes involved in this process. This sustained action over time may offer the opportunity for early intervention to prevent or reduce the accumulation of Aβ before extensive neuronal loss and cognitive decline occur, leading to durable benefits in terms of slowing disease progression and preserving neuronal function.
The modulation of the amyloidogenic processing of AβPP has been a key strategy to control Aβ production. As two crucial enzymes catalyzing the intramembrane proteolysis of AβPP, β-secretase and γ-secretase have been the most promising targets for AD drug discovery [89]. Singer et al. performed one of the first preclinical studies using a siRNA-mediated knockdown of BACE1 through lentiviral delivery into the hippocampi of 10-month-old AβPP mice. This resulted in decreased Aβ pathology and amelioration of behavioral deficits [90]. To minimize immunogenicity, targeted delivery of self-made exosomes from dendritic cells loaded with BACE siRNA was used by Alvarez-Erviti et al. This approach efficiently reduced BACE1 levels in wild-type mice [91]. However, BACE1 knockout mice showed hypomyelination and increased seizures [92], indicating that its suppression might not be the best option as AD therapy. Furthermore, BACE inhibitors have shown promise in preclinical studies and early-stage clinical trials as a potential treatment for AD. However, in later stages of the trials they showed safety concerns and limited efficacy in some cases, highlighting the complexity of targeting Aβ metabolism as a therapeutic approach for this multifactorial disorder [93].
Accumulation of neurotoxic Aβ oligomers (AβOs) generated via amyloidogenic processing of AβPP, may also be a valid target using the AAVs approach. Recently, Selles et al. showed the feasibility of immunotherapy using viral vector-mediated gene delivery of a single-chain variable-fragment antibody (scFv) that selectively targets a subpopulation of AβOs and prevents the inhibition of long-term potentiation (LTP) in hippocampal slices and memory impairment in mice [94]. Loss of AβPP has been reported to cause neurodegeneration, decreasing neuronal plasticity and neurons susceptibility to cellular stress and reducing synaptic signaling activity [95]. To this end, expression of neuroprotective AβPP fragments may also be a feasible strategy in AD. A product of AβPP cleavage by the α-secretase is amyloid precursor protein-alpha (sAβPPα) that has neuroprotective properties [96, 97]. Tan et al. used a lentiviral vector to express human sAβPPα in the hippocampus of 3-month-old APP/PS1 mice to evaluate the potential role of sAβPPα in preventing the age-related onset of AD-associated neuropathology’s and cognitive deficits. They found that sAβPPα expression prevented deficits in spatial reference and working memory, as well as a partial rescue of the LTP deficit, even in the absence of an effect on Aβ accumulation and plaque load [62]. These findings suggest that the positive effects of sAβPPα may extend beyond its impact on Aβ pathology. Specifically, it raises the question as to whether sAβPPα could be beneficial in addressing impairments caused by tau-induced pathology, independent of Aβ. Toward this end, Baltissen et al. studied the potential of sAβPPα to reduce aberrant kinase activity and rescue tau pathology and synaptic deficits in THY-Tau22 mice. They used an AAV9 to express sAβPPα in the hippocampus of 9-month-old THY-Tau22 mice with established tau pathology. After 3 months they found a decrease in tau pathology that was linked to a restoration of normal activity of GSK3β and CDK5, two of the major kinases that phosphorylate tau. These results clearly demonstrated that sAβPPα holds therapeutic potential to mitigate tau-induced pathology [98].
Neprilysin is a membrane-bound metallo-endopeptidase and a key Aβ-degrading enzyme [99]. Genetic approaches to upregulate neprilysin were first trialed by Marr et al. who injected a lentivirus encoding human neprilysin into the hippocampus of AβPP transgenic mice. They found that Aβ deposits and neurodegeneration were significantly reduced [100]. Preclinical in vivo validation has shown that expression of neprilysin by AAV [101], or HSV [102] in transgenic mouse models of AD led to the amelioration of Aβ pathology. Intracranial administration of AAVs is clearly an invasive procedure which requires surgery. To avoid the risks associated with the intracranial injection, Iwata et al. developed a novel AAVs vector (AAV9) to achieve neuronal gene expression throughout the brain following peripheral administration. Through intracardiac administration, this newly engineered AAV caused a widespread gene expression of neprilysin throughout the brains of APP23 mice. This increase in expression of neprilysin reduced Aβ pathology and improved cognitive function [103]. Notably, the exogenous neprilysin primarily localized in late and early endosomes, which are regions where newly generated Aβ tends to accumulate. This localization may explain the effective clearance of Aβ from the brain [103]. One important aspect to consider for a neprilysin-based gene therapy is that peptides other than Aβ are degraded by neprilysin, including neuropeptide Y which may have biological implications. However, the increased processing of neuropeptide Y by neprilysin may also have neuroprotective effects [104].
Although pre-clinical studies have shown promising results, there are not yet any clinical studies using AAVs gene therapy to target Aβ production or degradation. The only active clinical trial targeting AβPP gene is still ongoing and contemplates intrathecal injection of a small interfering RNA (ALN-APP) through liposomes to decrease AβPP mRNA in the central nervous system (CNS) and consequently Aβ (NCT05231785). The purpose of this study is to evaluate the safety, tolerability, pharmacodynamics, and pharmacokinetics of single intrathecal dose of ALN-AβPP in adult patients with early-onset AD. The study is being conducted in two parts: single ascending dose phase and multiple dose phase in patients with early-onset AD. The planned enrollment for this study is up to 60 patients.
Targeting tau
The hyperphosphorylation of the protein tau (MAPT) has been associated with many neurological diseases, including AD. Several studies strongly suggest that elevated tau protein levels are causally linked to AD pathogenesis [19, 20]. Therefore, using gene therapy to regulate tau levels and phosphorylation could be a strategy to find a therapy for AD.
In AD, the MAPT gene is not mutated, and the aberrant accumulation of tau seems to be caused by an excessive production of the protein itself [105]. In fact, mice that overexpress wild type tau have cognitive decline and malfunctioning of synapses [106]; on the other hand, decreasing the endogenous levels of tau in AD mice prevented the deficits induced by Aβ [107]. Based on this and other findings, Kim and colleagues after an extensive cross-species screening strategy identify three proteins (USP7, RNF130, or RNF149) involved in the regulation of tau levels in the brain. Using an AAV-shRNA to efficiently knocking down the three proteins in PS19 mice, they found a reduction in pathological tau species (ptau and tau oligomers) and an amelioration of the disease phenotype [108]. Protein phosphatase 2A (PP2A) is responsible for ∼70% of tau dephosphorylation activity [109], and it is significantly decreased by ∼30% in AD brains [110]. Planel et al. have demonstrated that starvation-induced PP2A inhibition contributes to tau hyperphosphorylation even when tau kinases such as GSK-3β are inhibited [111]. PP2A activity is modulated by heat-stable inhibitors 1 (I1PP2A) and 2 (I2PP2A) with differential efficacy at distinct tau phosphorylation sites [112]. Lentiviral vectors expressing I2PP2A siRNA were designed to enhance PP2A activity in tau transgenic mice. As expected, hippocampal injection of I2PP2A siRNA reduced tau hyperphosphorylation at multiple residues, decreased GSK-3β activation, improved neuronal spine density, and rescued memory deficits in tau transgenic mice [63]. However, PP2A has multiple substrates impacting on cellular functions, so the side-effects of unregulated PP2A expression could be considerable. Different studies have shown that using tau monoclonal antibodies reduces tau pathology in transgenic animal models [113]. However, passive immunotherapy presents several potential limitations, such as low possibility to cross the blood-brain barrier, inflammatory reactions associated to the treatment, requirement for repeated dosing, patient’s compliance, and high costs. To overcome these issues, several groups have engineered antibodies as fragments, i.e., single chain variable fragment (scFv), to be delivered using viral vectors. Interestingly, Vitale et al. demonstrated the in vivo feasibility and efficacy of targeting pathological tau in the brain, by employing intramuscular (IM) delivery of vectorized anti-tau scFvMCI. Two different tau transgenic models received a single IM injection of AAV1-scFvMC1, showing a significant reduction of tau pathology and more interestingly even if the scFvMC1 was internalized by the microglia they could not find any inflammatory reactions in the brain [114]. However, understanding the consequence of tau knockdown at adulthood is fundamental to halt pathological tau while preserving healthy tau. Interestingly, Velazquez et al. showed that acute reduction of tau levels in the hippocampus of adult mice, using an AAV expressing a doxycycline-inducible short-hairpin RNA targeted to tau (AAV-shRNATau), impairs both motor coordination and spatial learning and memory, supporting the theory that tau loss of function can be involved in the clinical manifestation seen in AD patients [115]. Even if tau represents an interesting target for gene therapy, there are currently no clinical trials that use tau as a target for gene therapy.
Targeting APOE
APOE is one of the most extensively studied genes associated with AD. There are three main isoforms, APOEɛ2, APOEɛ3, and APOEɛ4. The different structure of the isoforms influences their ability to bind lipids and Aβ [116]. APOE is a type of cholesterol carrier which plays a crucial role in lipid metabolism. Several studies have found a strong correlation between the presence of APOEɛ4 allele and the increased risk of contracting late-onset AD [117], while possessing APOEɛ2 allele decreases the risk of contracting AD at all. Since APOE has such a key role in the development of AD, it is considered an attractive target to be used in gene therapy.
APOE has been implicated in Aβ clearance [118], but surprisingly in APOE knockout mice Aβ deposits are considerably low [119]. A viral delivery approach, such as transduction with AAV vectors encoding the three APOE isoforms, differentially alters Aβ deposition and clearance in an isoform-dependent manner [120]. Furthermore, Hu et al. conducted a study where they injected APOE-targeted replacement (TR) mice with an AAV8-APOEɛ4 or with an AAV8-APOEɛ2. They found that overexpression of APOEɛ4 decreased APOE lipidation and enhanced Aβ accumulation, whereas APOEɛ2 had opposite effects [121]. Rosenberg et al. evaluated the CNS distribution and safety of APOEɛ2 gene therapy for AD in non-human primates. They found that intracisternal injection of the AAVrh.10hAPOE2-HA was the least invasive and effective delivery method [122]. One fascinating idea is that CRISPR/Cas9 genome-editing system could be used to convert the APOE genotype of APOEɛ4 carriers to APOEɛ3 or APOEɛ2, thus ameliorating the toxic effects caused by APOEɛ4 and conferring the protective benefits of the other isoforms. In human-derived iPSCs, CRISPR/Cas9 was effective in altering the genome to produce isogenic lines homozygous for all three major APOE alleles [123]. Another study showed that in iPSC-derived neurons, CRISPR/Cas9 correction of APOEɛ4/ɛ4 to an APOEɛ3/ɛ3 genotype decreased cytotoxicity, tau secretion, and tau phosphorylation [124]. Moreover, Lin et al. showed that in iPSC-derived organoids converting APOEɛ4 to APOEɛ3 seems to attenuate Aβ pathology by improving astrocytic and microglial clearance of Aβ [125]. Although there are many challenges with using CRISPR/Cas9 in vivo, its potential as a specific and efficacious therapeutic is unlimited.
Currently there is an open label, dose-ranging study (NCT03634007) designed to assess the safety and toxicity of intrathecal administration of AAVrh.10hAPOE ɛ2 (LX1001). This serotype rh.10 AAV gene transfer vector expressing the cDNA coding for APOEɛ2, will be directly injected into the CNS/CSF of AD patient’s homozygote for APOEɛ4 (APOEɛ4/ɛ4). This trial started in 2018 and represents the first attempt at a personalized genetic approach to AD. The rationale of injecting APOEɛ4/ɛ4 patients with a virus expressing APOEɛ2 is to hopefully convert the gene APOEɛ4/ɛ4 to APOEɛ2/ɛ4 and see if the presence of APOEɛ2 allele may protect the patients against the toxic effects caused by APOEɛ4. This approach of identifying and correcting specific monogenic abnormalities has been successfully applied to other diseases [126] and researchers are hopeful that this will open new avenues to find new therapeutic strategies to treat AD.
CONCLUSIONS
Despite extensive research efforts over the years, the treatment options for AD remain limited. While gene therapy itself is not a new concept, expanding knowledge of AD etiology and recent developments in gene therapy systems provides new promise for this currently incurable disease. Gene therapy is a complex process involving variable factors such as the temporal and spatial specificity, gene regulation and the most common problem faced in neurodegenerative disease, gene delivery mechanism. In the specific case of AD, the blood-brain barrier limits the passage of gene therapy vectors, making it difficult to target specific brain regions affected by the disease. Moreover, the widespread impact on various anatomical regions involved in learning and memory poses a significant obstacle for gene therapy in AD. Therefore, it is necessary to develop protocols that focus on either targeting the regions affected in the early stages of the disease or achieving widespread gene delivery across multiple anatomical regions. On the other hand, gene therapy offers the potential for long-term benefits by providing sustained therapeutic effects. By modifying or replacing faulty genes, it may slow down or halt the progression of AD, leading to improved cognitive function and quality of life for patients. Furthermore, gene therapy can be combined with other treatment modalities, such as drug therapies or lifestyle interventions, to create a comprehensive and synergistic approach for AD treatment. Vector-based genetic therapies that focus on targeting pathways such as Aβ, tau, APOE, and neurotrophins have demonstrated promising results in mouse models. However, substantial evidence of their effectiveness in human clinical trials is yet to be established. Before these therapies can be applied in clinical settings, critical challenges need to be addressed. These challenges include managing the risks associated with vectors and delivery methods, overcoming the significant therapy costs, and identifying multiple drug targets. Only by overcoming these hurdles the potential of genetic therapies for AD could be fully achieved.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank Dr. Laura Maria De Plano for the assistance in creating Figs. 1 and 2 with BioRender.com.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.