
Abstract
Background:
The agrammatic variant of primary progressive aphasia (PAA), primary progressive apraxia of speech (PPAOS), or a combination of both (AOS-PAA) are neurodegenerative disorders characterized by speech-language impairments and together compose the AOS-PAA spectrum disorders. These patients typically have an underlying 4-repeat tauopathy, although they sometimes show evidence of amyloid-β and tau deposition on PET, suggesting Alzheimer’s disease (AD). Given the growing number of pharmacologic treatment options for AD, it is important to better understand the incidence of AD pathology in these patients.
Objective:
This study aimed to evaluate the frequency of amyloid-β and tau positivity in AOS-PAA spectrum disorders. Sixty-five patients with AOS-PAA underwent a clinical speech-language battery and PiB PET and flortaucipir PET imaging.
Methods:
Global PiB PET standardized uptake value ratios (SUVRs) and flortaucipir PET SUVRs from the temporal meta region of interest were compared between patient groups. For 19 patients who had died and undergone autopsy, their PET and pathology findings were also compared.
Results:
The results showed that although roughly half of the patients are positive for at least one biomarker, their clinical symptoms and biomarker status were not related, suggesting that AD is not the primary cause of their neurodegeneration. All but one patient in the autopsy subset had a Braak stage of IV or less, despite four being positive on tau PET imaging.
Conclusions:
Inclusion criteria for clinical trials should specify clinical presentation or adjust the evaluation of such treatments to be specific to disease diagnosis beyond the presence of certain imaging biomarkers.
INTRODUCTION
Primary progressive aphasia is a form of neurodegeneration that presents with language difficulties as the primary symptom. The agrammatic variant of primary progressive aphasia (PAA) often occurs concomitantly with progressive apraxia of speech (AOS+PAA), which is characterized by motor-speech difficulties, and may also occur in isolation from aphasia, in which case it is termed primary progressive AOS (PPAOS). These disorders are often grouped together as one form of neurodegeneration (i.e., non-fluent variant of PPA [1]), which we collectively refer to as AOS-PAA spectrum disorders. 1 Patients with AOS-PAA spectrum disorders typically have one of the following underlying pathologies: corticobasal degeneration, progressive supranuclear palsy, or Pick’s disease [2–5].
Amyloid-β (Aβ) deposition on positron emission tomography (PET) has been previously observed in these disorders [6–9]. This indicates a likely co-pathology of Alzheimer’s disease (AD), despite patients not presenting with typical AD symptoms; although, these studies did not assess tau uptake on PET scans. Given the growing number of pharmacologic treatment options for patients with AD, it is important to better understand the incidence of AD pathology in the AOS-PAA spectrum disorders, in which it is unlikely that AD is the driving pathology of the clinical symptoms [2–5]. The goal of the present study was, therefore, to examine the frequency of Aβ and tau deposition on PET in a large cohort of patients with clinically defined AOS-PAA spectrum disorder and determine if biomarker status affects clinical presentation. We hypothesized that Aβ and tau positivity would not be uncommon within AOS-PAA spectrum patients and that biomarker status would not affect clinical presentation.
MATERIALS AND METHODS
Sixty-five patients were recruited by the Neurodegenerative Research Group at Mayo Clinic to participate in an NIH-funded study investigating neurodegenerative speech and language disorders, which was approved by the Institutional Review Board of Mayo Clinic. A diagnosis of either PAA (n = 6), AOS+PAA (n = 36), or PPAOS (n = 23), all of which fall under the umbrella of AOS-PAA spectrum disorders, was given by consensus after clinical scores, writing samples, and video recordings of each patient were reviewed by at least two speech-language pathologists. Video recordings included a thorough speech-language evaluation by a speech-language pathologist that included the Apraxia of Speech Rating Scale (ASRS-3) [10, 11], the Western Aphasia Battery-Revised (WAB) [12], and the Boston Naming Test (BNT) [13], among other clinical tests, to assess the presence and severity of aphasia and apraxia of speech. Patients also were evaluated on conversational speech, narrative picture description, and supplementary motor speech tasks (vowel prolongation, speech alternating motion rates (e.g., rapid repetition of ‘puhpuhpuh’), speech sequential motion rates (e.g., rapid repetition of ‘puhtuhkuh’), and word and sentence repetition tasks). Memory was also assessed using the Camden Memory Test by a neuro-psychometrist, independent of the speech-language diagnosis [14].
All patients underwent a Pittsburgh Compound B (PiB) PET scan to assess Aβ, as well as tau-PET scan using [18F]flortaucipir to assess tau uptake at the same visit. A global PiB standardized uptake value ratio (SUVR) of≥1.48 and a temporal lobe tau meta-ROI SUVR of≥1.25 [15, 16] were used to determine Aβ and tau positivity, respectively.
Clinical profiles of AOS-PAA spectrum patients were compared across biomarker status groups; categorical variables were analyzed using Fisher’s Exact Tests, and continuous variables were analyzed using an Analysis of Variance, adjusting for age at the time of imaging where applicable.
Sixteen of the patients in the present study have died and undergone an autopsy. Standard neuropathological evaluations were performed by a neuropathologist (DWD) following current diagnostic protocols [17], and Braak stage [18] and Thal phase [19] were determined, as well as primary and secondary pathological diagnoses using published diagnostic criteria. All participants underwent apolipoprotein E (APOE) genotyping, as previously described [20].
RESULTS
Eight patients (12%) were both Aβ and tau positive, suggesting underlying AD pathology. An additional ten patients (15%) were Aβ positive but tau negative, and ten others (15%) were tau positive but Aβ negative (Fig. 1). Furthermore, Aβ SUVRs increased with age more so than tau SUVRs, and the two biomarkers were related to each other (Fig. 1).

Plots of PiB and tau PET by age at imaging, and between PiB and tau SUVRs. The dashed lines show the threshold of what is considered PiB and tau positive for each imaging modality. Each point above the dotted line represents a patient who is positive on that imaging biomarker. In the third graph, each point within the shaded quadrant represents a patient who is positive on both PiB and tau PET biomarkers.
When comparing clinical characteristics by biomarker status, there were no significant differences among groups (Table 1). Those who were negative for both biomarkers tended to be younger than those who were positive on one or both biomarkers.
Participants characteristics at baseline by biomarker status. Data shown are median (Q1, Q3), or n (%). For categorical variables, p-values are from Fisher’s Exact Test. For continuous variables, p-values are from Fit an Analysis of Variance adjusting for age at imaging where applicable
Data are shown as Median (Q1, Q3). *Value is different from other clinical groups (p < 0.001). Note: ASRS-3, Apraxia of Speech Rating Scale- Version 3; WAB AQ, Western Aphasia Battery- Revised Aphasia Quotient; BNT, Boston Naming Test.
Additionally, none of the participants who were Aβ-negative but tau-positive were APOE ɛ4 carriers.
Nineteen of the patients in this study underwent autopsy, and their pathology findings are shown in the data in Table 2. The most common primary pathologies were progressive supranuclear palsy (n = 8) and corticobasal degeneration (n = 6), with two cases having FTLD with TDP-43 inclusions, one having Pick’s disease, one having globular glial tauopathy, and one having unspecified 3R-4R frontotemporal tauopathy. The Braak stages ranged from I-V with a median of III and Thal phase ranging from 0–3; the National Institute on Aging–Alzheimer’s Association AD levels [21] are also reported in Table 2 and reflect the likelihood of AD co-pathology. We did not see any relationship between the tau-PET SUVR and Braak stage, with the three patients who were positive on tau-PET having Braak stages of I-II, while 11 who were negative for tau-PET had Braak stages of III-V at death. All six patients that were Aβ-PET positive had Aβ deposition at autopsy, with Thal phase of 3 in four patients and Thal phase of 1/2 in the other two patients. None of these six patients were tau-PET positive. Of the 13 patients that were Aβ-PET negative, five had evidence for Aβ deposition at autopsy.
PET and pathology findings for nineteen patients
1These patients with FTLD-TDP also had genetic progranulin mutations, as well as executive dysfunction and mild behavioral changes [3]. PiB, Pittsburgh Compound B; NIA-AA, National Institute on Aging-Alzheimer’s Association; PSP, progressive supranuclear palsy; CBD, corticobasal degeneration; FTLD-TDP, frontotemporal lobar degeneration tar DNA binding protein 43; GGT, globular glial pathology.
DISCUSSION
The results of this study show that Aβ and tau positivity is not uncommon in patients whose disease falls within the AOS-PAA spectrum, with 42% showing positivity in at least one AD biomarker. When comparing the patients in this study by biomarker status, there were no significant differences in clinical presentation. The interpretation of the positive AD biomarkers in this cohort is uncertain. Biomarkers did not appear to influence the clinical presentation, as there were no differences on tests of AOS, aphasia, or memory when comparing patients by tau and Aβ status at the group level. As we have previously shown [5], and as is clear in this study, these patients do not typically have AD as a primary pathology, and instead most commonly have a 4R tauopathy. Typical AD PiB and tau SUVRS tend to be much higher than those observed in our cohort: for example, the PiB SUVR range in typical AD has been previously reported as 1.80–4.66, and the tau SUVR range in typical AD, 1.3–3.27 [22]. These values are much higher than what was observed in our AOS-PAA spectrum cohort. That said, concomitant low-intermediate levels of AD pathology can be observed. Aβ-PET has a specificity of 100% but sensitivity of 50% in detecting diffuse Aβ plaques in patients with 4R tauopathies [23], and we observed similar results in our somewhat overlapping autopsy cohort. Hence, we can be relatively confident that Aβ deposition is present in the brains of the Aβ-PET positive patients. However, the utility of tau-PET to detect AD-type tau in these patients is more questionable, with Braak stages of IV or below generally undetectable by tau-PET in 4R tauopathies [23].
Similarly, in our autopsy subset, none of the patients with Braak IV showed positive tau-PET. These patients also rarely have AD co-pathology with a Braak stage greater than IV. Therefore, in most of our tau-PET positive patients it is likely that the elevated uptake reflects measurement variability, off-target binding, or inflammation, as some possibilities. Further, many of the tau PET values were only just above the cut-point, reflecting borderline scores. A higher tau SUVR cut-off of 1.29 [24] is likely more specific for the presence, or clinical meaningfulness, of AD pathology. Only five of the 65 total patients in the present study showed convincingly high tau above this 1.29 threshold and positive Aβ-PET values, shown in Fig. 2; thus, it is still possible that we are detecting the presence of concomitant intermediate-high likelihood AD in these cases, although autopsy evaluations of these patients will be needed to verify. It should be noted that patient 4 in Fig. 2 had a high PiB SUVR as well (2.99) but also had a different clinical presentation. While his overall clinical presentation was consistent with AOS-PAA spectrum, he also had some features of the logopenic variant of PPA, including phonological errors and impaired word retrieval, which is most commonly associated with underlying AD pathology; he also reported a family history of early onset AD. Thus, he may be a unique case in which AD co-pathology is driving some of his symptoms.
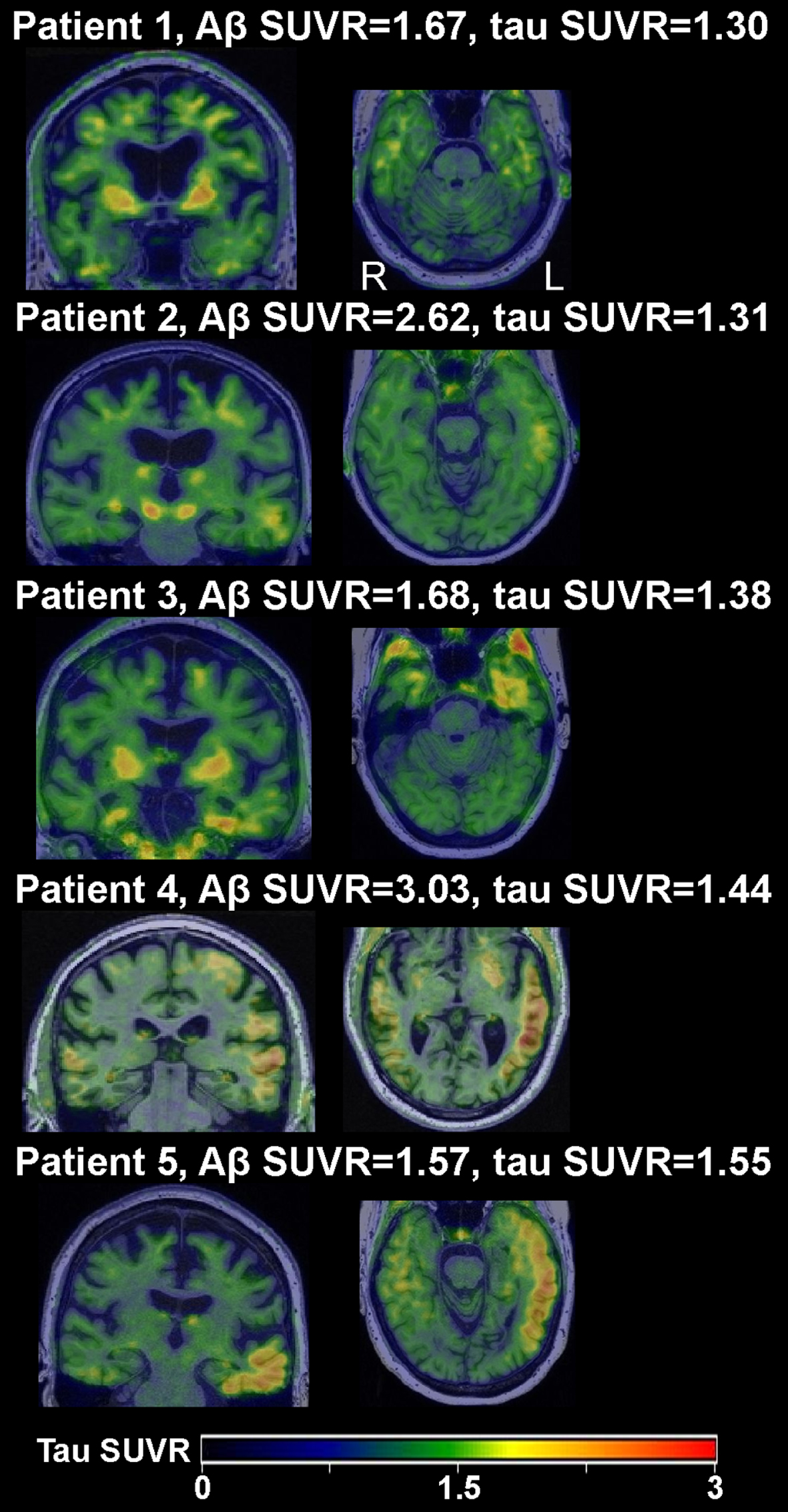
Tau-PET scans of patients whose temporal meta-ROI exceeded the 1.29 SUVR threshold.
In the present study, only uptake in the temporal meta-ROI was used in determining tau positivity on PET imaging, as this is the region most typically associated with AD dementia [18]. However, tau accumulation often corresponds to regional cortical atrophy [25–27], which for AOS-PAA spectrum disorder patients is typically in the inferior frontal and premotor cortices more so than in temporal regions in earlier stages of the disease [28–30]. It is possible that the patients who were tau-negative in the temporal meta-ROI may show increased uptake of both non-AD-type and AD-type tau in other cortical regions that correspond better to their clinical presentations [31]. The pathological findings in the autopsy subset revealed that patients who were negative on tau-PET imaging often had Braak stages of III or higher when looking at the whole brain.
A limitation of this study is that we were unable to compare the PiB and tau PET SUVRs within AOS-PAA spectrum disorder patients with an age-matched cohort of patients with typical AD; this was due to the fact that patients with typical AD do not undergo the same clinical battery as those with AOS-PAA spectrum disorder.
These findings are particularly important because the increasing number of targeted pharmacological options and ongoing clinical trials set enrollment criteria based on Aβ and/or tau PET positivity. As such, many patients with AOS-PAA spectrum disorders meet enrollment criteria, despite these clinical trials being aimed at patients with primary AD pathology. However, it is unlikely that these treatments will benefit these patients since their symptom-driving pathology is not AD. Including these patients may be problematic because clinical trial outcomes are often targeted to features of typical amnestic AD. Therefore, these targeted clinical trials should not only consider the biological variables but also clinical presentations. Alternatively, if patients without primary (or even atypical) AD are enrolled due to the presence of these AD biomarkers, the outcome measures should reflect the clinical impairments (i.e., change in speech/language impairments in AOS-PAA spectrum disorder patients). We plan to evaluate the significance of these biomarkers in the future in a larger autopsy-validated cohort.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This work was supported by National Institute on Deafness and Communication Disorders grants R01-DC010367 awarded to Keith A. Josephs, R01-DC14942 awarded to Keith A. Josephs and Rene L. Utianski, and R01-DC012519 awarded to Jennifer L. Whitwell.
CONFLICT OF INTEREST
Dr. Jennifer L. Whitwell is a Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review.
All other authors have no conflict of interest to report.
DATA AVAILABILITY
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
We avoid the use of the term nonfluent variant of PPA because we do not feel that patients with isolated PPAOS should be termed as having aphasia, when aphasia is, by definition, absent. Hence, we use “AOS-PAA spectrum disorder” to characterize these syndromes.