
Abstract
As a non-classical post-translational modification, O-linked β-N-acetylglucosamine (O-GlcNAc) modification (O-GlcNAcylation) is widely found in human organ systems, particularly in our brains, and is indispensable for healthy cell biology. With the increasing age of the global population, the incidence of neurodegenerative diseases is increasing, too. The common characteristic of these disorders is the aggregation of abnormal proteins in the brain. Current research has found that O-GlcNAcylation dysregulation is involved in misfolding or aggregation of these abnormal proteins to mediate disease progression, but the specific mechanism has not been defined. This paper reviews recent studies on O-GlcNAcylation’s roles in several neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, Huntington’s disease, Machado-Joseph’s disease, and giant axonal neuropathy, and shows that O-GlcNAcylation, as glucose metabolism sensor, mediating synaptic function, participating in oxidative stress response and signaling pathway conduction, directly or indirectly regulates characteristic pathological protein toxicity and affects disease progression. The existing results suggest that targeting O-GlcNAcylation will provide new ideas for clinical diagnosis, prevention, and treatment of neurodegenerative diseases.
Keywords
INTRODUCTION
O-GlcNAcylation is a non-classical post-translational modification (PTM), therein, O-GlcNAc transferase (OGT) enzyme attaches a single β-N-acetylglucosamine (GlcNAc) to the Serine/Threonine (Ser/Thr) hydroxyl of the protein in the O-glycosidic linkage, while the O-GlcNAcase (OGA) enzyme removes the GlcNAc residue. Almost all human organ tissues are enriched for O-GlcNAcylation, especially the brain [1]. The regulation of gene expression [2, 3], cell signaling [4, 5], nutrient metabolism [6–8], stress responses [9–11], and other important cellular functions are all impacted by this PTM. In the neurological system, O-GlcNAcylation is also necessary for neuronal survival and growth, synaptic transmission, and cognitive processes including learning and memory [12–14].
With the global population aging more and more, neurodegenerative diseases (NDs) are attracting more and more attention. The etiology and pathogenesis of NDs are complex, including oxidative stress, mitochondrial dysfunction, neurotoxins, and inflammatory damage. The common pathological feature of multiple NDs is the accumulation of abnormal proteins in the brain, such as amyloid-β (Aβ), tau, α-synuclein (α-syn), etc., which is thought to underlie neurodegeneration. Meanwhile, in NDs, O-GlcNAcylation dysregulation has been noted [15–20], and numerous experimental results have indicated that elevated O-GlcNAcylation may act as a protective factor in NDs pathology. Although the important connection between O-GlcNAcylation and NDs has been discovered, the precise molecular mechanism still needs to be elucidated in more detail. In this review, the biological functions of protein O-GlcNAcylation and its particular mechanisms in NDs are discussed, and we provide new ideas for the application of O-GlcNAcylation in clinical diagnosis and targeted intervention in NDs.
OVERVIEW OF THE O-GLCNACYLATION
The concept of O-GlcNAcylation and O-GlcNAc cycling
In 1984, Torres and Hart first found O-GlcNAc modified protein in mouse lymphocytes [21], then O-GlcNAcylation was generally discovered in bacteria, filamentous fungi, viruses, plants, protozoa, multicellular animals, and almost all other species. A 2021 meta-analysis showed that more than 5,000 proteins could undergo the O-GlcNAcylation [1]. The distribution of these proteins in cells are mainly in the nucleus and cytoplasm, and a small part is distributed in mitochondria, but in all of the human body’s main organ tissues [1]. Among them, pancreas, brain, heart, and skeletal muscle tissues show to have higher concentrations of O-GlcNAcylated proteins [22, 23], which is connected to the organ distribution of the enzymes that control O-GlcNAcylation. The large number of O-GlcNAcylated proteins suggests important and broad biological functions of this PTM in human body. The dysregulation of O-GlcNAcylation also leads to deleterious downstream effects and has been linked to the development of various diseases, including as NDs, diabetes, cancer, etc.
Unlike classical glycosylation modifications, O-GlcNAcylation linked a single GlcNAc group to the Ser/Thr residue of the protein in an O-glycosidic bond. O-GlcNAc modification is a dynamic, reversible, and inducible protein modification process, this dynamic cycling is regulated by intracellular substrates and two enzymes. The hexosamine biosynthesis pathway (HBP) produces uridine diphosphate-N-acetylglucosamine (UDP-GlcNAc), which is used as a donor substrate in this modification. HBP is a glycolysis pathway branch, requiring about 2%–3% intracellular glucose to complete the process [24], consisting of several enzymatic steps. Additional nutrients can also join into HBP by supplying a sugar donor. Despite the fact that just a little quantity of glucose enters the route, the HBP flux can play an important regulatory role as a nutrient receptor for cellular glucose metabolism, which can be influenced by other metabolic pathways [25]. Significantly, OGT or OGA, the two unique enzymes, is required for the addition or removal of O-GlcNAc respectively (Fig. 1).

A schematic diagram of the HBP and O-GlcNAc cycling. After glucose enters the cell by a glucose transporter, hexokinase (HK) phosphorylates it to glucose-6-phosphate (Glc-6-P), then is transformed by phosphoglucose isomerase (PGI) into fructose-6-phosphate (Fru-6-P). Subsequently, Fru-6-P reacts with glutamine via fructose-6-phosphate amidotransferase (GFAT) to generate glucosamine-6-phosphate (GlcN-6-P), then forming of N-acetylgluosine-6-phosphate (GlcNAc-6-P) with the participation of glucosamine 6-phosphate N-acetyltransferase (GNA1). Following that, GlcNAc-6-P is transformed into N-acetylgluconine-1-phosphate (GlcNAc-1-P) under the catalysis of N-acetylglucosamine-phosphate mutase (AGM1), the final reaction consumes uridine triphosphate (UTP), and producing uridine diphosphate-N-acetylglucosamine (UDP-GlcNAc), UDP-GlcNAc enters the O-GlcNAc cycle to complete the O-GlcNAc modification of proteins in the presence of O-GlcNAc transferase (OGT), O-GlcNAcase (OGA) performs the reverse. PUGNAc, NAG-thiazoline, NBut-GT, Thiamet-G, MK-8719, and ASN 90 are inhibitors of OGA. In addition, amino acid, lipid, and nucleotide metabolism all enter HBP at opportune locations to participate in the synthesis of UDP-GlcNAc. The intermediate metabolites can also enter into other sugar metabolic reactions, such as glycogen synthesis and glycolysis.
Two regulatory enzymes of the O-GlcNAc cycling
Structure and function of OGT
OGT, encoded by a single gene at the Xq13 in the human, has a highly conserved coding sequence and is critical for cell viability and mammalian growth [26–28]. The OGT protein contains two main regions: a region composed of multiple tetratricopeptide repeats (TPRs) and a multi-domain catalytic region. TPR consists of 34 amino acid motifs that can stabilize interactions with specific proteins and play a role in determining target substrate selectivity [29]. There are three natural variants produced by alternative splicing in mammals—nucleocytoplasmic OGT (ncOGT; 116 kDa), short OGT (sOGT; 75 kDa), and mitochondrial OGT (mOGT; 103 kDa), the most striking difference is the range of TPR [30–32]. The ncOGT and sOGT are the two major OGT types in cells, mainly in the nucleus and cytoplasm, and the nuclear localization signal (NLS) regions of OGT (DFP motif) is required for its nuclear import [33] (Fig. 2).
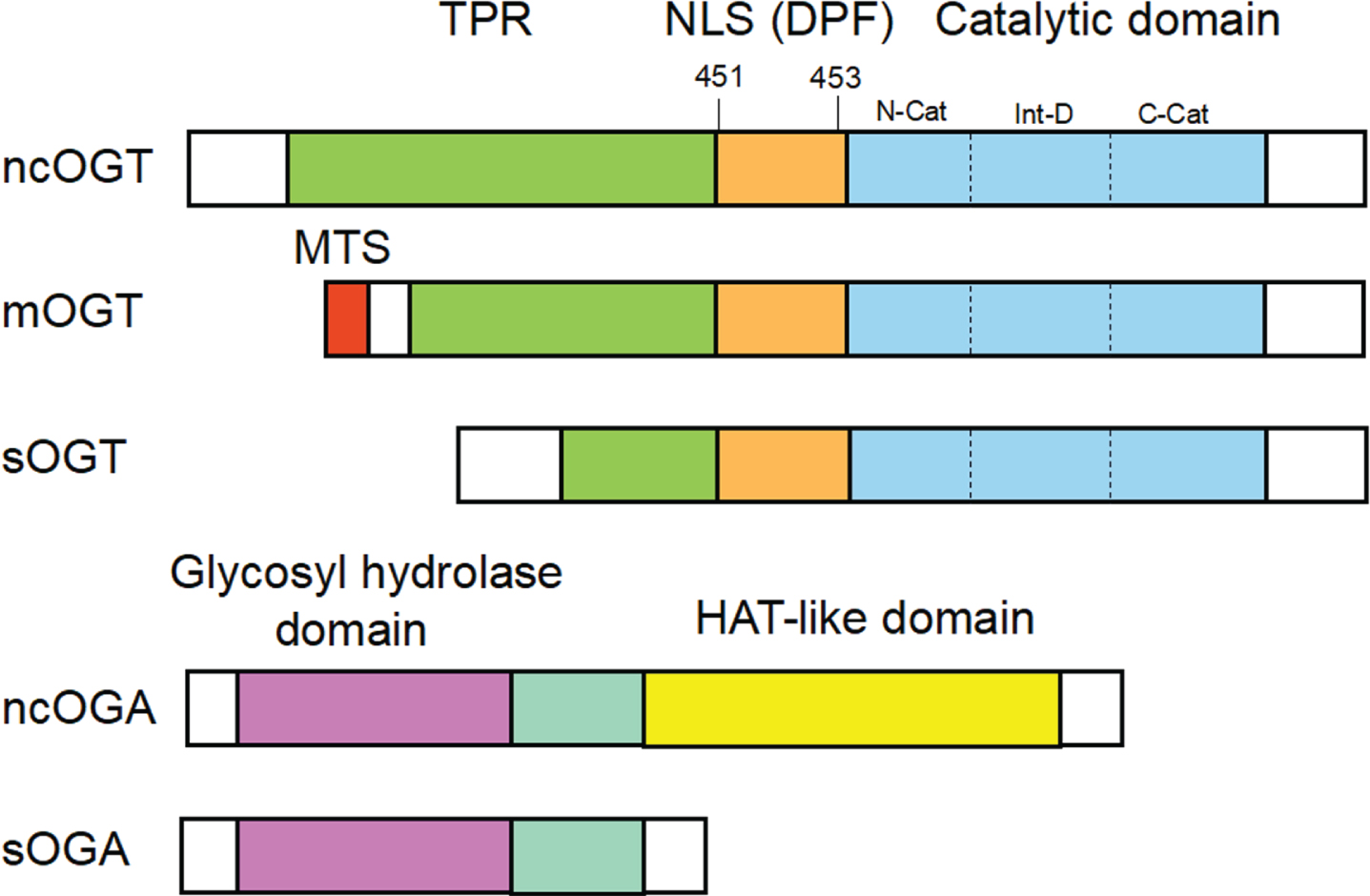
A schematic diagram of OGT and OGA structures. Mammals have three different types of OGT: nucleocytoplasmic OGT (ncOGT), mitochondrial OGT (mOGT), and short OGT (sOGT), they mainly consist of tetradecapeptide repeats (TPRs) in the NH2-terminus and the catalytic domain in the COOH-terminus. The ncOGT contains 13 TPRs, the mOGT contains 9 TPRs, and the sOGT contains 3 TPRs, which distinguish the three. The nuclear localization signal (NLS) regions of OGT is required for its nuclear import. Nucleocytoplasmic OGA (ncOGA) and short OGA (sOGA) are two isoforms of OGA, the former contains the glycosyl hydrolase domain in the NH2-terminus and the histone acetyltransferase (HAT)-like domain in the COOH-terminus, while the latter lacks the HAT-like domain.
Structure and function of OGA
The human OGA gene is found on chromosome 10q24 and encodes the OGA enzyme, which has an NH2-terminal hydrolase domain and a COOH-terminal domain (CTD). The CTD shares similarities with the histone acetyltransferase (HAT) family, although the CTD is usually classified as a histone acetyltransferase, it is short of the P-loop construction of AT enzymes needed to bind coenzyme A, showing that this domain may perform alternative functions [34]. There are two OGA forms. Nucleocytoplasmic OGA (ncOGA), one full-length protein of the OGA forms produced by alternative splicing, is mostly found in the cytoplasm. Whereas short OGA (sOGA) is largely found in the nucleus, which lacks the CTD (Fig. 2). OGA is also important for cell physiology, and OGA loss leads to mice embryonic lethality [35], delayed brain differentiation and neurogenesis [36].
O-GlcNAcylation and other post-translational modifications of protein
Protein’s PTMs can increase the functional diversity of the proteome through adding functional groups covalently or regulating proteolysis of subunits. In cellular compartments, O-GlcNAcylation is widespread and interacts with several PTMs widely, the most attention has been paid to phosphorylation and O-GlcNAcylation, the interaction between the two is involved in multiple biological aspects of the protein [37, 38]. The two have some things in common: they both modify the Ser/Thr residues of the protein and have a rapid cycle upon cell activation. But without the numerous kinases and phosphatases, O-GlcNAc modification involves just two specific enzymes as described above. Originally, Hart et al. [39] proposed a “Yin-Yang” concept to describe the connection between O-GlcNAcylation and phosphorylation. Then, Zeidan et al. [40] further summarized four different interplays between phosphorylation and O-GlcNAcylation, they proposed that there may be complicated crosstalks involving site-specific selection or simultaneous occupancy of the two PTMs. Moreover, they can also regulate each other through the production of large functional complexes, rapidly modifying the target protein substrates. Recently, Wang et al. [41] found epidermal growth factor (EGF) promoted OGT phosphorylation at Y976 and the binding of OGT to pyruvate kinase M2 (PKM2), stimulating PKM2 O-GlcNAcylation increased and enzymatic activity reduced, which reflects the tip of the iceberg of the wonderful relationship between them. In this paper, we focus on the subtle relationship between glycosylation and phosphorylation of tau in Alzheimer’s disease (AD), as detailed in the relevant sections below.
In addition to phosphorylation, O-GlcNAcylation and other PTMs, like ubiquitylation, methylation, and acetylation, also have some connections. It was shown that O-GlcNAcylation protects C. elegans from infection with Staphylococcus aureus and that this protective process is mediated through upregulation of proteins in the ubiquitination pathway [42].
Physiological function of O-GlcNAcylation
Regulation of gene expression
O-GlcNAcylation participates in various steps of transcription, which is precisely regulated by basal transcription and epigenetic chromatin remodeling mechanisms. The initiation of gene expression in eukaryote is regulated by three RNA polymerases (RNA pol), all of which have corresponding transcription factors to identify core promoters and construct the transcriptional pre-initiation complex (PIC) or initially transcribing complex (ITC) [43]. The TATA-box binding protein (TBP) is the common factor and is required for the formation of functional PICs [44]. Studies have shown that TBP was O-GlcNAcylated at T114, the O-GlcNAcylation here blocked the combination between TBP and B-TFIID TATA-box binding protein associated factor 1 (BTAF 1), which regulated TBP dynamics, thus causing major alterations of transcriptome and metabolic reorganization, influencing the expression of 408 genes [45]. In addition, RNA pol II’s CTD is O-GlcNAcylated [46], which is essential for the formation of PIC. RNA pol II is phosphorylated by the transcriptional kinase cyclin-dependent kinase 9 (CDK9) to promote transcriptional elongation [47, 48], after inhibiting it, both gene transcription and cellular proliferation are affected, moreover, homeostasis regulation of mRNA depend on OGT, too. This also explains the toxic effect of the combined targeting CDK9 and OGT on cancer cells expressing high CDK9 and OGT [49, 50].
Alternatively, O-GlcNAcylation affects gene transcriptional activity through modification of transcription factors. The network of pluripotency transcription factors (PTFs) regulates the pluripotency of embryonic stem cells (ESCs), it has been shown that O-GlcNAc modifies three core components of the PTFs network—OCT4, SOX2, and NANOG [26, 51]. Recent studies have shown SOX2, one of these components, is involved in pancreatic cancer cell self-renewal, and the stability of SOX2 is also dependent on O-GlcNAc modification [52]. Hence, given the importance of O-GlcNAcylation in gene expression, we could try to treat cancer by controlling the level of O-GlcNAcylation.
Regulation of cell signaling
Many signaling pathways are cross-regulated with O-GlcNAcylation signals, such as nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) signaling [53], protein kinase A (PKA)-cAMP response element binding protein (CREB) pathway [54], and insulin signaling [55], etc., affecting the expression of the downstream proteins, hence impacting all aspects of human physiology (Fig. 3). It is worth noting that dysregulation of OGT activity leads to a variety of cellular dysfunction, including various immune cells in addition to various tissue cells [56]. For example, downregulation of O-GlcNAc levels by OGT inhibitors hindered the differentiation and activation of monocyte derived dendritic cells (moDCs), which involves the participation of mitogen-activated protein kinase (MAPK) cascade and mTOR/AKT signaling axis, and thus affects the function of DCs [28].
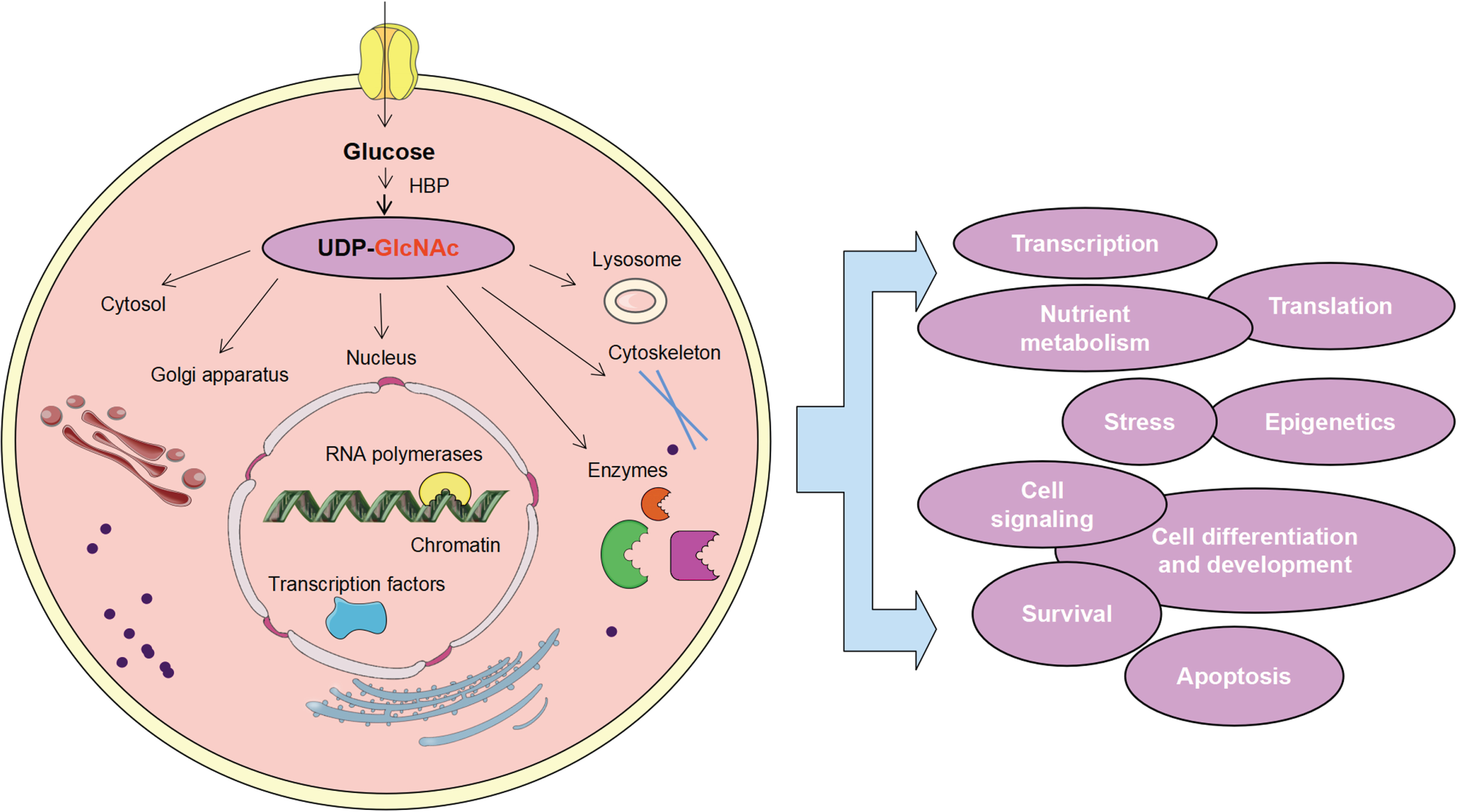
UDP-GlcNAc-mediated O-GlcNAcylation is engaged in various cellular processes. After glucose enters cells, UDP-GlcNAc is generated by the HBP, and O-GlcNAc was detected in various organelles, including lysosomes, cytoskeleton, nucleus, and Golgi apparatus, etc. O-GlcNAc modification plays an extensive role in the control of cell gene transcription, translation, epigenetics, nutrient metabolism, stress response, cell signaling, cell differentiation and development, cell survival and apoptosis, etc.
Regulation of nutrient metabolism
The sugar substrate for O-GlcNAcylation is UDP-GlcNAc, which is abundant in cells and closely related to the metabolism of nutrients, and its synthesis is affected by the glucose, amino acids, lipid, and nucleotides levels (Figs. 1 and 3). Meanwhile, O-GlcNAcylation also mediates multiple metabolisms at multiple levels. O-GlcNAc modification are involved in several cellular metabolic pathways including glycolysis, gluconeogenesis, pentose phosphate pathway, and lipogenesis [7, 57–59], O-GlcNAcylation affects pathways activity by directly modifying or indirectly regulating metabolism-related enzymes, for instance, phosphoglycerate kinase 1 (PGK 1) [7], glucokinase (GCK) [60], glucose-6-phosphate dehydrogenase (G6PD) [58], fatty acid synthase (FASN) [8], acetyl-CoA carboxylase 1 (ACC1) [61], etc.
Besides, O-GlcNAcylation also regulates metabolic flux by modifying the activity of metabolic transcriptional regulators. After the transcription factor SP1 is O-GlcNAcylated [2], its autostability and transcription of aquaporins (AQP 3) increase, which promote glycerol and glucose uptake to compensate Warburg-like glycolysis during the implantation window, maintaining the physiological demands of the endometrial cells, it suggests that the O-GlcNAcylation regulates metabolic reprogramming [62]. In addition, the mOGT modifies a variety of mitochondrial proteins, and dysregulation of mOGT influences mitochondrial function and cell bioenergetics through interactions with proteins [63].
Regulation of cellular stress and survival
O-GlcNAcylation is an important relieving component during stress (Fig. 3). After acute cold stimulation for 4 h, the levels of O-GlcNAcylation and OGT increase in liver tissues of piglets, and corresponding anti-apoptotic factor Bcl-2 increases, giving expression to the protective effect of O-GlcNAcylation during acute stimulation [11]. Miraculously, under the chronic stimulation of cardiac hypertrophy, O-GlcNAcylation signaling inhibited the PGC-1α and its downstream genes, leading to the disorder of cardiomyocyte metabolism and accelerating cell apoptosis [9]. One speculation for this contradiction is that prolonged elevated O-GlcNAcylation levels lead to sustained overall protein O-GlcNAcylation, which is thought to make metabolism disturbance persistent [64].
During oxidative stress, the increase of O-GlcNAcylation in human retinal microvascular endothelial cells (HRECs) reduces the cell damage induced by glyoxal through reducing the generation of reactive oxygen species (ROS) and decreasing cell apoptosis [65]. Furthermore, O-GlcNAcylation can impede proteasome’s action, affecting cell apoptosis [66], whilst inhibiting O-GlcNAcylation promotes the production of autolysosomes, which aids in the clearance of hazardous protein aggregates [67]. Thus, O-GlcNAcylation is also involved in the regulation of cell survival and death.
OVERVIEW OF THE NEURODEGENERATIVE DISEASES
NDs are a group of central nervous system (CNS) diseases with no clear etiology, insidious onset, chronic and continually worsening courses, they are aroused by the loss of selective neurons and (or) myelin sheath. Various NDs are usually defined by specific protein accumulation and anatomical changes, but there are often many similar pathologies processes linked with developing neuronal malfunction or mortality in different diseases, including proteotoxic stress and its concomitant ubiquitin-proteasome and autophagosome/lysosome system abnormalities, programmed cell death and neuroinflammation [68–70], etc. The pathological changes that accompany NDs are often irreversible, and the abnormal proteins that define NDs may exist before the clinical features, the disease courses often have developed to the middle or late stages, while treatment only alleviates the progression of the diseases. Misregulation of O-GlcNAcylation has been shown to be present in several kinds of NDs, and this condition is intimately linked to the development and accumulation of pathological proteins, revealing that these molecular pathological events play a key role for designing timely and feasible clinical diagnosis and prevention intervention schemes to delay disease progression.
THE ROLE OF O-GLCNACYLATION IN NEURODEGENERATIVE DISEASES
O-GlcNAcylation is highly enriched in the human brain, and O-GlcNAc cycling is essential for glucose homeostasis [71], mitochondrial function [72], and neuronal integrity [13]. Various NDs have been connected with dysregulated O-GlcNAcylation levels, details are as follows, and many proteins directly associated to the diseases are modified by O-GlcNAc and are modified in the wrong way, which is closely related to diseases progression and severity.
O-GlcNAcylation and Alzheimer’s disease
In terms of neurodegenerative diseases, AD is the most common, it is rapidly becoming one of the most serious worldwide diseases of the twenty-first century. An estimated 4.7 million people aged 65 or older have AD worldwide, and the number could reach 130 million by 2050 [73]. To date, the specific etiology and pathophysiology of AD remain unknown, and many factors can promote AD, such as genetic factors, environmental factors, immune factors, and more. The major pathogenic hallmarks of AD are extracellular amyloid plaques produced by the aggregation of Aβ and intracellular neurofibrillary tangles caused by the aggregation of tau protein [74]. In recent years, researchers have discovered that the O-GlcNAc levels are lower in AD patients compared to age-matched controls, which was involved in AD disease progression by regulating proteotoxicity, glucose metabolism and mitochondrial function, and neuroinflammation, etc.
O-GlcNAcylation and amyloid-β
The generally recognized “amyloid cascade hypothesis” in the pathogenesis of AD proposes that Aβ deposition in the brain is the key stage that eventually causes neuronal death and AD development [75]. Amyloid-β precursor protein (AβPP) experiences two metabolic pathways after being secreted and transported to the plasma membrane: non-amyloidogenic pathway and amyloidogenic pathway [76]. AβPP is split by α-secretase and γ-secretase at the plasma membrane in the non-amyloidogenic route, and does not produce Aβ, which accounts for about 90% of both pathways. In the amyloidogenic route, AβPP is taken up by endocytosis and transported to endosomes and lysosomes, where it is subsequently broken down by β-secretase and γ-secretase. AβPP is cleaved by β-secretase into a soluble secretion sAβPPα and a C-terminal fragment (CTF), the latter is further cleaved into an Aβ peptide by γ-secretase (Fig. 4). It appears that the tiny lysed Aβ oligomers are neurotoxicity [77]. Therefore, normal endocytosis, transporting, and cleavage of AβPP are thought to be essential for regulating Aβ production. Jean-Louis et al. [78] found that prostaglandin J2 (PGJ2), which mediated neuroinflammation, induced α- and β-secretase to process mature full-length AβPP by increasing O-GlcNAcylation, while decreased Aβ secretion may be related to transport disorders caused by dystrophic-like neurites, this also connects neuroinflammation and AD. Moreover, OGA inhibitors can reduce the endocytosis of AβPP and promote AβPP plasma membrane localization, thus decreasing amyloidosis [79]. Subsequent studies also discovered that AβPP was tend to be endocytosed from the lipid raft microregion of plasma membrane, and O-GlcNAcylation enhanced AβPP translocation to the non-lipid raft microregion, reducing the internalization amount of AβPP and the production of Aβ [80].
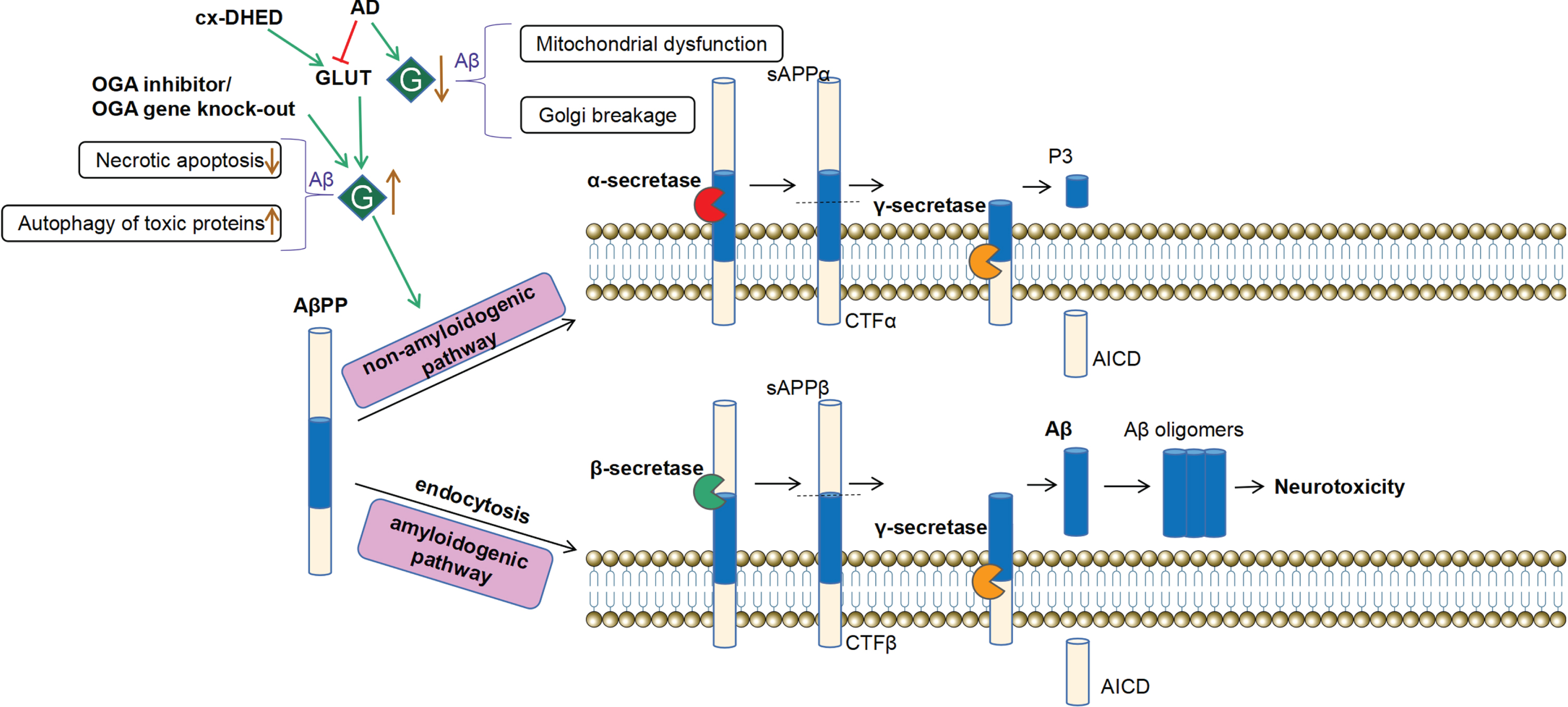
O-GlcNAcylation regulates the neurotoxic production process of amyloid-β (Aβ). The amyloid-β precursor protein (AβPP) has two metabolic routes. At the plasma membrane, α-secretase and γ-secretase degrade successively AβPP in the non-amyloidogenic route, and does not produce Aβ. AβPP enters the amyloidogenic pathway in endosomes and lysosomes through endocytosis. After being cleaved by β-secretase and γ-secretase to create Aβ, Aβ fails to undergo normal degradation and then aggregates to oligomers, which are toxic to cells. Alzheimer’s disease (AD) patient brains have reduced O-GlcNAcylation and increased Aβ toxicity, leading to mitochondrial dysfunction and Golgi breakage, etc. OGA inhibitor or OGA gene knock-out boosts the level of O-GlcNAcylation, reduces Aβ formation and aggregation, decreases necrotic apoptosis of nerve cells and increases autophagy of toxic proteins, etc. Besides, O-GlcNAcylation activity can be restored by the upregulation of the glucose transporter (GLUT), which could be produced by intraperitoneally injecting carboxy-dehydroevodiamine·HCl (cx-DHED).
Although the human brain accounts for only 2% of body mass, its metabolism accounts for 20% of systemic glucose intake under basal conditions [81]. In neurons, glucose is a significant source of energy supply, and impaired glucose metabolism contributes to AD pathophysiological alterations, such as oxidative stress and mitochondrial dysfunction, etc. [82]. O-GlcNAcylation is directly linked to glucose metabolism and acts as a metabolic sensor linking glucose metabolism and neuronal activity. Several lines of evidence suggest that in the AD brains, glucose metabolism is impaired and O-GlcNAcylation level changed [15–20]. Gu et al. [83] found that excessive activation of calpain I caused hydrolyzation of glucose transporter 3 (GLUT3) in AD brains, resulting in poor glucose uptake and impaired metabolism, then protein O-GlcNAcylation decreased. On the other side, Kim et al. [84] constructed AD animal 5xFAD mice models, compared to the control mice, intraperitoneal injection of acetylcholinesterase inhibitor carboxy-dehydroevodiamine·HCl (cx-DHED) restored the expression of GLUT1 and GLUT3 in the 5xFAD mice brains, upregulated O-GlcNAcylation levels, lessened accumulation of amyloid plaques, and behavioral tests showed a recovery from the memory impairment. These indicated that an adequate supply of glucose contributes to normal O-GlcNAcylation levels and recovery of AD.
However, Ahmad et al. [85] used the C. elegans models of AD to test the hypothesis by adding glucose to the conventional feed and found that they delayed the original AD-mediated Aβ-associated paralysis, promoted abnormal body shapes and movement. In this study, the inhibition of OGA increased O-GlcNAcylation and therefore decreased Aβ toxicity, but in the context of high glucose levels, this protective mechanism did not exist. Another study claimed that the downregulation of energy metabolism occurred as a defense mechanism against decreased nutrient and oxygen availability in the microenvironment in early AD [86]. Thus we can speculate that too high glucose levels increase the toxicity of Aβ oligomers against the body. Meanwhile, different laboratories seem to show conflicting results on the effects of glucose metabolism on nerve cells survival, which may be related to the different stages of AD, the different levels of glycometabolism and the differences in O-GlcNAcylation’s impact on the different models.
Alternatively, mitochondrial dysfunction caused by Aβ is a major early characteristic of AD. Pinho et al. [20] found an overall O-GlcNAcylation reduction in vitro and in vivo AD models, this change was related to mitochondrial network disruption and biological dysfunction, and O-GlcNAcylation and cell viability in vitro could be restored with Thiamet-G. Contrary to this result, Park et al. [87] found that Aβ might cause mitochondrial fragmentation and malfunction resulted from increasing O-GlcNAc modification at Thr585 and Thr586 sites on the dynamin related protein 1 (Drp1) of SH-SY5Y cells. This explains partly the reason why neurodegeneration is caused by the unbalanced mitochondrial dynamics. And we need to analyze the effects of O-GlcNAcylation on specific proteins to elucidate the specific pathogenic mechanisms involved in neurodegeneration, providing more convincing evidence for precision treatment.
Aberrant O-GlcNAcylation is involved in neuronal death, which is also a vital pathological aspect of AD. Park et al. [19] built 5xFAD mice with OGA haploinsufficiency, compared to the control, 5xFAD; OGA+/– mice inhibited necrotic apoptosis of neurons and improved cognitive function in mice. Meanwhile, this study demonstrated that inhibition of OGA reduced microglia and astrocytes activation, suppressed proinflammatory cytokines expression, and ameliorated Aβ pathology. Consistently, Zhu et al. [88] demonstrated that OGA inhibitor stimulated autophagy in the brain via the mTOR-independent route and significantly reduced associated toxic proteins in AD mice models. It is worth noting that there also is evidence that upregulation of O-GlcNAcylation promoted cell death. According to Choi et al. [89], O-GlcNAcylation at Ser56 and Ser57 of Aβ-induced cell death regulatory protein transcription factor c-Fos elevated in vivo and in vitro, then upregulated c-Fos stability and transcriptional activity, induced apoptotic protein Bim expression, thus accelerating neuronal cell death. Therefore, in the deeper study, we should pay more attention to the activity of OGA enzyme and the O-GlcNAcylation at specific sites of proteins in the disease, which even be contrary to the situation of the overall protein.
Besides, abnormal production of Aβ leads to a transformation of cellular and molecular pathways, one of which is the disturbance of calcium homeostasis. Sec31A is required for the smooth transport of secreted proteins in the endoplasmic reticulum-golgi apparatus network, Aβ toxicity causes intracellular Ca2 + increase, which triggers O-GlcNAcylation decrease of Sec31A, affecting the membrane transport events of newly synthesized proteins, and causing abnormal Golgi morphology, which be rescued by Thiamet G [90]. Golgi breakage accelerates the transport of AβPP and increases Aβ production, making AD more serious and creating a vicious cycle [91]. Thus, we conclude that the pathogenic process of Aβ interacts with dysregulated O-GlcNAc levels.
O-GlcNAcylation and tau protein
The formation of neurofibrillary tangles (NFTs) is another feature of AD. Research showed that NFTs can more quickly accelerate the progression of cognitive dysfunction and neurodegeneration than Aβ [92–94]. The NFTs are made up of abnormally hyperphosphorylated tau. Tau is a protein that assembles microtubules to stabilize neurons normally. In the brains of AD patients, tau proteins are hyperphosphorylated and away from microtubules, and phosphorylated tau monomers self-assemble into oligomers, forming paired helical filaments (PHFs) and ultimate NFTs, which cause neurodegeneration and cognitive impairment (Fig. 5). Diseases caused by hyperphosphorylated tau aggregation are collectively known as tauopathy. As phosphorylation is involved in the formation of pathogenic tau, the two PTM types will be detailed in this section.

O-GlcNAcylation regulates the neurotoxic production process of Tau. Tau protein is hyperphosphorylated in Alzheimer’s disease (AD) human brain and then separates from microtubules, aggregating into subsequent neurofibrillary tangles (NFTs) and causing neurotoxicity. AD brains have less O-GlcNAc-modified tau, increased phosphorylated tau, which mediates the occurrence of diabetes-associated cognitive dysfunction (DACD) and leads to synaptic dysfunction in the disease. Increased O-GlcNAcylation reduces protein-protein interactions and toxic substances decrease. In addition, phosphorylation modification alters the “β-sheet” structure of native Tau225 - 246, which is hardly altered by O-GlcNAcylation. The PHF R3-R4 region still shows a “C conformation” under the modification of O-GlcNAcylation, but it shows a different “H conformation” under the action of phosphorylation.
Compared with normal brain, O-GlcNAcylated tau in the brains of AD patient is lower [18, 95]. Increasing O-GlcNAcylation of tau by the Thiamet-G can inhibit tau hyperphosphorylation [96], slow down neurodegeneration and prevent cognitive decline [97]. There may be negative correlation between the hyperphosphorylation and O-GlcNAcylation of tau [17, 18], or O-GlcNAc modification may also directly hinder tau aggregation [98], the process may involve enhanced monomer solubility or destabilization of soluble aggregates [99]. Additionally, there are also studies of their different effects on protein conformation. Rain et al. [100] utilized molecular dynamics (MD) simulations to study the conformational impact of phosphorylation and O-GlcNAcylation on the important fragment of tau peptide (Tau225 - 246). They found that phosphorylation induced the strong salt bridge formation between adjacent residues, disrupting the native “β fold” structure of Tau225 - 246, whereas O-GlcNAcylation had a little structural effect and the conformation was similar to the peptide’s natural form. They further conducted MD simulations of the R3-R4 repeat domain of tau and found that the native PHF maintained a “C” shaped architecture [101]. The O-GlcNAcylated tau retained the “C conformation”, while the protein structure changed to a more open “H conformation” upon phosphorylation. Due to phosphorylation at Ser356 and subsequent loss of a crucial salt bridge between Lys353 and Asp358, this conformational shift occurred. Moreover, in the aging metabolic syndrome mouse models, aberrant tau phosphorylation and impaired cognitive function linked to decreased O-GlcNAc signaling were also demonstrated [102]. These experiments highlighted the gap in the effects between the two PTMs on the protein conformation and function, also indicated the important roles of PTMs in AD. We think that phosphorylation may impair brain function by more disrupting protein structure or increasing protein aggregation directly. Furthermore, O-GlcNAcylation also indirectly contributes to reversing neurodegeneration. Balana et al. [16] discovered that O-GlcNAcylation activated the anti-amyloid action of some small heat shock proteins (sHSPs), which reduced the formation of amyloid aggregates by blocking protein-protein interactions between sHSPs and other proteins.
Park et al. [103] found that chronic repeated hypoxia exposure induced AD-like pathological changes and learning and memory disorders in zebrafish brains, while glucose, glucosamine and Thiamet-G restored cognition after anoxic injury in zebrafish. Huang et al. [104] used a neuron model derived from human pluripotent stem cells (PSCs) and found that low-glucose induced abnormal O-GlcNAc levels led to AD-like phenotypes through mitochondrial dysfunction. Thiamet-G treatment alleviated the progression of AD by reducing tau phosphorylation and Aβ aggregation. In summary, numerous findings supported the idea of using OGA inhibitors to slow progression and reduce symptoms of NDs. Here, we are supposed to consider carefully the effects of the altered O-GlcNAc cycling level on disease onset and progression, especially activity of OGA.
In addition to AD, O-GlcNAcylation is also involved in other tau diseases (tauopathies). In the mouse models of tauopathy, the OGA inhibitor ASN90 had significant therapeutic effects on the production of PHFs and NFTs in the cortex and hippocampus, preventing development of tau lesions [105]. Xia et al. [106] created a tauopathy model by injected adeno-associated virus carrying hTau cDNA (AAVhTau) into the mouse hippocampus, they detected that dihydroartemisinin (DHA) induced O-GlcNAcylation and reduced phosphorylation, thus compensating for the learning and memory defects in tauopathy mice. In addition, chronic hyperglycemia could induce tau hyperphosphorylation in vitro and in vivo by downregulating O-GlcNAcylation that OGT participates in, thus mediating the occurrence of diabetes-associated cognitive dysfunction (DACD) [107]. As mentioned in this study, chronic hyperglycemia first affects OGT activity, and then affects the downstream O-GlcNAcylation. At the same time, it shows that blindly giving high sugar also has adverse effects, which causes us to think about the different effects of high metabolism and low metabolism on the O-GlcNAc cycle. These tauopathies are identical to AD and are protected by O-GlcNAcylation.
Tau-mediated memory deficits are associated with synaptic dysfunction. Lu et al. [108] found that in vitro, the histone deacetylase sirtuin type 1 (SIRT1) made CREB deacetylate and inactivate, which inhibited the expression of OGT and reduced the O-GlcNAcylation of tau, which improved the phosphorylation of tau at Ser199 and Ser214. Yin et al. [14] built a mouse model of SIRT1 deficiency (SIRT1 flox/Cre+), they found that SIRT1 gene loss reduced OGA expression, leading to enhanced O-GlcNAcylation of tau in vivo and increased site-specific phosphorylation of tau at Thr205, Ser396, and AT8 (Ser202/Thr205), and phosphorylated tau distribution changed in the synaptosomes, causing early synaptic dysfunction. Notably, instead of increasing overall tau phosphorylation, SIRT1 deficiency resulted in higher amounts of O-GlcNAcylation and phosphorylation at certain places. This evidence suggests that the SIRT1 gene involved in metabolic regulation may be related to both OGT and OGA expression. More crucially, it offered the possibility that tauopathies may be treated by targeting certain gene-related components like SIRT1.
O-GlcNAcylation and Parkinson’s disease
Parkinson’s disease (PD) is the second largest ND, characterized by the death of dopaminergic neurons in the substantia nigra (SN) and amyloid aggregation of Lewy bodies (LBs) in the cytoplasm (Fig. 6). The primary filamentous element of LBs is the α-syn that contains 140 amino acid sequences. α-syn normally exists as a soluble monomer and is cytotoxic when misfolded or aggregated into pre-formed fibrils (PFFs). The α-syn aggregation process can be roughly divided into three phases: (i) the initial lag stage, during which monomers accumulate to form aggregated nuclei (nucleation); (ii) the elongation stage, or fibrils growth stage; and (iii) the stationary stage, indicating that the expansion of the maximum fibrils has reached the steady state [109, 110].

O-GlcNAcylation regulates the neurotoxic generation process of α-synuclein (α-syn). The α-syn aggregation process depends on a nucleation dependent pattern, eventually forming to Lewy bodies (LB) and becoming toxic to the cells. The O-GlcNAcylated α-syn protein-protein interactions are decreased and the aggregation process is inhibited, and elevated O-GlcNAcylation levels also suppress neuroinflammation and restore dopamine function.
So, both the physical and chemical processes that prevent α-syn aggregation can affect its neurotoxicity. Scientists has found that appropriate O-GlcNAcylation prevented the gathering and fibrosis of α-syn. Levine et al. [111] have chemically synthesized a α-syn version modified by site-specific O-GlcNAc, found that O-GlcNAcylation at T72, T75, and T81 individually inhibited the nucleation process of protein aggregation, O-GlcNAc modification at T75, T81, and S87 inhibited the extension phase. The O-GlcNAcylated α-syn at T72, T75, and T81 triple sites also reduced aggregation. Moreover, there is a negative relationship between the internalization degree of α-syn PFFs and the level of O-GlcNAc proteins [112]. Wu et al. [113] used MD simulations to reveal the mechanism of α-syn monomer oligomerization, they found that O-GlcNAcylation had a spatial effect, which reduced the likelihood of residues around the modification site forming hydrogen bonds with other α-syn monomers, thereby reducing or delaying oligomer formation. Ryan et al. [114] found that truncated non-amyloid-β component (NAC) fragments modified by O-GlcNAc inhibited the pathological self-aggregation of α-syn by binding O-GlcNAc to the self-aggregation center of α-syn.
Permanne et al. [105] used the OGA inhibitor ASN90 to promote O-GlcNAcylation in α-syn transgenic Line61 mouse models, they observed pS129 α-syn aggregation in the hippocampus reduced and astrocyte proliferation decreased in the cerebral cortex, dyskinesia progression was slowed down. Of course, the more detailed upstream and downstream molecules involved in still need to be further tested.
Numerous brain processes are coordinated by dopamine neurons, such as reward-related learning and memory, movement selection and others. The midbrain dopamine system’s survival and maintenance, according to this article, depended on O-GlcNAcylation. Elevated O-GlcNAcylation enhanced dopamine function, yet without detrimental consequences [13]. In fact, the O-GlcNAcylation regulation in particular nerve cells appears to be more complex than we expect, and there is a gap in the responses of different neurons to O-GlcNAcylation.
O-GlcNAcylation and amyotrophic lateral sclerosis
The most prevalent motor neuron disease, amyotrophic lateral sclerosis (ALS), is marked by the slow degeneration of motor nerve cells in the brain and medulla spinalis. The false accumulation of neurofilament (NFs) is a hallmark of ALS. As observed in the ALS transgenic mouse models, O-GlcNAcylation levels reduced in the spinal cord compared to control mice. The OGA small inhibitor NButGT upregulated O-GlcNAcylation level of neurons [115]. In ALS rat models, the O-GlcNAcylated tail domain of neurofilament M (NF-M) is significantly reduced [116]. It suggested that ALS neurodegeneration might be related to decreased O-GlcNAcylation levels in neurons, and consistent with other neurodegenerative disorders, the change may be pharmacologically reversed by OGA inhibitors.
Another characteristic pathological marker of ALS is the production of ubiquitination inclusion bodies formed by TAR DNA-binding protein-43 (TDP-43) in the neuron cytoplasm [117]. TDP-43 is involved in multiple steps of RNA metabolism, including mRNA transcription, splicing, or transport, as well as microRNA metabolism [118]. In addition, the aggregation of TDP-43 is also observed in other NDs such as frontotemporal lobe degeneration (FTLD). Zhao et al. [119] found that OGT mediates O-GlcNAcylation of TDP-43 at T199 and T233 sites, which inhibited the hyperphosphorylation and aggregation of TDP-43, ameliorated the locomotion defects of drosophila overexpressing hTDP-43. The modification of TDP-43 may prevent TDP-43 proteinopathy by regulating its alternative splicing function for RNA. This also points out a way for us to solve the problem of genetic diseases.
The CNS is especially vulnerable to oxidative stress, which is thought to be a prevalent cause of NDs, including ALS. Hsieh et al. [120] introduced an oxidative stress sensor—nonselenocysteine-containing phospholipid hydroperoxide glutathione peroxidase (NPGPx), which was lowly expressed in the spinal cord of human ALS. When oxygen radicals increased, NPGPx inhibited OGA activity to promote O-GlcNAcylation and protect neurons from oxidative stress damage, and Thiamet-G treatment improved neurons degeneration in NPGPx knockout mice. This paper points to the role of redox signaling pathway interacting with O-GlcNAcylation in NDs.
O-GlcNAcylation and Huntington’s disease
Huntington’s disease (HD) is the most common hereditary neurodegenerative condition that is inherited in a phenotypic dominant manner. The mutation of huntingtin (HTT) protein is one of the characteristics of HD. According to Kumar et al. [67], O-GlcNAcylation regulated the basic autophagy process, and inhibiting O-GlcNAcylation increased autophagic flux and reduced toxic HTT aggregates in the eyes of HD drosophila, and partially restored striated muscle morphology and vision in HD drosophila models. This study explored how O-GlcNAcylation controls autophagy.
Nuclear pore complexes (NPCs), which are made up of nucleoporins (NUPs), mediate the nucleoplasmic transit of macromolecular substances between the nucleus and the cytoplasm. Grima et al. [121] found that Thiamet-G treatment reduced nucleocytoplasmic transport defects caused by mislocalization and aggregation of NUPs in primary cortical neurons transfected with HTT 82Q, rescuing cell death. In addition, O-GlcNAcylation also prevents breakdown of NUPs by reducing ubiquitination, thus maintaining NPC homeostasis [122]. These conflicting conclusions about O-GlcNAcylation suggest its different effects on different pathological processes of ND, this suggests that we must specifically analyze the upstream and downstream molecules, even for the same phenotype, we cannot ignore the internal and possible opposite molecular effects, which requires our further research and exploration.
O-GlcNAcylation and Machado-Joseph’s disease
Machado-Joseph’s disease (MJD), commonly referred to spinocerebellar ataxia type 3 (SCA 3), is a neurodegenerative disease characterized by ataxia, caused by polyglutamine extension in ataxia-3, and the high concentrations of extended mutant ataxin-3 protein forms the inclusion body [123, 124]. Pereira Sena et al. [125] found the OGT levels were increased in the MJD models, pharmacological inhibition of OGT decreased mutant ataxin-3 accumulation, alleviated movement impairment in the MJD zebrafish, suggesting that inhibition of O-GlcNAcylation reduced the pathophysiology and phenotype of this condition. Further studies are required to understand the links between MJD and O-GlcNAcylation levels.
O-GlcNAcylation and giant axonal neuropathy
With an autosomal recessive mode of inheritance, giant axonal neuropathy (GAN) is a ND characterized by the disorders of giant axons and cytoskeletal components [126]. The aberrant gigaxonin protein of GAN is produced by the GAN gene alteration, which causes abnormal accumulation and aggregation of the intermediate filament (IF) protein of neural axons in the CNS, impairing neuronal function and viability. Chen et al. [127] identified S272 and T277 sites as specific O-GlcNAc modification sites of gigaxonin protein, which were necessary for connecting with IF protein and promoting their renewal. This has important implications for optimizing the treatment of the rare disease.
CONCLUSIONS
The presence of numerous O-GlcNAcylated proteins in brain suggests that this PTM is highly associated with neurological diseases. Through intermodulation with other PTMs, as a glucose metabolism sensor, mediating synaptic plasticity and synaptic transmission, regulating protein processing and abnormal production, involved in oxidative stress response and signaling pathway conduction, affecting the transport of secreted proteins and so on, O-GlcNAcylation regulates directly or indirectly the characteristic pathological protein toxicity in NDs, affecting neuronal function and survival, thus participating in the pathological progression of various diseases. There is no doubt that the intervention of this modification can protect the disease through multiple biological pathways and achieve multiple benefits. For another, studies found that the clinical functional benefit of intervention O-GlcNAcylation was disproportional to abnormal protein pathology changes [105], which showed the substrate diversity and influence universality of the PTM, the precise mechanisms of the O-GlcNAc modification in controlling cell biological processes remain largely elusive, too. Besides, up to now, there is still disagreement over the O-GlcNAcylation’s roles in AD, HD, and there are contradictory findings of both increased and decreased O-GlcNAcylation, which might have something to do with the different models used in the experiments or the different development stages of the disease in the animals. However, more of these reports showed that upregulation of O-GlcNAcylation had protective effects against NDs. Hence, the more concrete regulatory mechanisms of O-GlcNAcylation need to be further investigated. Moreover, in recent years, multiple agonists and inhibitors targeting two enzymes in the O-GlcNAc cycling have been designed and developed, especially OGA inhibitors for most [105, 128–131], in order to alleviate disease by increasing the level of O-GlcNAcylation. Some scientists synthetized radioligands for OGA that could enter the human brain to quantify OGA of brain for the assessment of disease status [132]. It is significant to highlight that the effect (increase or decrease) of the inhibition on toxic proteins may depend on the target sites of the protein, the specific use, and the duration of treatment. The protective effects produced by the OGA inhibitor may not only be the effect of increased O-GlcNAcylation levels, which may also involve other protective downstream molecular effects of OGA, in contrast, in such disease cases what could be compromised is the activity of OGA. The precise molecular mechanisms need to be further studied. Meanwhile, selective inhibitors with high affinity that can easily cross the blood-brain barrier still need to be further developed.
In conclusion, O-GlcNAcylation has great effects on development and progression of NDs. There are broad prospects to use the PTM as a clinical target for prevention, diagnosis, and treatment. Given that O-GlcNAcylation is prevalent in the brain, the current exploration still cannot reveal its mystery. NDs usually have chronic and progressive disease courses, in the future we should consider in vivo environmental factors for the different stages of disease, distinguishing the overall proteins and specific proteins and the specific sites of O-GlcNAcylation involved, more attention to the downstream molecular changes of O-GlcNAcylation, to observe more detailed and accurate, more targeted and persuasive pathological mechanism. We need to be aware that a crucial treatment strategy for NDs may involve controlling O-GlcNAc cycling, rather than just up- and down-regulating O-GlcNAcylation, including both enzymes and their respective downstream pathways. Again, the long-term effects of modulators should be even more therapeutic. Here, we should combine multidisciplinary research methods of chemistry, biology and structural techniques and appropriate monitoring and simulation techniques, such as immunoprecipitation-nanoflow liquid chromatography-high resolution mass spectrometry (IP-nLC-HRMS) technique [133], elucidating the function of O-GlcNAcylation in protein structure and its influence on interactions with other proteins, we can successfully and fully illuminate the complex functional mechanism of O-GlcNAcylation.
Footnotes
ACKNOWLEDGMENTS
We express gratitude to our departments for providing support to this work.
FUNDING
This work was supported by the Department of Human Resources and Social Security of Shanxi Province. Grant Number: [2019] 91st.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.