
Abstract
Background:
Alzheimer’s disease (AD), the most common form of dementia, remains long-term and challenging to diagnose. Furthermore, there is currently no medication to completely cure AD patients. Rapamycin has been clinically demonstrated to postpone the aging process in mice and improve learning and memory abilities in animal models of AD. Therefore, rapamycin has the potential to be significant in the discovery and development of drugs for AD patients.
Objective:
The main objective of this systematic review and meta-analysis was to investigate the effects and mechanisms of rapamycin on animal models of AD by examining behavioral indicators and pathological features.
Methods:
Six databases were searched and 4,277 articles were retrieved. In conclusion, 13 studies were included according to predefined criteria. Three authors independently judged the selected literature and methodological quality. Use of subgroup analyses to explore potential mechanistic effects of rapamycin interventions: animal models of AD, specific types of transgenic animal models, dosage, and periodicity of administration.
Results:
The results of Morris Water Maze (MWM) behavioral test showed that escape latency was shortened by 15.60 seconds with rapamycin therapy, indicating that learning ability was enhanced in AD mice; and the number of traversed platforms was increased by 1.53 times, indicating that the improved memory ability significantly corrected the memory deficits.
CONCLUSIONS:
Rapamycin therapy reduced age-related plaque deposition by decreasing AβPP production and down-regulating β-secretase and γ-secretase activities, furthermore increased amyloid-β clearance by promoting autophagy, as well as reduced tau hyperphosphorylation by up-regulating insulin-degrading enzyme levels.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by a progressive decline in cognitive function, typically manifested as memory loss [1]. Of the disseminated forms of AD, symptoms usually begin to manifest after the age of 65, and the number of AD patients worldwide is expected to reach 81 million by 2040 as the global population ages [2]. However, the deaths of people suffering from AD are estimated to increase by 100 percent in developed countries between 2001 and 2040, and over 300% in India, China, and their neighbors in South Asia and the Western Pacific [2]. Overall financial costs associated with healthcare for people with dementia in the United States were estimated at $800 million for only 13 years [3].
According to previous studies, the pathogenesis of AD is based on the doctrines of Aβ toxicity, Tau protein hyperphosphorylation, neuroinflammation, cholinergic damage, excitatory amino acid toxicity, and cardio-cerebral cascade [4]. Despite intensive and extensive research into the pathogenesis of AD, the etiology and pathogenesis of AD are still unclear, and no drugs are available to cure or delay AD in the clinic. According to Scientific American, the failure rate of AD drug development is as high as 99.6%, which is 18.6 percentage points higher than the failure rate of cancer drug development. To date, only six AD therapeutics have been approved by the US Food and Drug Administration (FDA), which include four acetylcholinesterase inhibitors (donepezil, tacrine, resigning, galantamine) [5], a noncompetitive blocker of the NMDA receptor (memantine) [5], and an Aβ antibody drug (aducanumab) [6].
Rapamycin, an immunosuppressive drug approved by the FDA [7], was initially developed to prevent organ rejection in organ transplant recipients. Recently, it has been recognized as a potential anti-aging drug for slowing the aging process in animal model studies [8]. A recent study found that rapamycin could reverse or delay age-related decline and disease [9, 10]. Rapamycin could suppress the mammalian target of rapamycin (mTOR) protein kinase activity in the rodent brain following peripheral administration [7], which triggers a signaling cascade stimulating neuronal autophagy [11]. Long-term peripheral administration of rapamycin in a mouse model of AD with tauopathy has demonstrated that rapamycin could prevent tau-induced neuronal loss, synaptic toxicity, reactive microglial cell proliferation, and astrocyte hyperplasia, as well as the activation of innate neuroimmunity [12]. In addition, long-term inhibition of mTOR by rapamycin was shown to prevent cognitive deficits associated with AD and reduce levels of Aβ42, a major toxicant in AD, in a PDAPP transgenic mouse model [13]. Compared to control mice, 3×Tg-AD mice given rapamycin therapy showed a significant reduction in the latency to cross platform positions and a significant increase in the number of platform position crossings in 3×Tg-AD mice, indicating that rapamycin administration could rescue the early learning and memory deficits in 3×Tg-AD mice [14]. Preventative administration of rapamycin induces autophagy and significantly decreases plaques, tangles, and impairments in cognition in 2-month-old 3×Tg-AD mice [15]. Regardless of whether the rapamycin intervention was administered before the start of disease signs or following the onset of pathology and symptoms, ameliorated effects were demonstrated in these animal models of AD [16–18]. Furthermore, genetic suppression of mTOR could reduce Aβ deposition and tau pathology, rescue memory deficits, and improve cognitive functioning [19]. Given the large number of studies showing that rapamycin attenuates normal aging and AD-like disorders in preclinical models, and the fact that rapamycin is an FDA-approved drug; therefore, rapamycin may be a potentially promising medication for the prevention in patients with AD or patients with mild cognitive impairment.
MATERIALS AND METHODS
Review protocol
The article was conducted and registered with PROSPERO (ID: CRD42023466106) following PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses), and the Cochrane Collaboration. The PRISMA checklist has been provided in Supplementary Figure 1.
Search strategy
The PRISMA guidelines were strictly followed for this systematic review and meta-analysis [20], and the results of the meta-analysis were investigated to explore the effects and mechanisms of rapamycin intervention in the AD mouse model. Based on the consideration of the database authority, six electronic databases were systematically searched, including PubMed, Web of Science, Embase, Cochrane Reviews, China National Knowledge Infrastructure (CNKI), and Wanfang Data Information Website from the time of publication of the first relevant article to March 1, 2023, and the MeSH terms “Alzheimer’s disease” and “rapamycin” as the subject terms were searched in the full-text. The MeSH terms “Alzheimer’s disease” and “rapamycin” were used as subject terms for full-text search in the databases. Available languages in the database were restricted to English and Chinese. The retrieved articles were imported into EndNote (Thomas Reuters, US), which was utilized to remove duplicates, and all remaining results were screened for further qualification.
Inclusion and exclusion criteria
Three authors (Jie Cai, Danni Xie, and Zhenwei Zhai) independently judged the quality of the article by evaluating the title, abstract, and full text based on relevant training.
The following eligibility criteria were applied: 1) Intervention drug: Rapamycin intervention individually; independent experimental group of rapamycin in case of co-medication; 2) Animal model: capable of expressing the pathology of AD; 3) Cognitive function index test method: Morris Water Maze (MWM) test; 4) Indicators and data: Escape latency and number of platform crossings in the target quadrant of the MWM test; complete experimental data, including definitive experimental group and animal counts; 5) Language: Articles in English or Chinese as publication language.
Studies were excluded if they were: 1) Interventional drugs: no administration of rapamycin; 2) Animal models: non-AD mouse model, indirect animal model induced by AD pathology; 3) Indicators and data: efficacy evaluations limiting to biochemical and physiologic outcomes; 4) Outcome measure: without MWM test or non-specific evaluation indicators of MWM test; 5) Duplicate publications; 6) Literature type: reviews, conferences, commentary articles, etc.
Data collection
Three authors (Jie Cai, Danni Xie, and Zhenwei Zhai) performed the initial literature screening by reading the titles and abstracts and reviewing the full text of relevant studies to assess their suitability for meta-analysis. In addition, experimental data from eligible articles were extracted.
The extracted information included 1) Basic parameters of the studies: authors and year of publication, animal species, type of AD animal model, animal sex, animal weight, animal age, rapamycin dosage, time of administration, and route of administration. 2) The main outcome indicators: escape latency, and number of crossing the target platform the MWM test for testing the learning and memory abilities of animals [21]. 3) Secondary indicators: Pathological characteristics (Aβ40, Aβ42, and tau).
The number of animals in each treatment and control group, and the standard deviation (SD) or standard error of the mean (SEM) were extracted. Indicator data were expressed as SD or SEM, which were converted uniformly to SD for ease of analysis by the software. When behavioral experiments were conducted consecutively, only data from the last day were extracted. When multiple independent groups were included in a study (e.g., different dosages were administered), these data were extracted and treated as separate experiments, and the corresponding control groups were also treated separately. Graph-based data were calculated using Plot Digitizer software.
Quality assessment
Three authors (Jie Cai, Danni Xie, and Zhishan Zhu) independently assessed the methodological quality of the included animal studies according to the Systematic Review Centre for Laboratory Animal Experimentation (SYRCLE) risk of bias tool, which is based on the Cochrane Risk of Bias tool and has been adjusted for aspects of bias that are particularly relevant to animal studies [22]. Specifically, SYRCLE’s risk of bias tool has been adjusted to the specificity of animal experiments. This adaptation involves adjusting indicators related to animals, such as randomization and blinding procedures, and considering factors such as sample size, animal housing conditions, and blinding of experimenters [23, 24]. The SYRCLE’s risk of bias tool has broader applicability, enabling a more comprehensive assessment of bias risks in animal experiments [22, 25].
The SYCLE bias risk assessment tool includes the following ten components: 1) randomized sequence generation; 2) baseline characteristics; 3) allocation concealment; 4) randomized housing; 5) blinding of participants and staff; 6) randomized outcome evaluation; 7) blinding of outcome evaluations; 8) incomplete outcome data; 9) selective reporting; and 10) other biases.
The quality assessment of the article will be conducted by three independent authors (Jie Cai, Danni Xie, and Zhishan Zhu). If disagreements arise during the review process, a full discussion will be conducted with the fourth author (Tao Sun) in order to reach a consensus. The possibility of bias was considered unclear when the paper mentioned “randomization” but did not specify the particular method; it was judged as low risk if the method was clarified (such as the random number method); when no “randomization” was depicted, it was judged as high risk. Furthermore, if animals were familiarized with the environment before behavioral experiments, it was regarded as a low risk of bias. If there was no mention, it was considered to have an unclear risk of bias. Other sources of bias were judged as low risk if researches indicated no interest conflict, and studies were seen as unclear when there was no statement of conflict of interest.
Data synthesis and statistical methods
According to the Cochrane Handbook for the Systematic Evaluation of Interventions, the overall validity of the data was recognized as continuous. The pivotal effect of rapamycin was determined by comparing the experimental group with the corresponding control group concerning the main effectiveness indicators of cognitive function (escape latency, number of crossing the target platform) and pathologic features specific of AD, including Aβ40 and Aβ42 levels.
The mean and SD of the required data were collected with Plot Digitizer software and subsequently meta-analyzed by Review Manager 5.3 software. The Q statistic and I-square (I2) were used to evaluate respectively statistical significance and study heterogeneity. According to the Cochrane Handbook for Systematic Review of Interventions, heterogeneity was considered to exist when p < 0.1 in the Q-test. For I2 statistic, I2 = 0% indicates no heterogeneity; 0 < I2≤40% means low heterogeneity; 30% < I2≤60% represents moderate heterogeneity; and I2≥50% is considered high heterogeneity. The I2 value was recognized as being within the acceptable range when it was less than 50% [26]. The p-value < 0.05 was recognized with statistical significance. High heterogeneity studies at I2 > 50% would need to be analyzed using a random effects model, while otherwise the fixed effects model would be adopted. The 95% confidence intervals (CI) were calculated using Mean Difference (MD) when the units of the indicator were uniform, while otherwise Std. Mean Difference (SMD) was conducted for the analysis to eliminate errors due to units.
RESULTS
Included studies
A total of 4,277 articles were obtained from the four English databases and two Chinese databases, and the relevant references were reviewed. After the search was completed by the two authors, the duplicates were automatically deleted first by the Endnote software, and the unrecognized duplicates were manually deleted, resulting in a total of 1,066 duplicate articles. The retained 3,211 articles were screened in the next step. Conference, review, short survey, or commentary articles were excluded after reviewing titles and abstracts; In addition, articles without reports on the administration of rapamycin in AD animal models were also excluded, and a total of 2,970 articles were excluded from this step. Finally, the 241 articles remaining from the preliminary screening were scrutinized in full text, of which 228 were excluded.
Reasons for exclusion specifically included the following elements: 1) Experiment type: non-AD animal tests; cellular tests, clinical trials (89); 2) Pharmacologic intervention: non-rapamycin administration, or combination (82); 3) Measurement of outcome: no MWM test or no measure of escape latency or number of platform crossings in MWM test (31); 4) Unspecific sample size (6); 5) Unavailability of full text (20).
Eventually, 13 articles were included for analysis (Fig. 1).
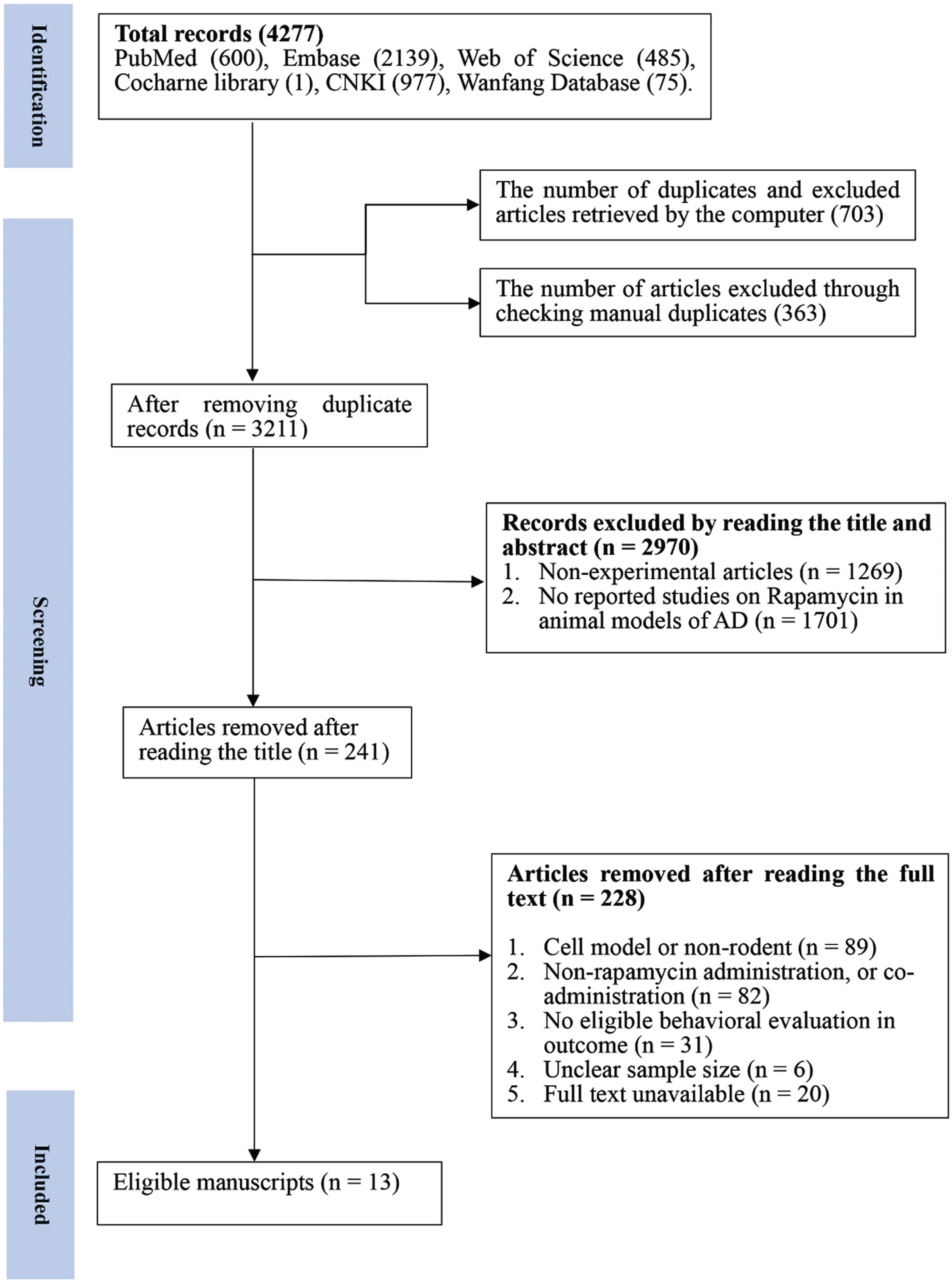
Flow diagram of the study selection process. 13 eligible articles were selected from 4,277 documents for a comprehensive analysis according to the predefined inclusion and exclusion criteria.
Study characteristics
13 articles, including seven in English and six in Chinese, were included by screening duplicate articles, reading title articles, abstract articles, and full-text articles (Table 1).
Characteristics of the included studies
MWM, Morris Water Maze; i.g., intragastric; p.o., oral administrations; i.p., intraperitoneal injection; i.v., intravenous injection.
Meanwhile, meticulous examination of the included articles revealed that the sex of the animals used in the included articles was predominantly male (7/13), followed by a mix of males and females (2/13), with only one article employing female mice, which could be a source of heterogeneity. In addition, the remaining articles (4/13) did not reveal the sex of the animals involved in the experiments. The administration method of nine animal experiments in the included articles was intragastric, six animal experiments were oral administrations, three animal experiments were intraperitoneal injection, and only one animal experiment was intravenous injection.
Nine of the included articles used transgenic mice as AD animal models, including single-transgenic mouse models, double-transgenic mouse models, triple-transgenic mouse models, and five-transgenic mouse model. In addition, there are two articles using D-galactose as an animal model of AD, one article using a senescent rat model, and one article using the chemical agent Zn to induce an animal model of AD.
Quality of included studies
The quality of the study methodology and article reporting was assessed using improved Sycle risk of bias tool based on the Cochrane tool [22]. The quality of the 13 included articles was independently assessed by two authors (Jie Cai and Danni Xie), and the disagreement was resolved with the third author (Tao Sun) through serious discussion (Fig. 2). For baseline characterization, the risk of bias was determined to be low when the animals were already familiar with the environment before the behavioral trials. Three studies (Lin, A. L. 2013; Lai, C. 2022; Spilman, P. 2011) demonstrated randomized housing and were decided to be low risk of bias, whereas the other six studies failed to indicate randomized housing and thus were categorized as unclear risk of bias. Two studies (Lai, C. 2022; Xiaoli, Cui. 2021) were categorized as high risk of bias due to inconsistent animal sample sizes in experimental evaluations and the absence of specific explanations provided by the material. Studies without explicit reporting about allocation concealment, the blinding of caregivers and researchers, the blinding of outcome assessment, and selective reporting were recognized as being at unclear risk of bias. For the other sources of bias, studies without conflicted interests were determined to pose a low risk.
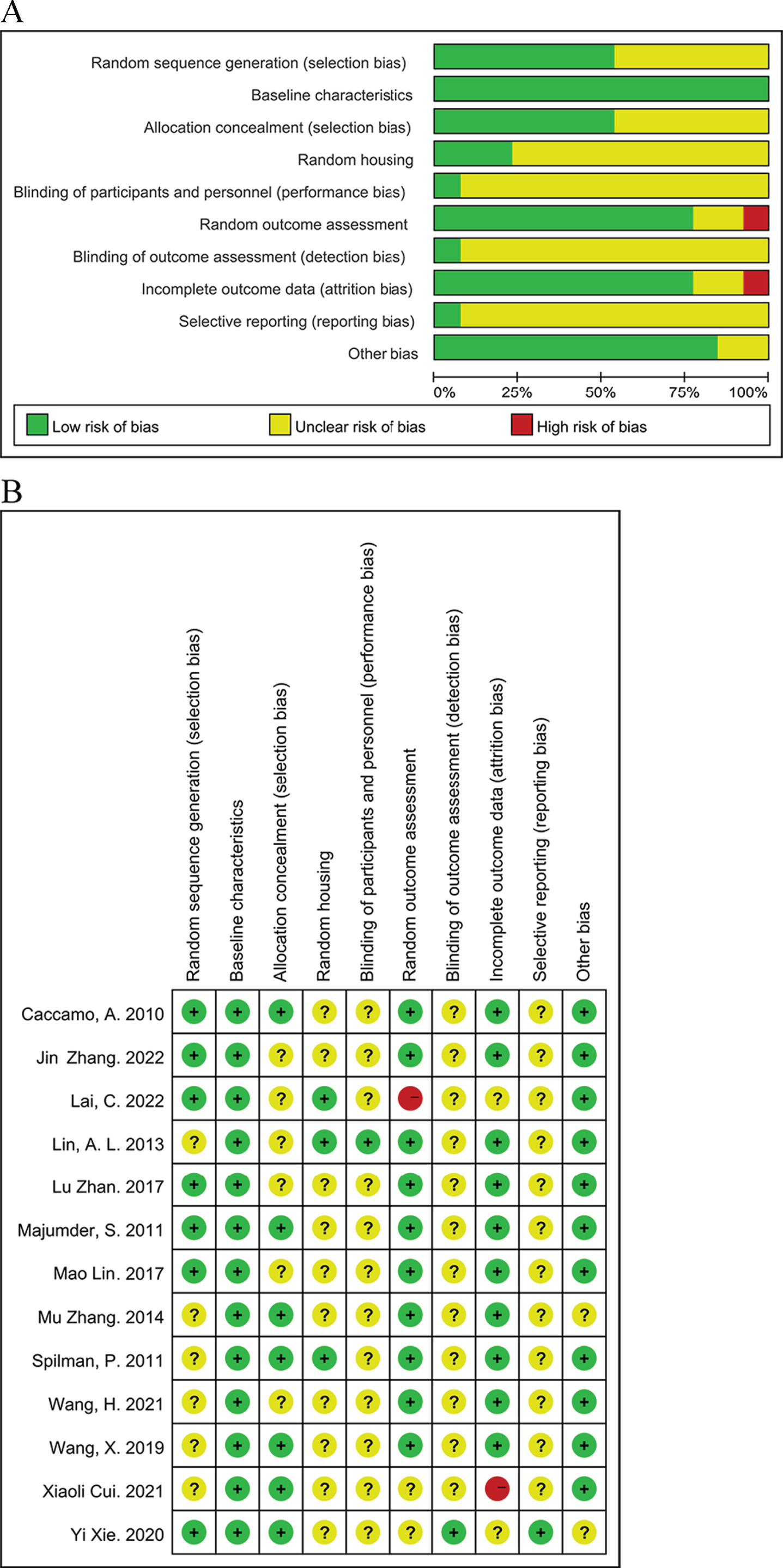
Assessment of literature quality using the SYRCLE risk of bias tool based on the Cochrane tool. (A) Risk of bias graph, (B) Risk of bias summary.
Behavioral test analysis
The MWM test represents one of the most prevalent behavioral tests of spatial learning and long-term spatial memory and has currently been used extensively for research on AD [27]. The escape latency in spatial detection trials is interpreted as an indicator of spatial learning ability, and the number of crossing the target platform represents the degree of memory consolidation after training.
Learning ability
There are 18 groups from 13 studies involving 288 animals, with a sample size of 170 in experimental groups and 118 in control groups. Escape latency was utilized to determine the effect of rapamycin on learning ability in AD animal models (Fig. 3). The data analysis revealed a significant improvement in the learning ability of the animals with rapamycin therapy compared to the control group; According to the overall estimation results, the escape latency (MD = –15.60, 95% CI: [–18.42, –12.79], Z = 10.87, p < 0.00001).
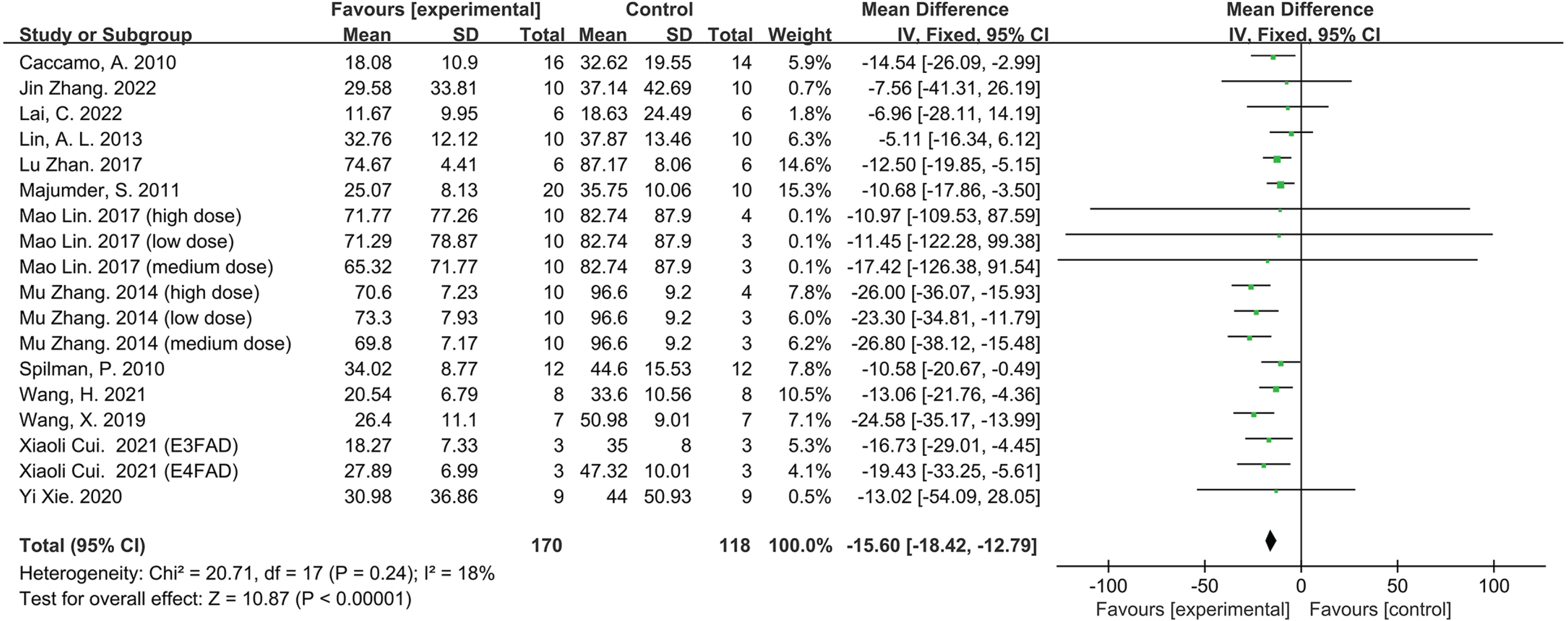
Forest plot displaying the effect of rapamycin on learning ability of the experimental compared with the control group, as determined by measuring the escape latency.
Memory ability
There are 19 groups from 13 studies involving 304 animals, with a sample size of 178 in experimental groups and 126 in control groups. The number of crossing platforms was applied to identify the effect of rapamycin on memory in AD animal models (Fig. 4). Analysis of the data showed that the memory capacity of the AD animals was significantly ameliorated with Rapamycin therapy compared to the control groups; According to the overall estimation results, the number of crossing platforms (MD = 1.53, 95% CI: [1.29, 1.78], Z = 12.22, p < 0.00001).
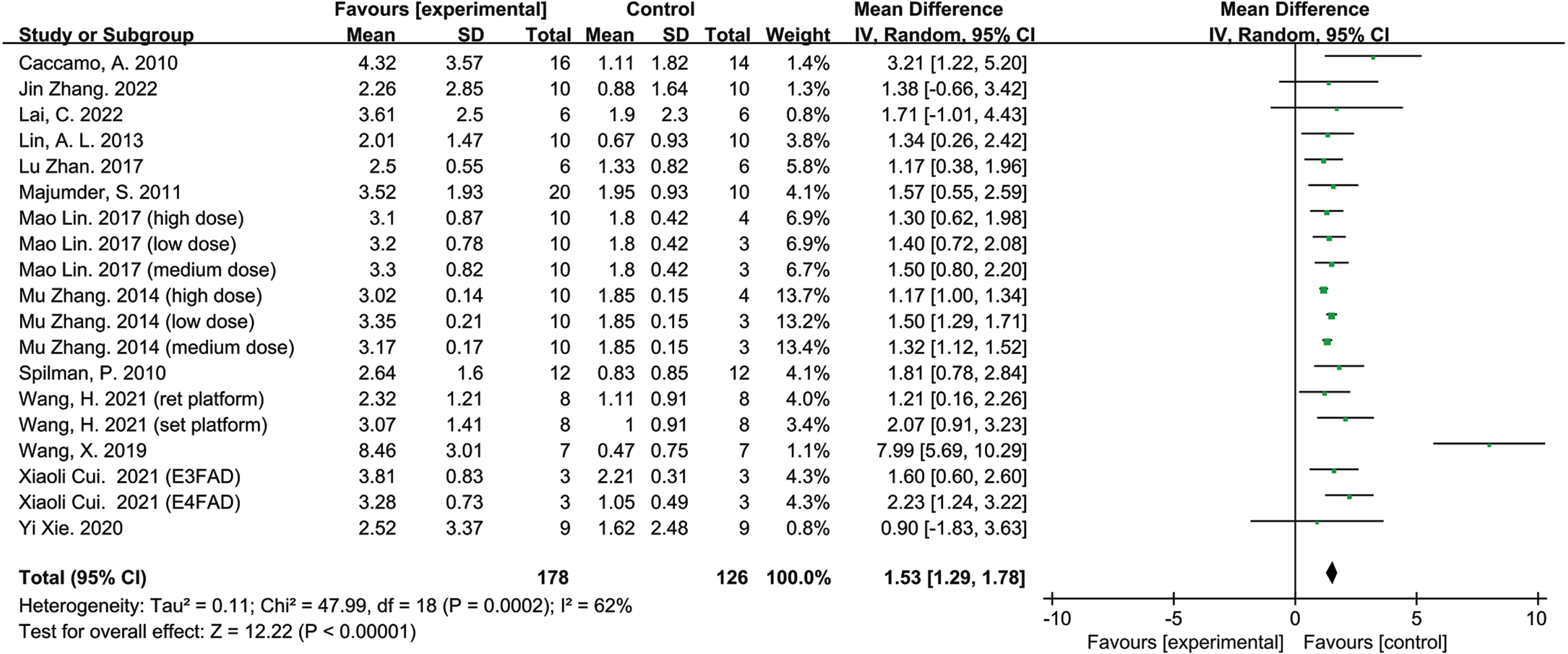
Forest plot displaying the effect of rapamycin on memory ability of the experimental compared with the control group, as determined by measuring the number of crossings over the target platform location.
Pathological characteristics
Aβ40, Aβ42, and tau, as markers of AD pathology, were analyzed to establish the effect of rapamycin on the pathological features of AD animal models (Fig. 5). According to the Q-test and I2 test, Aβ40 heterogeneity test resulted in χ2 = 8.66, df = 2, (P = 0.01); I2 = 77%, Aβ42 resulted in χ2 = 1.20, df = 2, (p = 0.55); I2 = 0%, and tau heterogeneity test resulted in χ2 = 6.17, df = 4, (P = 0.19); I2 =35%.
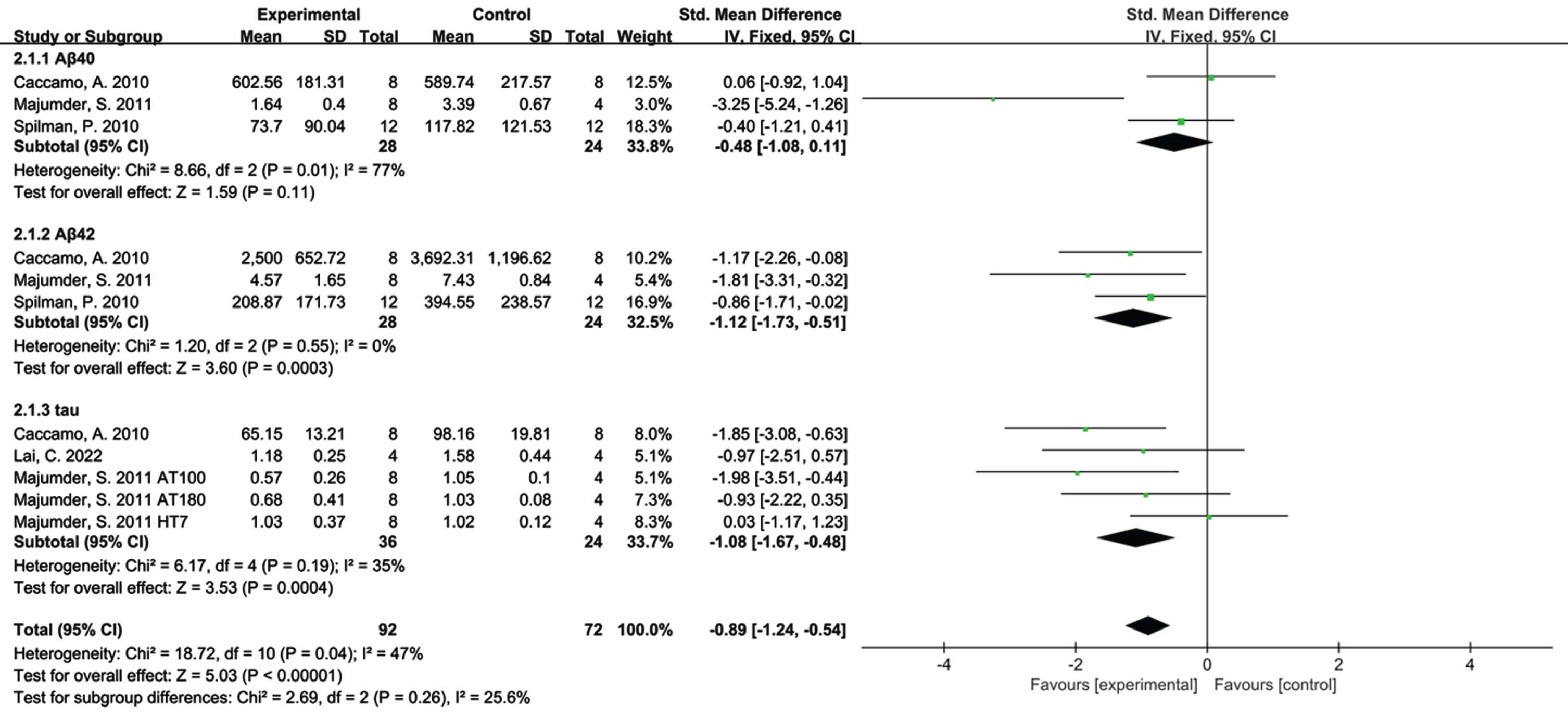
Forest plot depicting subgroup analyses based on the effect of rapamycin on Aβ40, Aβ42, and tau levels.
Based on subtotal estimation, rapamycin therapy was found to decrease Aβ40 (SMD=–0.48, 95% CI: [–1.08, –0.11], Z = 1.59, p = 0.11), significantly reduce Aβ42 (SMD=–1.12, 95) % CI: [–1.73, –0.51], Z = 3.60, p = 0.0003), and Tau (SMD=–1.08, 95% CI: [–1.67, –0.48], Z = 3.53, p = 0.0004) levels compared with the control group.
In addition, the overall heterogeneity was: χ2 = 18.72, df = 10, (P = 0.04); I2 = 47%. That demonstrates statistical heterogeneity existing among the sample estimates but within acceptable bounds.
Subgroup analysis refers to the process of dividing the study population into different subgroups according to some characteristic of the study population (e.g., modeling method), and then separately estimating the effect values for the different groups and conducting comparisons between the subgroups [28]. Adhering to the principles of subgroup analysis, the 13 included studies satisfied the quantitative requirements, followed by the scientific analysis and description of each characteristic. The heterogeneity and drug mechanisms of the included articles were investigated by analyzing the differences existing in the various studies in terms of study participants and intervention protocols. The studies were divided into two or more groups following one of the above factors to observe whether there was a statistical difference between the combined effect values of the subgroups [28].
To specifically elucidate the pathological mechanisms of rapamycin effect in AD animal models, subgroup analyses were conducted: 1) AD animal models; 2) single-transgenic animal models of AD, double-transgenic animal models of AD, triple-transgenic animal models of AD, and quintuple-transgenic animal models of AD.
AD animal models
To investigate the performance of AD animal models in learning ability (Fig. 6A), AD animal models were separated into two groups: transgenic AD animal models and non-transgenic AD animal models. According to the Q-test and I2-test, the heterogeneity of the transgenic AD animal model group was detected as χ2 = 18.88, df = 13, (P = 0.13); I2 = 31%, the non-transgenic AD animal model group was χ2 = 0.30, df = 3, (P = 0.96); I2 = 0%.

Effects of rapamycin on ameliorated cognitive function in transgenic and non-transgenic animal models of AD. (A) learning ability (B) memory ability.
According to the subtotal estimation results, rapamycin therapy was detected to significantly ameliorate the learning ability of the transgenic AD animal model group (MD = –16.42, 95% CI: [–19.52, –13.32], Z = 10.39, p < 0.00001) compared to the control group. And the non-transgenic AD animal model group (MD=–11.76, 95% CI: [–18.47, –5.05], Z = 3.44, p = 0.0006). In addition, the total heterogeneity was: χ2 = 1.53, df = 1, (P = 0.22); I2 = 34.5%, which indicates the presence of some statistical heterogeneity among the sample estimates but lies within the acceptable limit.
To investigate the performance of AD animal models in memory ability (Fig. 6B), AD animal models were separated into two groups: transgenic AD animal models and non-transgenic AD animal models. According to the Q-test and I2-test, the heterogeneity of the transgenic AD animal model group was detected as Tau2 = 0.12; χ2 = 47.61, df = 14, (P < 0.0001); I2 = 71%, the non-transgenic AD animal model group was Tau2 = 0.00; χ2 = 0.22, df = 3, (P = 0.97); I2 = 0%.
According to the subtotal estimation results, rapamycin therapy was detected to significantly ameliorate the memory ability of the transgenic AD animal model group (MD = 1.58, 95% CI: [1.31, 1.84], Z = 11.48, p < 0.00001) compared to the control group. And the non-transgenic AD animal model group (MD = 1.21, 95% CI: [0.52, 1.90], Z = 3.45, p = 0.0006). In addition, the total heterogeneity was: Tau2 = 0.11, χ2 = 47.99, df = 18, (P = 0.0002); I2 = 62%. It demonstrates that there is no statistical heterogeneity between the sample estimates.
Due to the significant impact of the article by Zhan Lu on the learning and memory abilities of the non-transgenic AD animal model, there were substantial differences observed upon its inclusion, while no significant variances were noted upon its exclusion (refer to the Supplementary Figure 2). Consequently, further conclusions regarding the non-transgenic AD animal model were not expounded upon. The effectiveness of rapamycin in ameliorating learning and memory deficits in non-transgenic AD animal models demands further study.
Statistical analysis revealed that the number of articles on non-transgenic animal models provided insufficient support for further subgroup analyses. Therefore, only transgenic AD animal models were further analyzed.
Transgenic AD animal models
Escape latency was used as a measure of learning ability in AD animal models, while transgenic AD animal models were categorized into four groups: single-transgenic AD animal models, double-transgenic AD animal models, triple-transgenic AD animal models, and five-transgenic AD animal models (Fig. 7A). According to the Q-test and I2-test, the heterogeneity of the single-transgenic AD animal model was tested as χ2 = 0.50, df = 1, (P = 0.48); I2 = 0%; According to the Q-test and I2-test, the test for heterogeneity in the double transgenic AD animal model was χ2 = 5.71, df = 7, (P = 0.57); I2 = 0%; According to the Q-test and I2-test, the heterogeneity of the triple transgenic AD animal model was tested as χ2 = 0.31, df = 1, (P = 0.58); I2 = 0%; According to the Q-test and I2-test, the heterogeneity of the five transgenic AD animal models was examined as χ2 = 0.08, df = 1, (P = 0.77); I2 = 0%.

Effects of rapamycin on ameliorated cognitive function in a transgenic AD animal model. (A) learning ability (B) memory ability.
In accordance with the estimation of subtotals, rapamycin was detected to significantly ameliorate the learning ability in transgenic AD animal models compared to the control group, in which single transgenic AD animal model (MD = –8.14, 95% CI: [–15.64, –0.63], Z = 2.12, p = 0.03), double-transgenic APP/PS animal model, (MD=–21.77, 95% CI: [–26.36, –17.19], Z = 9.31, p < 0.00001), triple-transgenic murine model for AD (MD = –11.76, 95% CI: [–17.85, –5.66], Z = 3.78, p = 0.0002), the five-familial AD (5×FAD) transgenic mouse model (MD = –17.92, 95% CI: [–27.10, –8.74], Z = 3.83, p = 0.0001).
In addition, the total heterogeneity was: χ2 = 18.88, df = 13, (P = 0.13); I2 = 31%, which indicates the presence of some statistical heterogeneity among the sample estimates but lies within the acceptable limit.
The number of crossing platforms was measured as the memory ability of AD animal models; meanwhile, the transgenic AD animal models were divided into four groups: single-transgenic AD animal models, double-transgenic AD animal models, triple-transgenic AD animal models, and five-transgenic AD animal models (Fig. 7B). According to the Q test and the I2 test, the heterogeneity of the single-transgenic AD animal model was tested as: Tau2 = 0.00; χ2 = 0.38, df = 1, (P = 0.54); I2 = 0%; The heterogeneity test for the double transgenic AD animal model was Tau2 = 0.13; χ2 = 39.96, df = 8, (P < 0.00001); I2 = 80%; the test for heterogeneity of the triple transgenic AD animal model was Tau2 = 0.69; χ2 = 2.06, df = 1, (P = 0.15); I2 = 51%; the heterogeneity of the five-transgenic AD animal model was tested as Tau2 = 0.00; χ2 = 0.76, df = 1, (P = 0.38); I2 = 0%.
According to the subtotal estimation results, rapamycin was observed to ameliorate significantly the memory ability of transgenic AD animal models compared to the control group, in which single-transgenic AD animal models (MD = 1.59, 95% CI: [0.84, 2.33], Z = 4.19, p < 0.0001), double-transgenic AD animal models (MD = 1.50, 95% CI: [1.18, 1.81], Z = 9.28, p < 0.00001), triple-transgenic AD animal models (MD = 2.16, 95% CI: [0.62, 3.70], Z = 2.74, p = 0.006), and quintuple-transgenic AD animal models (MD = 1.92, 95% CI: [1.21, 2.62], Z = 5.32, p < 0.00001).
Furthermore, the total heterogeneity was: Tau2 = 0.12; χ2 = 47.61, df = 14, (P < 0.0001); I2 = 71%, which demonstrates the existence of statistical heterogeneity within the sample estimates.
DISCUSSION
The 13 included articles demonstrated reduced avoidance latency and increased number of platform crossings in AD animals with rapamycin therapy, indicating that the learning and memory abilities of the AD animal model were ameliorated. Importantly, Aβ and tau levels were also revealed to be reduced in AD animal models treated with rapamycin compared to the control group. Furthermore, transgenic AD animal models have shown the ameliorative effects on learning and memory abilities after rapamycin therapy. Further subgroup analyses were not supported by the number of included articles on non-transgenic AD animal models. Therefore, the transgenic AD animal models alone were further analyzed, wherein the transgenic AD animal models contain single-transgenic AD animal models, double-transgenic AD animal models (APP/PS1), triple-transgenic AD animal models (3×Tg-AD), and five-transgenic AD animal models (5×FAD). Significantly, rapamycin therapy could ameliorate the learning and memory abilities in transgenic animal models of AD.
Included animal models of AD
Animal models of AD have been employed to simulate pathological changes and behavioral characteristics of brain tissue in AD patients. The development of animal models that mimic AD pathology as comprehensively and realistically as possible is significant both for translational research in clinical medicine as well as for preclinical drug screening. According to different pathologic and physiologic factors, AD animal models could be classified into spontaneous aging models, interventional models, and transgenic models [29]. Although spontaneous aging models of senescent ruminants provide research value, considering the high cost of maintenance and the difficulty of operation, the utilization of these models in research has been restricted [30]. The interventional model utilizes chemicals and drugs to induce AD symptoms and pathological changes in vivo [31]. Additionally, advances in genetic engineering have opened up new avenues for biological research, and transgenic animals of AD have contributed significantly to the study of human diseases [32].
However, neurodegenerative diseases are characterized by a long course and numerous neurological pathologic variations, whereas most cases of AD are sporadic with no clear pathogenesis or etiology [32]. In addition to the repetition of pathological events, the sequencing of different pathology events and behavioral changes, such as cognitive deficits, need to be taken into consideration when constructing animal models of AD [29]. Therefore, preclinical animal experiments are incredibly demanding for animal models of AD. Although no one mouse model is modeling all aspects of AD neuropathology, transgenic mice have been proven to be uniquely valuable from the standpoint of genetic background, difficulty of genetic manipulation, reproductive capacity, and economics of husbandry and management [33].
The transgenic AD animal model has the following strengths compared to other AD animal models: 1) The etiology of transgenic animals has been determined and the pathological symptoms are recognized, thus facilitating the study of AD mechanisms and the screening of drugs for its prevention and treatment; 2) Typically the single-transgenic animal model, double-transgenic animal model, and multiple-transgenic animal model of AD, which are dominated by mutations in genes such as APP and tau that are associated with the pathogenesis of AD, have been used as the main research subjects; 3) The rationale for numerous transgenic animal models of AD has strengthened support for the amyloid hypothesis, and several mutations in the APP or PS1/2 genes have been detected in early-onset FAD [34–37].
hAPP(J20)
The hAPP(J20) mouse, developed by Games et al. (1995) [38], was the first transgenic mouse model to exhibit AD pathology. The mutant gene in hAPP(J20) mice is APP and the mutations are APP K670_M671delinsNL (Swedish) and APP V717F (Indiana) [39]. The amyloid-β protein precursor (APP) transgenic animal model is derived from the integration, expression, and inheritance of the human-derived APP gene into the mouse genome [39].
The hAPP(J20) mouse encapsulates many characteristics of AD, including pathologically elevated Aβ levels in AD-vulnerable brain regions, amyloid plaque formation, neuroinflammatory dystrophy, synaptic dysfunction and loss, learning and memory deficits, and other behavioral abnormalities [40–44]. The hAPP(J20) mice were enabled to demonstrate more Aβ [39], including Aβ1–x, Aβ1–42, and Aβ1–42/Aβ1–x, compared to mice expressing equivalent amounts of wild-type human APP. The hAPP(J20) mouse progressively developed amyloid deposits in the brain over 8–10 months, and plaques were extensively distributed. Aβ was originally deposited in the hippocampal region of the brains of the hAPP(J20) mice at 1 month of age [45]. The hippocampus has been demonstrated to be associated with learning and memory abilities in the brain [46]. Massive neuronal loss was observed in the CA1 region of the hippocampus of the hAPP(J20) mice at 12, 24, and 36 weeks of age compared to controls [44], which could result in learning and memory deficits in animal models of AD [47].
Spatial memory and learning abilities develop deficits as mice age. J20 mice demonstrated spatial reference memory deficits by four months as measured by the radial arm maze [44] and the MWM test. PDAPP mice, as hAPP(J20) mice with mutation sites in APP V717F (Indiana), developed amyloid plaques at 6–9 months of age and exhibited spatial learning deficits with age [39]. The study data revealed that the plaques of adult mice were generally larger and denser than younger mice [32].
According to the studies included in the article, rapamycin therapy was discovered to regulate the mTOR signaling pathway, autophagy as well as cerebral blood flow (CBF), thus ameliorating cognitive dysfunction in hAPP(J20) mice. The mTOR is an essential signaling center combining the supply of nutrients or growth factors with cellular metabolism through the use of two distinct complexes, the mechanistic target of rapamycin complex 1 (mTORC1) and the mechanistic target of rapamycin complex 1 (mTORC2) [48, 49]. The mTOR could regulate autophagy through mTORC1 and mTORC2. Primarily, mTORC1 inhibited autophagy through the phosphorylation of Unc51-like kinase 1 (ULK1) and mAtg13 when nutrients and energy were sufficient [50]. Second, Act and PKC become phosphorylated and activated by activated mTORC2 [51, 52]. Since serine-threonine kinase (Akt) positively regulates mTORC1, Akt phosphorylated by mTORC2 would stimulate mTORC1 function, consequently inhibiting autophagy. mTOR or P70S6K, one of the most significant serine/threonine kinases in eukaryotic cells, could regulate protein synthesis, phosphorylation, and autophagy [53–55]. In the brains of postmortem AD patients, the expression of p-mTOR (S2448) and p-P70S6K (T389) was increased, associated with the accumulation of hyperphosphorylated tau in AD [56, 57]. It indicates that phosphorylation of mTOR at S2448 and P6S389K at T70 are critical targets for disease intervention [58].
Autophagy is the primary degradation pathway for cellular organelles and aggregated proteins [59] for recycling cellular components such as protein aggregates, misfolded proteins, and unnecessary organelles [60, 61]. Simultaneously, recent studies concerning AD mainly focused on the contribution of autophagy to AD pathology [62, 63]. Substantial evidence has highlighted the role of autophagy in a variety of age-dependent neurodegenerative diseases characterized by protein accumulation, including AD [64–66]. Similar to other cells, neurons could also accumulate toxic substances in the aging process, which requires autophagy activation to maintain cellular homeostasis. Autophagy function decreases with age, potentially causing the accumulation of proteins in the brain [67, 68]. Importantly, autophagy induction represents a major catabolic process used by the cell for protein turnover, and mTOR was the negative regulator of autophagy induction [69–71].
The avoidance latency of hAPP(J20) mice treated with rapamycin decreased compared to controls, suggesting ameliorated learning ability, as viewed by data from the MWM behavioral tests. In addition, rapamycin could inhibit mTOR as well as decrease Aβ42 levels in the brain of hAPP(J20) mice, and studies have demonstrated that rapamycin increased autophagy levels in the brain of hAPP(J20) mice [18]. Interestingly, the memory ability of hAPP(J20) mice was also ameliorated, and CBF was restored with rapamycin therapy upon the onset of the disease [16].
According to the included article, 2.24 mg/kg of rapamycin therapy for 13–16 weeks resulted in significant ameliorated learning and memory ability in the hAPP(J20) mice, a single-transgenic animal model of AD.
APP/PS1
The mutated genes in APP/PS1 mice are APP and PSEN1, and the mutation sites are APP K670_M671delinsNL (Swedish) and PSEN1 L166P, which was primarily developed to mimic AD pathology arising in patients at earlier ages [72]. APP/PS1 mice contain a human transgene carrying the Swedish mutation in APP and PSEN166 containing the L1P mutation, both of which are in the control of the Thy1 promoter [72].
AD pathology was generated in the APP/PS1 mouse model: Aβ deposition and tau hyperphosphorylation with neurofibrillary tangles (NFTs) structure generation at earlier ages compared to a single transgenic mice model. NFTs, another characteristic lesion of AD, is positively correlated with the degree of dementia. The main component of NFTs is abnormal and hyper-phosphorylated tau protein forming double-helical filaments [73]. Amyloid plaques of APP/PS1 mice appeared in the cerebral cortex from about four months onwards and in the hippocampus from about six months onwards, and the size and number of plaques presented trends of increasing with age [72].
Rapamycin was demonstrated that may have regulatory effects in the pathogenesis of APP/PS1 through its actions on the cholesterol biosynthesis pathway and cytoplasmic ribosomal proteins [74]. One hundred significantly altered proteins were identified in the hippocampus of APP/PS1 mice and were mainly enriched in cholesterol biosynthesis pathways and cytoplasmic ribosomal proteins [74]. Whereas the presence of persistent excess cholesterol could induce atherosclerosis and AD. Rapamycin therapy has been shown to rescue 57 proteins in the hippocampus and 167 proteins in the temporal lobe and has inhibited regulators of cholesterol biosynthesis, including the farnesyl pyrophosphate synthase (Fdps) and the mevalonate kinase (Mvk). Within the cholesterol biosynthesis pathway, the Fdps was upregulated in the transition from WT to APP/PS1 mice, while Mvk was overexpressed in the pathogenesis of APP/PS1. Notably, both Fdps and Mvk were inhibited by rapamycin therapy. In detail, the proteins involved in the pathogenesis of AD, ribosomal proteins, such as: Ribosomal protein 16, Ribosomal protein17, Ribosomal protein l18, Ribosomal protein l24, Ribosomal protein l36a, Ribosomal protein s6, and Ribosomal protein s8 significantly upregulated in the hippocampus; Significantly high levels of ribosomal proteins were also detected in the temporal lobe, such as the: Ribosomal protein17, Ribosomal protein122, Ribosomal protein127, Ribosomal protein 136a, Ribosomal protein s6ka2, and Ribosomal protein s27a.
With rapamycin therapy, most of the proteins in the hippocampus, including ribosomal proteins, were downregulated, and extensive inhibition was detected in the temporal lobe. Furthermore, the severe ribosomal dysfunction in APP/PS1 mice was inhibited by rapamycin. Recent studies have demonstrated that ribosomal proteins, such as Rpl7 and Rpl26, have been associated with the activation of mTORC2, one of the synthesizers of mTOR. Based on our results, rapamycin reversed the pathogenesis of APP/PS1 mice and ribosomal proteins that perform important roles in mTOR regulation, such as Rps6ka2, by inhibiting mTOR [74].
In addition, rapamycin might also effectively alleviate synaptic plasticity defects in APP/PS1 mice by activating mitochondrial autophagy, thereby modulating the pathogenesis of APP/PS1. In the AD brain, mitochondrial dysfunction primarily manifests in decreased potential and increased permeability of the mitochondrial membrane, resulting in oxidative stress in the cells [75]. Studies have indicated that oxidative stress is also one of the major causes of AD [76]. The damaged mitochondria released cytochrome C and initiated a cystatinase cascade reaction that eventually induced neuronal apoptosis [75]. Neuronal apoptosis has been demonstrated to cause cognitive dysfunction in AD mice [77]. The number of presynaptic mitochondria decreased in AD, and the morphology of synaptic mitochondria around Aβ plaques was destroyed, and Aβ oligomers could directly interact with mitochondria, promoting oxygen radical production, apoptosis, and mitochondrial structural disruption. In addition, studies have demonstrated that impairment of synaptic plasticity correlates with AD [75]. Considerable evidence demonstrates that soluble oligomers of Aβ and tau could be propagated in different regions of the brain, which would directly lead to synaptic dysfunction and loss [75].
Rapamycin could not only enhance learning and memory abilities in APP/PS1 mice but also enhance synaptic plasticity and expression of synapse-associated proteins, block cytochrome C-mediated apoptosis, reduce oxidative status, and restore mitochondrial function [75]. The rapamycin therapies resulted in mice with prolonged stay in the destination quadrant and increased number of traversals across the platform (p < 0.05) compared with the model group, as measured by the MWM behavioral test [78]. Notably, the ameliorated escape latency of double-transgenic AD mice in the middle dose group was more evident than the other dose groups when rapamycin was given by gavage using high, medium, and low doses [78]. However, the other included study showed that low-dose rapamycin therapy by gavage resulted in more ameliorated escape latency and number of platform crossings in double-turned AD mice than in the other dose groups [79].
According to the included articles, 1–2.24 mg/kg of rapamycin therapy for 4–8 weeks could significantly ameliorate the learning memory ability of APP/PS1 mice.
3×Tg-AD
3×Tg-AD has emerged as one of the most extensively used models of AD for investigating the development of tau and amyloid pathology due to its advantage of overexpressing tau [80]. The mutant genes in the 3×Tg-AD mice are APP, PSEN1, and MAPT; The mutation sites are APP K670_M671delinsNL (Swedish), MAPT P301 L, PSEN1 M146 V, which are associated with FAD [80, 81].
The progressive accumulation of NFTs within neurons resulting from massive neuronal death represents the significant neuropathologic feature of AD [82]. Meanwhile, the translation products of overexpressed transgenes are present in the central nervous system, including the hippocampus [32]. Furthermore, the hippocampus, with its essential function in learning and memory, would be particularly susceptible to damage in the early stages of AD [83]. Amyloid plaques and NFTs could be produced simultaneously in the brains of 3×Tg-AD mice [32]. The extracellular Aβ deposits appeared in the prefrontal cortex and became more extensive at 12 months. The formation of Aβ in the 3×Tg-AD brain gradually increases with age, and the effect on Aβ42 levels is particularly pronounced. The study demonstrated that Aβ precedes NFTs formation in 3×Tg-AD mice [80]. Importantly, the 3×Tg-AD model has a unique feature in that plaques and tangles appear simultaneously in advanced stages due to overexpression of the Tau mutant form (MAPTP301 L) [80]. Indeed, the intracellular hyperphosphorylation of Tau has been shown to be present in neurons from 15-month-old mice of 3×Tg-AD [84]. Configurational alterations and aggregates of hyperphosphorylated tau were detected in the hippocampus of 3×Tg-AD at 12 to 15 months of age [80, 81]. Notably, aging-associated synaptic dysfunction precedes the manifestation of plaques and tangles. Moreover, protrusion dysfunction could result in defects in protrusion plasticity [80]. Importantly, indicators of synaptic plasticity in the hippocampal CA1 region were intimately related to spatial learning and memory [84], and deficiency in synaptic plasticity was correlated with the accumulation of intraneuronal Aβ [85].
Rapamycin therapy could reduce Aβ and tau levels by restoring mTOR signaling and increasing autophagy induction levels, which in turn ameliorated cognitive dysfunction in 3×Tg-AD mice. Accumulation of Aβ and tau increased the mammalian targets of mTOR signaling, while reduction of mTOR signaling could reduce Aβ and tau levels [14]. Interestingly, the reduction of Aβ and Tau levels after rapamycin administration was not attributed to altered production of Aβ and Tau. Since the steady-state levels of APP, C99, and C83 were similar between the treated and untreated 3×Tg-AD mice. The levels of APP, C99, and C83, essential substances for Aβ production, were positively correlated with Aβ content. The facts demonstrated that rapamycin did not affect the processing of APP and thus did not affect the production of Aβ. Furthermore, inhibited transgenic Tau expression would not change the accumulation of NFTs in the AD animal models; therefore, rapamycin also had no effect on tau production [14].
However, the levels of Atg7 and Atg5-Atg12 complexes were significantly increased in the brains of 3×Tg-AD mice treated with rapamycin. Agt7 and the Atg5/Atg12 complex were essential related proteins for autophagy induction [86]. Moreover, previous studies have demonstrated that reducing mTOR function would directly increase autophagy induction [87]. Rapamycin could significantly reduce intracellular Aβ42 levels by increasing autophagy induction, in contrast, rapamycin was unable to significantly reduce the intracellular Aβ42 levels in the presence of the autophagy inhibitor 3-methyladenine, indicating that autophagy induction would be necessary for reduction of Aβ levels mediated by rapamycin. Therefore, rapamycin ameliorated cognitive deficits of 3×Tg-AD mice by increasing the clearance of Aβ and tau through increased autophagy induction rather than by decreasing the generation of Aβ and tau levels.
According to the analysis of data from the included articles, rapamycin therapy of 2.24–14 mg/kg for 10–12 weeks has been revealed to significantly ameliorate the learning and memory ability of 3×Tg-AD mice.
5×FAD
The mutant genes in the 5×FAD mouse are APP and PSEN1. 5×FAD mice overexpress human APP and PSEN1 proteins under the control of the mouse Thy1.2 promoter, with a total of five chain mutations of AD, respectively the Swedish (K670N/M671L), Florida (I716V), London (V717I) mutations in APP, and the M146L and L286V mutations in PSEN1 [88].
The 5×FAD model is characterized by the demonstration of rapid onset of AD disease. Of the original three strains generated in the 5×FAD mouse model (Tg6799, Tg7031, and Tg7092), the Tg6799 strain expressed the highest level of mutant APP expression, which was studied most frequently of the three strains [89]. The E4FAD, E3FAD, and E2FAD mouse models were hybridizations respectively, among the widely used 5×FAD mice (Tg6799 line) with the APOE4, APOE3, and APOE2 targeted replacement mice [90]. There were significantly more plaques in the E4FAD mice than in the E3FAD and E2FAD models at four and six months. Specifically, E4FAD mice exhibit the greatest age-dependent defects relative to E3FAD and E2FAD animals. Evidence indicated that soluble Aβ42 was detectable in these mice at 1.5 months of age, and its levels increased dramatically with age [89]. The level of Aβ40 also increased with age, but at a slower rate and a much lower level than Aβ42, with Aβ42/Aβ40 ratios as high as 25% in juvenile animal models of AD [89]. However, tau hyperphosphorylation and formation of NFTs were not observed in 5×FAD mice [91]. In addition, astrocyte hyperplasia and microglia hyperplasia were seen in 5×FAD mice at two months of age [92].
Rapamycin has been demonstrated to attenuate amyloid deposition and tau protein-associated pathological damage in AD transgenic mice through enhanced autophagy, thereby ameliorating learning and memory deficits in AD animal models [88]. Rapamycin could inhibit the phosphorylated mammalian target of rapamycin p-(mTOR) expression and increase the expression of autophagy marker light chain 3-1 (LC3-1) in EFAD mice, indicating enhanced autophagy levels. The neuroprotective effect of rapamycin was associated with the APOE genotype, and the effect was particularly pronounced in the E4FAD group. Compared with E3FAD mice, E4FAD mice had more pronounced decreases in the autophagy marker light chain 3 (LC3), an increase in functionally heterogeneous autophagic vesicles and material to be degraded, and elevated levels of Aβ, indicating that APOE4 exacerbated autophagic dysfunction and Aβ deposition in mice [88]. In addition, APOE4 mice exhibited the blood-brain barrier (BBB) loss of integrity and CBF deficits, which were associated with cognitive function and brain Aβ levels. Importantly, rapamycin restored the integrity of the CBF and BBB in APOE4 mice after six months of receiving therapy. These restorations were associated with reduced CypA levels in the vascular system. CypA, a pro-inflammatory cytokine, has been previously demonstrated to be damaging to the vascular system of APOE mice with aortic aneurysms and atherosclerosis [93]. CypA could result in BBB catabolism through activation of the NF-κB-matrix-metalloproteinase-9 (MMP9) pathway [93]. However, rapamycin could suppress endothelial CypA levels, thereby restoring the CBF and maintaining the integrity of the BBB [93].
According to the data analysis of the included articles, rapamycin therapy at 1 mg/kg for six weeks was revealed to be able to significantly ameliorate the learning and memory capacity of 5×FAD mice.
Pathological mechanism
The accumulation of Aβ, the major component of neuroinflammatory plaques, has been recognized as a molecular driver of AD pathogenesis and progression. Aβ is produced from APP via sequential cleavage by β-secretase and γ-secretase and has been recognized as a major cause of neurotoxicity in AD [94]. The BACE1 cleaved APP1 on APP generating sAβPP and C99 sites for the first time. Subsequently, γ-secretase cleaved C99 to release Aβ, of which Aβ1–40 and Aβ1–42 were the most common isoforms. Whereas aggregation of Aβ could potentially form plaques and oligomers that have been recognized as the more toxic form of Aβ [95, 96]. In addition, Aβ causes other AD pathologic features, including neuroinflammation, oxidative stress and mitochondrial dysfunction, and neuronal death and dysfunction [18]. Decreasing Aβ levels could eliminate cognitive deficits in multiple models of AD. Therefore, the balance between Aβ production and effective clearance is significant for Aβ homeostasis [97].
Tau is a microtubule-binding protein with the function of promoting microtubule formation and stabilization. NFTs forming from hyperphosphorylated tau accumulation was the hallmark lesion of AD [98]. Hyperphosphorylation of tau was mediated by protein kinases or phosphatases, which are participating in neurofibrillary degeneration in AD [99, 100]. In addition, tau pathology has been demonstrated to be heavy reliance on Aβ accumulation [101, 102]; meanwhile, the toxic state of tau proteins also could impact the production of Aβ [103].
Growing evidence demonstrated the significance of Aβ-tau interactions in the pathogenesis of AD [104, 105]. Extracellular Aβ was detected to accumulate in the neocortex and hippocampus in 3×Tg-AD, and then tau formed fibroblastic tangles [80, 106]. The inhibited activity of BACE1, an enzyme required for Aβ production, has been a critical approach to blocking the amyloid cascade reaction [107]. Significantly, the release of BACE1 siRNA and rapamycin was demonstrated to reduce BACE1 expression, resulting in the promotion of autophagy and reduced Aβ deposition [108].
Research has demonstrated that autophagy may have a critical effect in reducing Aβ42 in the brains of transgenic animals with AD [18]. The study indicated that rapamycin could reduce Aβ42 levels and deposition and restore mTOR signaling to normal levels in the brains of 3×Tg-AD mice [14]; Spilman et al. demonstrated that inhibited mTOR pathway could reduce Aβ42 levels in vivo and block or delay the onset of AD in mice [18]; Majumder et al. demonstrated that rapamycin reduced the formation of Aβ plaques and NFTs, and demonstrated Aβ levels and deposition reduced with lifetime administration of rapamycin [15]. In addition, multiple shreds of evidence have shown that decreasing mTOR function would directly increase autophagy induction [87], and rapamycin could inhibit mTOR signaling in transgenic mice with AD [109]. Importantly, levels of autophagy-related proteins, Atg7 and the Atg5/Atg12 complex, were significantly increased in the brains of transgenic mice of AD treated with rapamycin therapy [110]. Moreover, the level of LC3-II was significantly increased in the brains of AD mice with rapamycin intervention compared with control mice, while the level of LC3-II, as an autophagy marker, was proportional to the degree of autophagy [88]. Therefore, the reason for the reduced Aβ is easily deduced to be most likely owing to the induction of autophagy by the mTOR pathway signaling inhibited by rapamycin. And studies have suggested that rapamycin may primarily alleviate AD symptoms through reduced deposition of Aβ40 and Aβ42 in the cortex [15].
Rapamycin, as a well-known inhibitor of mTOR, has been shown not only to decrease Aβ deposition but also to inhibit tau phosphorylation induced through autophagy [57, 112]. mTOR and Phosphoprotein 70 ribosomal protein S6 kinase (P70S6K) increased expression would be co-localized with NFTs and mediate tau phosphorylation [113–115]. The targets of mTOR and p70S6K were serine/threonine kinases, which have critical functions in the regulation of protein synthesis and degradation, age-dependent cognitive decline, and the pathogenesis of AD [113, 114]. Research firmly demonstrated that overactive mTOR resulted in increased tau levels through reduced autophagy induction [116]. Meanwhile, mTOR signaling was reduced, and autophagy induction was increased in P301S mice intervened with rapamycin, which ameliorated tau pathology in AD mice [116].
Oxidative stress
Oxidative stress refers to the process of oxidative damage caused by the imbalance between the production and scavenging of oxygen free radicals in vivo or in cells [117, 118], resulting in the accumulation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) in vivo or cells [119, 120]. Increased ROS level was detected in postmortem brain tissues of patients and animal models of AD [121, 122]. In addition, pathological tau impairs mitochondrial function, causing increased products of ROS and resulting in oxidative stress [123, 124]. Excessive metal ions (e.g., zinc) and excessive cholesterol production cause oxidative stress, which in turn further triggers inflammatory responses and then produces more free radicals, resulting in further oxidative stress, thus creating a vicious cycle [57, 125–127]. Importantly, the inflammatory response could result in neuronal degeneration, thereby causing neurodegenerative diseases [128, 129].
In AD pathogenesis, rapamycin is able to reverse the vicious cycle that develops between excess zinc, tau, and oxidative stress. Increased zinc levels enhance ROS production in mitochondria. Oxidative stress could increase zinc concentration and tau hyperphosphorylation. In addition, excess zinc and hyperphosphorylated tau proteins cause oxidative stress and neurotoxicity [130, 131]. Increased oxidative stress has been demonstrated to cause tau hyperphosphorylation and exacerbate neuronal death [126, 132]. Oxidative stress has been previously proven to be the potential mechanism of mTOR activation in AD [133]. While rapamycin intervention attenuated zinc-induced oxidative stress damage in rat hippocampus. ROS level was reduced in zinc-induced AD rats with rapamycin administration [58]. Zinc could induce oxidative damage to Mitochondrial DNA via the Nrf2/HO-1 pathway and the ROS generation associated with the Nrf2/HO-1 pathway could potentially provide significant effects in neurodegenerative processes [134–137]. However, Nrf2, a transcription factor, dramatically decreased in the brains of AD patients, which negatively regulates ROS levels to prevent oxidative stress damage [138]. Rapamycin reversed the reduction of Nrf1/HO-5 levels induced by zinc and also rescued oxidative stress induced by tau hyperphosphorylation associated with the active Nrf2/HO-1 signaling pathway in rats [139].
In addition, rapamycin might also reduce oxidative stress by modulating cholesterol, thereby reducing the inflammatory response and reversing neuronal damage. The oxidative stress and inflammation resulted in neurodegeneration in AD, generating a vicious cycle. Oxidative stress induced activation of astrocytes and microglia, subsequently releasing pro-inflammatory factors that resulted in the inflammatory response. In turn, astrocytes could promote and maintain the oxidative stress state with time in pathological conditions, and neuroglial activation resulted in the release of toxic free radicals, exacerbating neuronal damage [140]. An essential function of astrocytes was supporting neurons in providing energy substrates and producing cholesterol. However, the excessive cholesterol would be oxidized to more hydrophilic metabolites that could diffuse out of the brain via the BBB [141]. Different oxidized sterols generated as toxic accumulation due to the disruption of BBB integrity in the AD brain [142]. Importantly, rapamycin could inhibit oxidative stress by maintaining normal cholesterol levels via its impact on the cholesterol biosynthesis pathway and cytoplasmic ribosomal proteins [74]. Rapamycin could ameliorate astrocyte ROS production by activating autophagy, thereby inhibiting oxidative stress, reversing the inflammatory response of neurons, and slowing down the further progression of AD [143].
In addition, dysfunctional mitochondria have been recognized as one of the most prominent and earliest features of AD [144, 145]. Mitochondrial dysfunction could generate high levels of ROS, thus contributing to oxidative stress and potentially causing toxic effects on neuronal cells with longevity and deficient antioxidant defenses [146]. It is demonstrated that normal mitochondrial function could alleviate the degree of oxidative stress and reduce cytotoxicity to maintain neuronal health. Importantly, mitochondrial autophagy is capable of engulfing and degrading damaged mitochondria to sustain healthy mitochondrial function [147, 148]. Interestingly, rapamycin, considered an activator of autophagy [149], fostered the binding of mitochondria to autophagosomes and the fusion of autophagosomes and lysosomes in the hippocampus of AD mice. Eventually, rapamycin accelerated mitochondrial degradation and impeded mitochondria-dependent apoptosis in the hippocampus of AD mice [75].
Therefore, rapamycin activated mitochondrial autophagy to restore normal mitochondrial function, thereby restoring normal ROS levels, inhibiting oxidative stress, and ameliorating neuronal damage.
Cholesterol metabolism
Currently, numerous pieces of evidence have been presented in supporting the effect of lipids, particularly cholesterol, in the pathogenesis of AD. AD was demonstrated to be associated with abnormalities in cholesterol biosynthesis and catabolism in a metabolomic and transcriptomic study as well as a meta-analysis [150]. The failure of cholesterol homeostasis in the brain could cause synaptic dysfunction and cognitive decline, impacting neuronal function [151, 152]. Plasma cholesterol could not cross the BBB since the plasma and brain cholesterol pools are separated by two barriers, the BBB and the cerebrospinal fluid (CSF) barrier, and therefore brain cholesterol metabolism has been largely independent of peripheral tissues [153]. However, excess cholesterol could be oxidized into more hydrophilic metabolites, such as oxysterols that would diffuse out of the brain via the BBB [154]. While the proteins in plasma were found in the CSF of cognitively healthy individuals who carried APOE4 and who subsequently suffered from AD [155]. The fact that these proteins may have leaked through the BBB demonstrated that the integrity of the BBB has been disrupted before cognitive decline [156]. Disruption of BBB integrity could result in the generation of different oxysterols to toxic accumulation and cause neurodegeneration through enhanced oxidative stress and inflammation in the AD brain [157]. Therefore, abnormal level of cholesterol was oxidized, diffusing into the brain through the damaged BBB and mediating oxidative stress and inflammation to further damage neurons, ultimately causing cognitive dysfunction.
The brain, with a quarter of the non-esterified cholesterol pool in the body, is the most abundant organ in lipids [158, 159]. Therefore, the maintenance of stable homeostatic levels of cholesterol in the brain would be critical for neuronal function and brain development. Apolipoprotein E (ApoE) has a critical effect on lipids, including cholesterol metabolism, and the apolipoprotein E ɛ4 (APOE ɛ4) allele increases the risk of developing AD [160, 161]. In addition, the APOE ɛ4 allele represents the major genetic risk factor for AD [162]. Therefore, dysregulation of cholesterol homeostasis in the brain has received extensive attention in the past decades [163]. There are three main variants of APOE: APOE2, APOE3, and APOE4. APOE4 could significantly increase AD risk compared to the more common APOE3 variant [164]. Expression of cholesterol metabolism genes, including Fdps and Mvk, was regulated by oligo amyloid-β peptide in the cholesterol biosynthesis pathway [165]. Fdps was upregulated during APP/PS1 mice and reversed by rapamycin therapy. Furthermore, in the temporal lobe, Mvk was overexpressed in the progression of APP/PS1 pathogenesis and inhibited by rapamycin therapy [74]. Therefore, rapamycin could alleviate AD symptoms by modulating the cholesterol biosynthesis pathway.
In adult brains, most cholesterol synthesis primarily occurred in astrocytes [166]. Astrocytes in the brain contribute to the maintenance of glutamate, ionic, and water homeostasis and function to prevent oxidative stress [167]. In addition, astrocytes have another important function of supporting neurons in providing energy substrates and producing cholesterol [168]. Astrocytes store glucose in the form of glycogen and transport lactate through the lactate shuttle to provide energy via glutamate released in response to neurons [169, 170]. However, reactive astrocytes were capable of producing various pro-inflammatory mediators and potentially neurotoxic compounds, including nitric oxide (NO) that could cause oxidative stress [171]. While rapamycin inhibited oxidative stress and abnormal accumulation of cholesterol levels by reducing ROS production in astrocytes through the activation of autophagy [143]. In addition, rapamycin was identified as being present in brain tissue, which indicates that rapamycin crossed the blood-brain barrier, according to the research of Cloughesy and colleagues, who measured the amount of rapamycin in brain tumors [172]. Rapamycin could directly affect brain mTOR signaling; meanwhile, mTOR signaling inhibited by rapamycin enhanced the level of autophagy, thereby inhibiting oxidative stress [15]. Therefore, rapamycin could reduce ROS production in astrocytes by activating autophagy, inhibiting the abnormal accumulation of cholesterol levels and oxidative stress, and could cross the BBB and inhibit the mTOR signaling, thus reversing the vicious cycle between oxidative stress and cholesterol production [74].
Vascular dysfunction
Abnormal CBF has been one of the earliest features of AD progression, as reduced CBF is a phenomenon observed in the vast majority of dementia patients [173, 174]. In the early stages of AD, the cognitive function of healthy individuals would be immediately impaired when the CBF has been reduced by about 20% [175]. Furthermore, impaired blood flow in the AD mouse model resulted in increased Aβ deposition, which indicated that blood flow defects would worsen Aβ pathology [176]. Studies have shown that brain Aβ load in early AD patients was associated with reduced blood flow to various brain regions [177]. The operation of Aβ1–40 reduced resting CBF in APP transgenic mice [178], and vascular Aβ deposition resulted in reduced local CBF in APP/PS1 mice [179]. In the early stages of AD, abnormal Aβ downregulation in CSF appeared to be associated with reduced temporoparietal CBF, and this perfusion deficit worsened with the appearance of abnormal upregulation of tau in CSF [180]. Phosphorylated tau and total tau of cerebrospinal fluid in healthy brains were associated with reduced CBF in frontotemporal regions [181]. All these data demonstrate that reduced CBF may contribute to cognitive dysfunction and AD progression.
Cerebrovascular lesions could also cause inadequate supply of glucose and other nutrients and impede the removal of harmful substances from areas of the brain caused by the lesions. Importantly, glucose metabolism impairment and energy deficiency serve as hallmarks of AD. In addition, cerebral vascular lesions can induce impaired neurovascular regulation [182], especially cholinergic innervation [183] and disruption of the BBB [184]. These vascular defects ultimately alter the blood supply to the brain, leading to neuronal damage and cognitive impairment [185].
Numerous studies have shown that rapamycin could inhibit mTORC1, exerting significant and multifaceted protective effects in AD preclinical models [19]. mTOR attenuation has previously been shown to ameliorate microvascular endothelial vasodilator function and increase cerebral microvessel density, which ultimately rescues CBF and cognitive deficits in AD mouse models [16, 18]. Rapamycin therapy was found to increase CBF and restore microvascular endothelial function, reducing amyloid load and ameliorating cognitive function in a mouse model of AD [186]. Rapamycin benefited the removal of damaged or misfolded proteins, including tau proteins, by enhancing autophagy. In addition, recovery of cerebral microvascular endothelial function involved activation of endothelial NO synthase in the hAPP(J20) model with rapamycin therapy [16]. Endothelial NO synthase dysregulation and reduced NO bioavailability were also associated with the development of AD amyloid plaques [187] and neurovascular uncoupling [188], which were both mitigated by mTOR attenuation.
Rapamycin supplementation with food resulted in approximately 30–35% reduction of mTOR activity of the brain [14, 18]. In several independent models of AD, mTOR has been identified as a critical mediator of cerebrovascular dysfunction, including the hAPP(J20) mouse, models of vascular cognitive impairment, and canonical aging in rats [189]. In hAPP(J20) mice, maintenance of CBF and vascular integrity through mTOR attenuation depended on the restoration of NO synthase (NOS) activity [16]. It suggests that mTOR acted in resting CBF deficits of AD animal models at least partially by inhibiting NOS. The attenuation of mTOR could increase Aβ clearance by restoring the integrity and function of the cerebral vasculature and inducing autophagy. Previous studies have demonstrated that rapamycin could restore cerebral glucose uptake, CBF, and BBB integrity in young APOE4 transgenic mice, which would be efficient in ameliorating early learning memory deficits in mice [186, 190]. In addition, the fact that cerebrovascular dysfunction occurred in the early stages of AD indicated that the attenuated signaling of mTOR inhibited by rapamycin may potentially support the prevention and reversal of memory deficits in AD.
Therefore, rapamycin could ultimately rescue cognitive functional impairment by inhibiting mTOR signaling and thus NOS activity, restoring CBF deficits, the integrity of cerebrovascular function, as well as increasing the clearance of Aβ in animal models of AD [191].
Insulin signaling pathway
Insulin is a pivotal regulator of energy metabolism due to its original discovery as the treatment for diabetes mellitus [192]. Proteins involved in insulin signaling were detected in many brain regions, including those affected by AD, such as the hippocampus and temporal lobe [193]. A recent population-based longitudinal study demonstrated that compromised insulin response in midlife was associated with enhanced risk of AD [194], probably due to abnormalities of insulin signaling [195]. Cell culture-based studies have reported that exposure of neurons to soluble Aβ oligomers results in a decrease in insulin receptors [196]. In addition, another study demonstrated that insulin promotes Aβ translocation to the cell membrane and facilitates Aβ release [197]. Clinical literature indicated that insulin injection (1.0 mU/kg×min) could increase the expression and activity of BACE1, causing increased Aβ42 level production and consequently cognitive dysfunction [198, 199].
Insulin resistance has been recognized as a potential mechanism for the development of AD [200]. As an impairment of insulin signaling, insulin resistance represents a relative deficiency of insulin due to various factors that lead to high than normal levels of insulin in the body while its effective concentration and bioefficacy are reduced [79]. In the brains of AD patients, insulin receptor (INSR), insulin-like growth factor-1 receptor (IGF1R), and insulin receptor substrate (IRS1) expression of proteins are downregulated, and defective brain insulin signaling is considered to be a contributing factor to cognitive impairment in AD patients. Insulin-induced autophagosomes activate β- and γ-secretase by enrichment of secretase proteins in autophagosomes, thereby increasing Aβ production [201]. Autophagy inhibitor 3-MA reversed the increased Aβ accumulation induced by insulin resistance, indicating that the accumulation of autophagosomes would result in the exacerbation of Aβ production under insulin-resistant conditions [202]. Importantly, rapamycin therapy could increase Aβ clearance by promoting autophagy and reduce tau hyperphosphorylation by upregulating insulin-degrading enzyme levels. In addition, rapamycin may contribute to ameliorated insulin sensitivity in AD patients by inhibiting mTOR [203]. In preclinical studies, rapamycin showed potential neuroprotective effects [204] and may reduce impairment caused by oxidative stress and inflammation in the brain by inhibiting mTOR and ameliorating insulin sensitivity [205, 206]. Previous studies have revealed that insulin impairment in transgenic AD mice has been mTOR-dependent [112].
Protein kinase B (PKB), alternatively named Akt, exists crosswise in multiple signaling pathways and can regulate neuronal apoptosis by affecting downstream factors [207]. PI3K has an essential role in insulin function, which is primarily accomplished through the activation of the Akt/PKB cascade. PKB, as a factor downstream of the insulin PI-3K/PKB signaling pathway, could directly regulate downstream glycogen synthase kinase-3β (GSK-3β) activity. It was shown that Aβ may result in a blocking effect on the insulin PI-3K/PKB signaling pathway and decreased the activity of PKB, which is located downstream of the insulin PI-3K/PKB signaling pathway, resulting in reduced levels of phosphorylation at the sre9 or sre2l sites at the end of GSK-3β. This would activate GSK-3β activity, modulate downstream factors, and induce apoptosis in neuronal cells. Importantly, activated Akt could induce glycogen synthesis by inhibiting GSK-3β, then proceed to protein synthesis via mTOR and downstream components, and further promote cell survival by inhibiting multiple pro-apoptotic factors, including FoxO transcription factor, GSK-3, and MST1 [208, 209]. Therefore, activated Akt promoted protein synthesis and rescued neuronal apoptosis, which was caused by the fact that Aβ decreased insulin PI-3K/PKB activity and thus activated GSK-3β activity. Interestingly, rapamycin could induce Akt phosphorylation through extremely significant positive effects [112]. It is indicated that rapamycin could rescue the activation of GSK-3β activity by inducing Akt phosphorylation caused by Aβ-induced decrease in insulin PI-3K/PKB activity.
Therefore, rapamycin may reduce oxidative stress and inflammation-induced damage in the brain by inhibiting mTOR and improving insulin sensitivity. Furthermore, rapamycin could rescue the Aβ-induced decrease in insulin PI-3K/PKB activity by inducing Akt phosphorylation and then regulate GSK-3β activity to promote neuronal cell survival, ultimately alleviating cognitive dysfunction.
Clinical implications of rapamycin therapy
Rapamycin is primarily used clinically to prevent solid organ transplant rejection [8] and secondly could inhibit the mTOR pathway and reduce accumulation of Aβ plaques and NFTs in animal models of AD [14]. A placebo-controlled pilot study was conducted to explore the safety and tolerability of rapamycin in elderly AD patients. The findings indicated that rapamycin attenuated the decline in age-related spontaneous activity. Importantly, studies have shown that rapamycin interventions were effective even in older animals [109, 210] and that patients with AD who did not receive rapamycin therapy until old age could achieve the effect of cognitive alleviation [109].
In fact, the depth and breadth of positive preclinical data about rapamycin probably exceeds any other current potential AD therapy [211]. Significant effects of rapamycin include reduced Aβ deposition, decreased accumulation of phosphorylated tau and NFTs, restored CBF and maintained BBB integrity, which ameliorated cognitive functions [14–16, 58]. Favorable outcomes have been observed in the AD mice models in the included articles, including hAPP(J20) mice, APP/PS1 mice, 3×Tg-AD mice, and 5×FAD mice. However, there are still concerns in the research field about the safety of rapamycin [14], including mouth sores, nausea, and gastrointestinal discomfort [14], while in most cases the severity has no clinical significance [212, 213]. In a clinical study, subjects received 1 mg of rapamycin therapy per day, producing a plasma level of 7.2±2 ng/ml. The majority of the 17 patients who completed 8 weeks of therapy had no significant adverse events during their participation in the therapy [214]. Based on the ongoing preclinical trials of rapamycin therapy for AD (NCT04629495, NCT04200911, and NCT06022068), rapamycin was administered at doses of 1 mg/day, 1 mg/day, and 7 mg/week, respectively (ClinicalTrials.gov(https://clinicaltrials.gov/)). Indeed, rapamycin has been extensively practiced clinically with referenceable dosage as well as safety information [16]. With limited side effects, rapamycin appeared to be well tolerated in elderly subjects and could be acceptably administered as a treatment for AD [211]. Side effects related to rapamycin were dose-dependent and reversible, which demonstrated that establishing safety dosage guidelines for clinical trials of rapamycin therapy for AD should prove readily achievable. In addition, the development of rapamycin as an emerging drug for AD therapy has the realities of high cost and low profitability. Since rapamycin was off-patent and accessible as a generic drug, in this situation, preclinical trials of rapamycin for AD therapy progressed slowly [211].
Importantly, Matt Kaeberlein and Veronica Galvan strongly recommend that clinical trials of rapamycin efficacy should ideally be conducted in early-stage AD patients and newly diagnosed AD patients in ideal situation [211]. Therefore, the efficacy and safety of rapamycin therapy for AD deserve to be explored intensely in further studies. It will facilitate the observation and reflection on the efficacy and limitations of rapamycin therapy for AD and promote better translation into a suitable drug for AD patients.
Strengths and limitations
Based on literature research, our article is the first detailed meta-analysis and systematic review of rapamycin therapy in AD animals. This study comprehensively evaluated the therapeutic effects of rapamycin therapy in animal models of AD, mainly in terms of behavioral tests and pathological features. Comprehensive analysis of the findings obtained from the 13 included articles demonstrated that rapamycin has the ability to ameliorate cognitive dysfunction by modulating the autophagy and mTOR pathways, as well as oxidative stress, etc. In addition, the MWM test was chosen as the sole cognitive function index test method because of the following advantages compared with other behavioral testing methods: 1) Reliable in a variety of cross-species studies including guinea pigs, rats and mice. 2) Ability to track various swimming strategies such as spatial, non-spatial and stagnation.
However, this study has some limitations. Relevant studies that meet the inclusion criteria are limited, and further research in this area deserves to be continued. The limitation may be due to the strict inclusion and exclusion criteria. Specifically, in order to explore the improvement effect of rapamycin as a single drug on cognitive impairment in AD animal models, rather than the synergistic effects of other drugs, combination drugs articles were excluded. Secondly, based on the generalizability of behavioral testing methods, only MWM test was selected as the main indicator of behavioral testing methods. Compared with other behavioral testing methods, MWM test has the following advantages: 1) Reliable in a variety of cross-species studies, including guinea pigs, rats, and mice. 2) Ability to track various swimming strategies such as spatial, non-spatial, and stagnation. Therefore, other types of behavioral testing methods were excluded, such as the Y maze and Barnes water maze. In addition, for the experimental indicators of MWM test, only articles simultaneously containing escape latency and number of platform crossings were included, therefore the articles only containing escape latency or number of platform crossings were excluded. In summary, the number of included articles meeting the requirements was limited objectively due to strict inclusion standards. Due to the limited number of included articles, the indicators measured in each article are different, which restricts subsequent subgroup analysis, thereby constraining a more comprehensive interpretation of the manuscript results.
Secondly, the methods involved in the experiments required to be clearly stated, such as randomized grouping, blinding, etc., which resulted in the existence of a certain degree of bias in the assessment of the results. The quality of included articles has been evaluated according to SYRCLE’s criteria, and we found that many experiment designs may lack rigor. Blinding and the reporting of complete data were unavailable in most included articles. Importantly, relatively larger studies have confirmed the benefits of performing randomization, allocation concealment, and blinding. Schulz et al. (1995) showed that the odds ratios were inflated by 30% in trials with unclear concealment. Additionally, trials lacking double-blinding produced significantly larger effect estimates, showing a 17% exaggeration in odds ratios [215].
Furthermore, there was a certain degree of heterogeneity among the included studies, which may be caused by factors such as the administration method, dosing cycle, and dosage of rapamycin. First, the included articles covered a variety of administration modes, administration durations, and drug doses, which presented challenges to the data analysis and result interpretation processes. In these studies, different routes of administration may result in different bioavailability. This may influence the results of our analysis to some extent and may constitute an important source of observed heterogeneity. Furthermore, the potential impact of differences in administration duration and drug dose on heterogeneity must be considered. Different treatment regimens may involve different doses and treatment periods, potentially leading to different effects observed in different studies and contributing to between-study heterogeneity. Another potential reason is that the drug safety evaluation of AD animal models was not reported in the included articles; furthermore, there is currently still no one AD mouse model that accurately captures the full pathological profile of AD patients, which results in the limitation of simulating AD pathology experimentally in AD animal models.
Conclusion
According to the comprehensive analysis of the included articles, rapamycin was found to significantly ameliorate learning and memory abilities in transgenic animal models of AD, mainly through the reduction of Aβ and tau levels, as well as the improvement of oxidative stress, cholesterol, cerebrovascular function as well as insulin resistance. Among the transgenic animal models, 2.24 mg/kg rapamycin was administered for 13–16 weeks in the single transgenic animal model of AD, and the human dose following conversion was 12.6 mg/70 kg; 1–2.24 mg/kg rapamycin was administered for 4–8 weeks in the double-transgenic animal model of AD, and the converted human dose was 5.6–12.6 mg/70 kg; 2.24–14 mg/kg rapamycin was administered for 10–12 weeks in the triple transgenic animal model of AD, and the converted human dose was 12.6–79.8 mg/70 kg; The administration of 1 mg/kg rapamycin for six weeks in the five-transgenic animal model of AD could ameliorate cognitive dysfunction and the converted human dose was 5.6 mg/70 kg. Through a comprehensive analysis of the converted doses, we decided to take the intersection (5.6–12.6 mg/70 kg/day) as the recommended dose of rapamycin to facilitate further clinical studies in this area. Based on the ongoing preclinical trials of rapamycin therapy for AD (NCT04629495, NCT04200911, and NCT06022068), rapamycin was administered at doses of 1 mg/day, 1 mg/day, and 7 mg/week, respectively. The above dosages have a certain correlation with our recommended dosages, indicating that our recommended dosage may be the potential therapeutic dose, which will benefit the prospective clinical studies of rapamycin therapy for AD.
AUTHOR CONTRIBUTIONS
Jie Cai (Conceptualization; Data curation; Formal analysis; Methodology; Software; Visualization; Writing – original draft); Danni Xie (Conceptualization; Software; Visualization; Writing – original draft); Fanjing Kong (Methodology; Software; Visualization); Zhenwei Zhai (Project administration; Software; Visualization); Zhishan Zhu (Methodology; Project administration; Visualization); Yanru Zhao (Project administration; Software); Ying Xu (Formal analysis; Funding acquisition; Resources; Software; Supervision); Tao Sun (Conceptualization; Data curation; Formal analysis; Funding acquisition; Methodology; Project administration; Resources; Supervision; Writing – review & editing).
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This work was supported by grants from the Sichuan Science and Technology Program (No. 2021YJ0178, 2020GFW194) and the Xinglin Scholar Research Promotion Project of Chengdu University of TCM (No. ZRQN2020008, MPRC2021034). The sponsor had no involvement in the research analysis or interpretation, and no contribution to drafting the manuscript.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article.