
Abstract
Background:
Alzheimer’s disease (AD) is a progressive neurocognitive disorder. There is no cure for AD. Maintenance on intracellular levels of nicotinamide adenine dinucleotide (NAD+) has been reported to be a promising therapeutic strategy for the treatment of AD. NAD+ precursors that represent candidate targets include nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR).
Objective:
This systematic review provides insights into the potential therapeutic value of NAD+ precursors including NMN and NR, for the treatment of AD using preclinical and clinical studies published in the last 5 years.
Methods:
The Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) protocol was followed to systematically search the literature using two databases.
Results:
We found 3 studies that used NMN to treat AD in preclinical murine models. However, human clinical trials using NMN as a therapeutic intervention in AD was not available in the current literature. We also found 4 studies that investigated the potential benefits of NR for the treatment of AD in preclinical models. We also found 2 human clinical trials that showed marked improvements in plasma and neuroimaging biomarkers, and cognitive measures following supplementation with NR.
Conclusions:
Results of preclinical and clinical studies confirm the potential benefits of NAD+ precursors for the treatment of AD. However, further clinical studies are required to confirm the increasingly important value of NAD+ precursors as effective pharmacological interventions in the clinic.
INTRODUCTION
Alzheimer’s disease (AD) is a complex multifactorial neurodegenerative disorder characterized by the accumulation of extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles containing hyperphosphorylated tau [1]. At present, no disease modifying therapeutic agents are available for the treatment of AD and existing therapeutic interventions provide symptomatic relief only [1].
Recent developments in AD drug discovery include immunotherapies which involve using artificial antibodies that target irregular Aβ plaques and clear Aβ deposits from the AD brain. The first passive anti-Aβ immunotherapy developed for AD was bapineuzumab. While the drug lowered Aβ burden in the AD brain, it failed to improve clinical outcomes in AD patients, although some benefits were observed in APOEɛ4 carriers [2]. Recent approaches include donanemab, aducanumab, and lecanemab; the latter two are both approved in the United States [3]. Donanemab and lecanemab attenuated memory decline by 20% and 27% and improved the quality of life by 40% and 57% respectively [4, 5]. Remternetug, another anti-Aβ immunotherapeutic that is currently undergoing clinical trials, significantly lowered Aβ burden in 75% of patients within 6 months compared to donanemab which took 18 months to lower Aβ in 72% of patients [6]. However, the use of Aβ-based immunotherapeutics is limited due to undesirable adverse effects such as encephalitis, poor clinical improvement, and limited effect on neurofibrillary tangles and other AD pathologies [7]. Other factors include variability and inconsistencies in the design of clinical trials using immunotherapies, which are crucial to ensuring reliable findings that can be translated to the clinic [7].
AD also consists of several metabolic features and is sometimes defined as a metabolic syndrome, since neurons cannot survive without an optimal energy supply [8]. The fact that cognitive decline, which is a major manifestation of AD is linked to metabolic dysregulation, suggests that metabolic disruption is a major contributor to AD [8]. Lowered levels of the pyridine nucleotide nicotinamide adenine dinucleotide (NAD+) have been reported in catabolic organs [9, 10] and plasma during aging [11], and aging is a major risk factor for AD and other neurocognitive disorders [9]. NAD+ is an essential redox and cofactor and promotion of cellular NAD+ anabolism could have beneficial neuroprotective effects [12]. NAD+ is fundamental for cellular energy metabolism and is necessary for electron transport through the mitochondrial electron transport chain, and ATP generation via oxidative phosphorylation [13]. NAD+ is also an essential substrate for ADP-ribosylation by the DNA repair poly-ADP-ribose polymerase (PARP) enzymes, cyclic ADP-ribose production for secondary messenger signaling by CD38 and CD157 NAD+ glycohydrolases, and protein deacetylation of the longevity genes known as sirtuins or silent information regulators of gene transcription [13].
Mitochondrial homeostasis is crucial for optimal neuronal function and neuronal viability. Mitochondrial function is mediated by mitophagy, whereby imperfect/nonfunctional mitochondria are cleared and recycled. Neuronal mitophagy protects against neuronal cell death and slows down pathogenic brain ageing associated with defective mitochondrial function [14–16].
NAD+ is also an essential substrate for ADP-ribosylation by the DNA repair poly-ADP-ribose polymerase (PARP) enzymes, cyclic ADP-ribose production for secondary messenger signaling by CD38 and CD157 NAD+ glycohydrolases, and protein deacetylation of the longevity genes known as sirtuins or silent information regulators of gene transcription [13].
SIRT1 mediates autophagy/mitophagy via numerous pathways including: 1) deacetylation of the autophagy related proteins-5 and -7 (ATG5 and ATG7), and light chain-3 (LC3) [17, 18]; 2) upregulation of beclin-1 (BECN1) expression, which facilitates the class III phosphatidylinositol 3-kinase nucleation complex autophagy [19]; 3) promoting the de-acetylation and translocation of LC3 from nucleus to cytoplasm [17]; 4) upregulation of LC3, and other autophagic protein including ATG12, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) and Ras-associated binding (Rab7) to stimulate both the acetylation and deacetylation of FOXO1 and FOXO3 [20–22]; and by upregulation autophagic proteins including PTEN-induced kinase 1 (PINK1), cytosolic E3 ubiquitin ligase (Parkin), and the BCL2 and adenovirus E1B 19-kDa-interacting protein 3 (NIX) [23–25], SIRT2, a cytoplasmic member of the sirtuin family can regulate mitochondrial function and mitophagy, via deacetylation of ATG5 [26]. The mitochondrial sirtuins, SIRT3, SIRT4 and SIRT5 can induces mitophagy via regulation of the mitochondrial SIRT3-PGAM5-FUNDC1- pathway [27–29], interaction with OPA1 and parkin [27, 30], and regulation of ammonia [27–31].
SIRT6 and SIRT7 are nuclear sirtuins that inhibit mTOR to initiate autophagy [27–32]. Apart from sirtuin, CD38, is involved in the regulation of autophagic fusion with lysosomes [33]. Overactivation of PARPs and CD38 family, can lead to NAD+ depletion and subsequently limiting the availability of NAD+ as a substrate and consequently reducing the activity of SIRT1 and therefore reducing mitophagy [14–25].
Under normal physiological conditions, NAD+ is produced de novo by the kynurenine pathway of tryptophan (TRYP) catabolism, primarily in the liver [34]. The process also catalyzes the conversion of TRYP to nicotinamide (NAM) which is then released into the serum. NAM and other NAD+ precursors including nicotinic acid (NA), nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR) can be used to synthesize NAD+ via the NAD+ salvage pathway [35]. In this process, NAM is produced as a by-product which is then converted to NMN via the catalytic activity of nicotinamide phosphoribosyltransferase (NAMPT) [36], and then transformed to NAD+ via the salvage pathway [35].
There is a wealth of studies supporting the role of NAD+ supplementation using NAD+ alone or its reduced form NADH, or NAD+ precursors including NAM, NA, NMN, and NR as an effective neuroprotective strategy to prevent cognitive decline and attenuate and/or slow down AD pathology [37–42]. Animal studies have shown that upregulation of NAD+ levels can mediate neuroprotection through several diverse mechanisms including lowered oxidative stress and impaired protein aggregation, reduced inflammation, impaired autophagy and restoration of mitochondrial function [43–45]. This has led to the hypothesis that upregulation of cellular NAD+ anabolism has multifactorial benefits albeit not restricted to increased energy alone. The current review summarizes evidence on supplementation and treatment with NAD+ precursors, and NMN and NR in particular, to attenuate and/or slow down AD pathology and maintain cognitive function.
CHOICE OF NAD+ PRECURSOR IS CRITICAL FOR THE DEVELOPMENT OF INTERVENTION STUDIES FOR AD
When selecting NAD+ precursors for the treatment of age-related neurocognitive disorders, it is important to note that NAD+ precursors have different boosting efficiencies, toxicity and dosage range. Niacin or NA has been reported to induce severe flushing, associated with the binding of NA to the G-protein coupled receptor (GPR) GPR109A receptor [46]. Therefore, NAD+ precursors that have little or no binding efficiency to GPR109A, yet can increase cellular NAD+ levels, such as NR and NMN, are more clinically beneficial.
NAM has been previously touted as a treatment for Type I diabetes, although several clinical trials have doubted this hypothesis [47–53]. In a comparative study, NAM but not NA was found to restore cellular NAD+ levels in streptozotocin-induced diabetes (reviewed in [54]). Treatment with NAM (100 mg/kg for 4 weeks) also induced improvements in metabolic function in a rodent model of obesity and type 2 diabetes (reviewed in [54]). NAM treatment enhanced hepatic NAD+ levels, and significantly improved glucose control [51]. However, chronic or high doses of NAM has been shown to be detrimental to hepatic function and may contribute to a fatty liver. This is likely due to a reduction in available methyl groups due to the enhanced conversion of NAM into 1-methyl-NAM (mNAM or MNA) via the catalytic activity of nicotinamide n-methyltransferase (NNMT) [55]. Supplementation with methionine, which serves as a methyl group donor, prevented the development of fatty liver due to high doses of NAM [56].
Age-related NAD+ depletion can downregulate sirtuin activity and SIRT1-mediated deacetylation of the tumor suppressor protein p53 [57]. NAM is an endogenous inhibitor of SIRT1 deacetylase activity. High levels of NAM have been reported to antagonize NAM cleavage reactions and/or mediate competitive inhibition at NAD+-binding sites leading to impaired sirtuin function and increased levels of NAD+. However, the effect of NAM on PARP-1 activity inhibition remains controversial. It has been reported that the Ki value for NAM-mediated inhibition of PARP-1 is between 90–600μM in vitro [58]. In mammals, oral NAM is catabolized in the small intestine and liver before it enters circulation. NAM is taken up by catabolic tissue in small amounts and rapidly converted to NAD+. Therefore, it is unlikely that the levels of tissue NAM can reach concentrations necessary to inhibit PARP-1 activity.
While supplementation with NMN has reported several benefits in a variety of disease models, there is growing body of evidence to suggest that NMN is effectively contained within the cells membranes and is not affected by high diffusion gradients. This suggests that NMN is unlikely to effectively traverse across most cell membranes [59]. Additional work is required to determine the range of disorders for which NMN may prove beneficial in the clinic. Recently, a randomized, multicenter, double-blind, placebo-controlled, parallel-group, dose-dependent clinical trial showed that whole blood NAD+ concentration was significantly and dose- dependently increased following oral supplementation with NMN treatment [60]. Oral administration of NMN up to 900 mg/day for 60 days was found to be safe and well tolerated and had significant improvements on the physical endurance and general health conditions of healthy adults [60]. Further work is needed to determine the effect of NMN supplementation on NAD+ and its metabolites in other tissues to determine a broader effect of NMN supplementation in health and disease.
NR have been reported to be well tolerated and orally bioavailable in mice and humans [61] and has been approved by the TGA and FDA. Higher levels of NMN, NAMN, NAM, NAAD, NAD+ and NADP+ were reported following oral gavage of NR compared to oral NAM which suggests increased activation of the NAD+ biosynthesis pathway following oral supplementation with NR [61]. Additionally, ADP-ribose levels were more significantly elevated compared to NA and NAM, providing evidence for increased activity of NAD+ consuming enzymes following NR supplementation compared to other NAD+ precursors [61]. As well, a randomized double-blinded placebo-controlled study showed that NR in combination with pterostilbene, a naturally occurring phytochemical found in blueberries, can increase NAD+ in a dose-dependent manner in whole blood lysates throughout the entire 8-week trial [62]. Taken together, this suggests that NR is a more potent precursor of NAD+ synthesis and NAD-dependent activities than amidated and acidic forms of niacin.
PRECLINICAL AND CLINICAL STUDIES FOR NMN AND NR IN AD
We used the Preferred Reporting Items for Systematic Review and Meta-Analysis (PRISMA) guidelines for our research strategy [63]. In the current study, we examined recent developments of using NMN and NR in AD preclinical and clinical pharmacotherapeutic trials to assess the efficiency of these NAD+ precursors on attenuating AD pathology and cognitive impairment and improving disease outcomes. We explored PubMed database using a set of keywords related to AD: ("Alzheimer’s” or “post*Alzheimer’s” or “Alzheimer’s disease” or “AD"), and NAD+ precursors: (“NR” or “nicotinamide riboside” or “NMN” or “nicotinamide mononucleotide") and AD preclinical and clinical trials with pharmacotherapeutics: (“preclinical studies” or “clinical trials” or “therapeutic” or “medication” or “drug”). Inclusion criteria was as follows: (1) written in English, (2) preclinical murine studies or human-based study sample, (3) clinical trial study design, (4) published after January 2015. We excluded review papers; studies that did not focus on brain and cognitive function or AD pathology (Fig. 1).
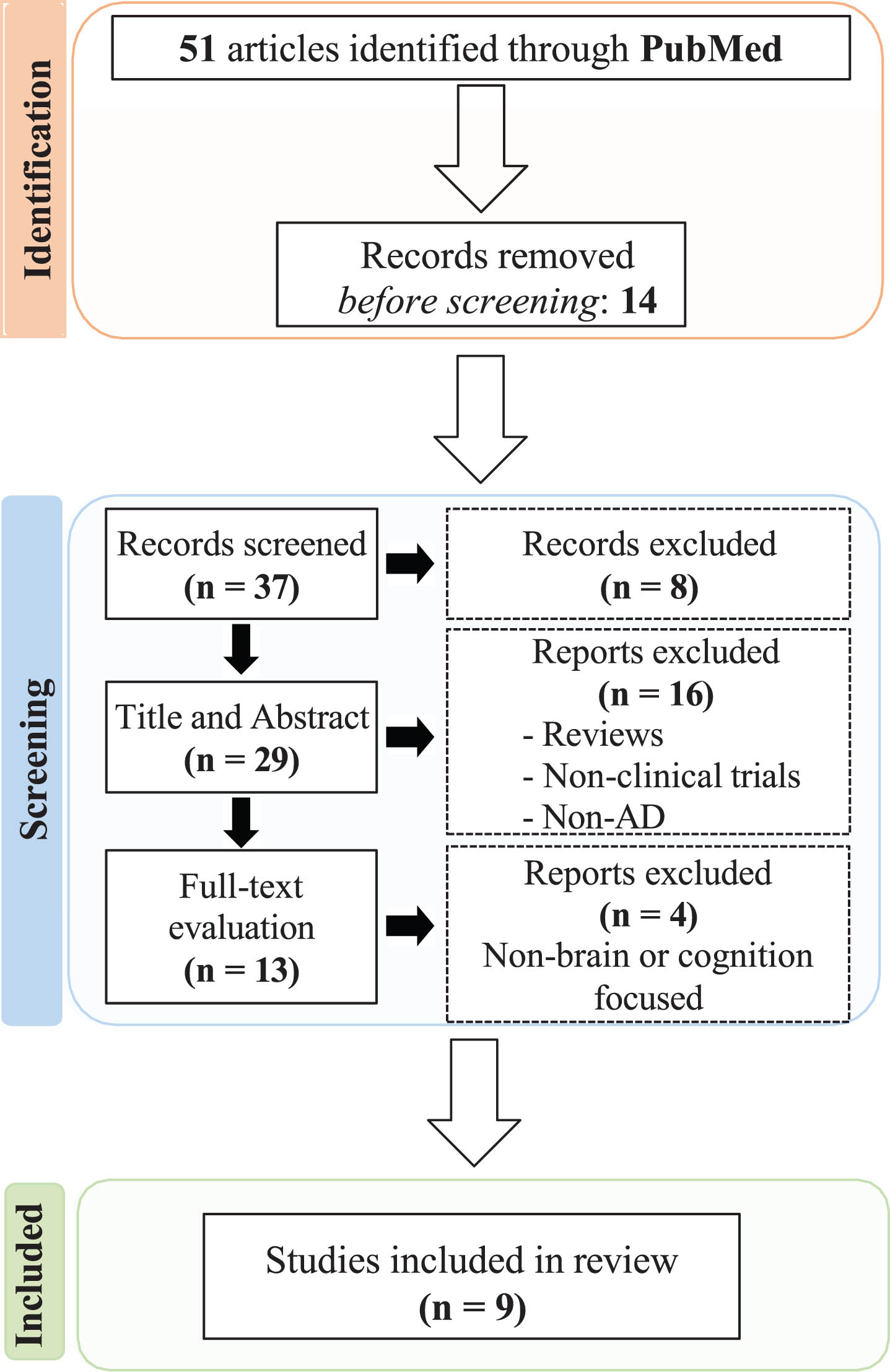
Systematic literature search and article screening results according to PRISMA.
We identified 51 papers, all of which were acquired by searching PubMed. We found that 14 papers were found irrelevant based on title reading and were excluded. For the remaining 37 reports, which were screened based on both title and abstract, 8 records were excluded for not meeting our search criteria. The remaining 29 papers were read in full and evaluated, 16 articles were excluded because they were non focused on AD and/or were review articles and not original research. Of the remaining 13 articles, 4 articles were excluded because they were non- brain or cognition focused. We included 3 full-text articles that assessed the protective mechanism of action of NMN in AD Tg mice models. There were no human clinical studies using NMN as a therapeutic intervention for AD. We found 4 studies that examined the potential neuroprotective effects of NR in AD Tg mice models and 2 human clinical trials using NR in AD patients. A schematic representation of papers identified and assessed are summarized in Fig. 1.
Nicotinamide mononucleotide
Preclinical studies
A majority of studies on the effect of NAD+ precursors in AD has been conducted in animal models. Mitochondrial dysfunction is an important pathological hallmark of AD and is associated with a significant decline in NAD+ and ATP levels. Therefore, NAD+ precursors represent an alternative strategy to maintain cellular NAD+ homeostasis and protect against bioenergetic failure and limit cell death. NMN is an important NAD+ precursor that is produced during the reaction of NAMPT [36].
One study administered NMN to APP(swe)/PS1(ΔE9) double transgenic (AD-Tg) mice (3–12 months) to determine its potential protective effects on mitochondrial respiratory deficits [64]. The study also examined levels of full-length mutant APP, NAD(+)-dependent substrates (SIRT1 and CD38) in homogenates and fission/fusion proteins (DRP1, OPA1 and MFN2) in mitochondria extracted from the brain. The study found that mitochondrial respiratory function was restored in NMN-treated AD-Tg mice [64]. Moreover, the levels of SIRT1 and CD38 declined with age and the effect was ameliorated with NMN treatment. The study also noted significant improvements in dynamics from mitochondrial fission to fusion proteins in the NMN-treated mice [64]. These findings suggest that NMN is a potential therapeutic compound that has a positive effect on brain mitochondrial morphological dynamics that may be beneficial in the ADbrain.
As well, it is well established that Aβ oligomers are the major neurotoxic forms of Aβ in the AD brain. Previous studies have shown that promotion of cellular NAD+ anabolism can reduce Aβ toxicity in AD. A recent study found that treatment with NMN significantly improved behavioral measures of cognitive impairments in APPswe/PS1dE9 (AD-Tg) mice compared to non-treated control AD-Tg mice [65]. AD-Tg mice treated with NMN demonstrated a significantly lower Aβ plaque burden, reduced synaptic loss, and a lower inflammatory response. The study hypothesized that the beneficial effects of NMN on Aβ production was mediated by NMN-mediated JNK activation [65]. Treatment with NMN led to a dynamic shifted directed to nonamyloidogenic amyloid precursor protein (APP) and suppressed amyloidogenic APP by altering the expression of APP cleavage secretase in AD-Tg mice [65]. This suggest that NMN can significantly lower multiple AD-associated pathological hallmarks partially due to inhibition of JNK activation.
Apart from its effect on some pathological hallmarks of AD and behavioral impacts, supplementation with NMN has been shown to play a positive effect on gut flora in AD [65]. It has been hypothesized that intestinal flora and its metabolites play an important role in the attenuation of neurodegenerative disorders including AD through a bidirectional interaction between the gut-brain axis (GBA). A more recent study evaluated the effect on NMN on gut flora in APP/PS1 transgenic (AD) mice through the 16 S ribosomal RNA (rRNA) high- throughput sequencing analysis of mouse feces following supplementation with NMN for 16 weeks [66]. The study found that treatment with NMN significantly altered the composition of the intestinal microbial community in AD mice. NMN also elevated the relative abundance of bacteria associated with the production of short-chain fatty acids, including Lactobacillus and Bacteroides [66]. These findings provide evidence for the promising role of NMN for the treatment of AD and emphasizes the critical role of NAD+ precursors including NMN on gut microbiota in AD pathology.
Nicotinamide riboside
Preclinical studies
NR is another NAD+ precursor that requires either a two-step or three-step process to produce NAD+. NR is converted to NMN by two nicotinamide ribose kinases, NMRK1 and NMRK2 (or NRK1 and NRK2) [67]. Alternatively, NR can be converted to NAM by purine nucleoside phosphorylase (NP), which is subsequently converted to NAD+ via NMN by NMNAT [68–70].
One study administered NR (2.5 g/kg food) to APP/PS1 transgenic AD mice and aged mice for 3 months [44, 45]. The study found that NR supplementation improved the short-term spatial memory of aged mice, and the contextual fear memory of AD mice. As well, NR supplementation attenuated the activation of astrocytes and reduced serum NAMPT in aged mice. NR supplementation also reduced Aβ burden and astrocytic migration to Aβ [45]. NR supplementation also inhibited body weight gain in both aged and APP/PS1 mice [45]. Therefore, NR demonstrated beneficial effects for both AD and aged mice, and the oral uptake of NR may be a promising strategy to attenuate the progression of AD.
Another study examined the effect of NR supple-mentation in a DNA repair-deficient 3xTgAD/Polβ+/– mouse that demonstrates enhanced features of human AD including phosphorylated Tau (pTau) pathologies, synaptic dysfunction, neuronal death, and cognitive impairment [71]. Treatment with NR lowered pTau pathology in both 3xTgAD and 3xTgAD/Polβ+/– mice but had a limited effect on Aβ burden. Treatment with NR lowered DNA damage, neuroinflammation, and apoptosis of hippocampal neurons and upregulated the enzymatic activity of SIRT3 in the brain of 3xTgAD/Polβ+/– mice [71]. The study also reported significant improvements in cognitive function using multiple behavioral batteries and improved hippocampal synaptic plasticity in 3xTgAD mice and 3xTgAD/Polβ+/– mice [71]. Therefore, promotion of cellular anabolism may have beneficial effects on mitigating neuroinflammation, pTau, DNA damage, synaptic dysfunction, and neuronal loss in AD.
Another study which treated APP/PS1 transgenic mice with NR for 5 months reported significant increases in brain NAD+ levels, downregulation of proinflammatory cytokines, and lowered activation of microglia and astrocytes [44]. Other beneficial effects of NR treatment included reduced NLRP3 inflammasome expression, lowered DNA damage and apoptosis, enhanced mitophagy, reduced cellular senescence, and increased cognitive performance and synaptic function in the AD mouse brains. As well, GMP-AMP synthase (cGAS) and stimulator of interferon genes (STING) were found to be significantly activated in APP/PS1 mutant mice and was associated with increased DNA damage and cellular senescence. Treatment with NR normalized cGAS-STING expression to near control levels [44]. Taken together, these findings suggest a role for NR on activation of cGAS-STING as an additional protective mechanism of action.
Apart from an earlier finding on the effect of NMN on the gut biota, supplementation with NR for 8 weeks reduced diversity and perturbated microbial compositions to normal levels in AD mice [72]. This included the species Oscillospira, Butyricicoccus, Desulfovibrio, Bifidobacterium, Olsenella, Adlercreutzi a, Bacteroides, Akkermansia, and Lactobacillus [72]. These findings suggest that the effect of NR on gut dysbiosis may represent an additional neuroprotective mechanism in AD.
Clinical studies
Depletion of cellular NAD+ levels in the brain during the ageing process is a major contributor to the underlying metabolic and cellular impairments associated with neurodegenerative diseases. Several therapeutic interventions have been explored to elevate brain NAD+ levels in various animal models, with promising results. The NAD+ precursor, NR has been reported to be generally safe and effective at promoting NAD+ levels in several peripheral tissues in humans (reviewed in [73]). However, there is still limited evidence supporting the role of NR to increase brain NAD+ levels and ameliorate neurodegenerative pathologies. A recent study investigated biomarkers from plasma extracellular vesicles enriched for neuronal origin (NEVs) from 22 healthy older adults who were enrolled in a randomized, placebo-controlled crossover trial (NCT02921659) of oral NR supplementation (500 mg, 2x /day, 6 weeks) [74]. The study found that oral NR supplementation increased NAD+ levels in NEVs and lowered the NEV levels of Aβ42, pJNK, and pERK1/2 (kinases involved in insulin resistance and neuroinflammatory pathways) [74]. These findings provide further evidence for the role of oral NR in augmenting neuronal NAD+ levels and improving AD related biomarkers in humans.
AD is associated with metabolic abnormalities linked to critical elements of neurodegeneration. A randomized, double-blinded, placebo-controlled phase-II clinical trial examined the effect of administration of combined metabolic activators (CMA) on the global metabolism of AD patients [75]. The single-dose CMA was composed of 12.35 g L-serine (61.75%), 1 g NR (5%), 2.55 g N-acetyl-L-cysteine (12.75%), and 3.73 g L-carnitine tartrate (18.65%) [75]. AD patients were administered a single oral dose of CMA or placebo daily for the first 28 days and then twice daily between day 28 and day 84. The study found a significant reduction in AD Assessment Scale-cognitive subscale (ADAS-Cog) score on day 84 vs day 0 (P = 0.00001, 29% improvement) in the CMA group [75]. In particular, the study reported improvement in cognitive functions in the CMA group compared to the placebo group in patients with a higher ADAS-Cog scores. Improvements in cognitive function were correlated with positive changes in hippocampal volumes and cortical thickness determined using neuroimaging analyses [75]. As well, proteins and metabolites associated with NAD+ and glutathione metabolism were also favorably altered following CMA treatment. CMA was found to be safe and well tolerated in AD patients [76]. These findings suggest that oral formulations composed of CMAs including NR can improve cognitive function and alter the pathological markers of AD.
CONCLUSIONS
An extensive body of preclinical research has highlighted the importance of NAD+ precursors as promising therapeutic interventions for variety of age-related disorders. Identification of the important role of NAD+ as a cofactor in cellular bioenergetic processes, and its unique role as a substrate in DNA repair and epigenetic control through protein deacetylation emphasis the importance of NAD+ in the pathobiology of AD and other forms of dementia. Further research on the role of NAD+ in immune activation, energy to metabolism, epigenetic control and cell viability in degenerative disorders may provide renewed hope for the development of effective therapeutics for the treatment of neurocognitive disorders. NAD+ precursors such as NR, and to a lesser degree, NA and NAM, have demonstrated protective effects against axonal degeneration in several animal models for Wallerian degeneration, and attenuated Aβ and tau pathology in transgenic AD mousemodels.
However, although the majority of studies have revealed promising results, the use of NAD+ enhancing agents may have some unwanted adverse effects. Given the complexity of the biological processes regulated by NAD+, one size fits all, approach to NAD+ therapeutics will likely limit the clinical translation of NAD+ therapy and may in fact cause harm under some disease contexts. Additionally, while several NAD+ precursors have been recently identified and examined in several disease models, a side-by-side comparison of these precursors in humans is not available in the current literature. It is highly likely that NAD+ precursors may exhibit different modes of action and target different processes under various pathologies [76].
NAD+ precursors have become ubiquitous in the commercial market. However, further research is needed to determine whether increasing levels of NAD+ may increase the risk of development of certain cancers, given the high energy requirement of most neoplasia. Clearly NAD+ is used in all cells, including cancer cells to produce energy. NAD+ supplementation should be used with caution and patients with a family history of cancer or have been or are currently diagnosed with cancer should get advice from a healthcare practitioner prior to consuming supplements that elevate NAD+ levels. While NAD+ may slow down and/or delay the onset and development of age- related diseases, strategies aimed to eliminating senescent cells from the body prior to NAD+ supplementation should be considered.
Owing to the multiple effects of NAD+ precursors, products that are limited in their methodology or ingredients should not be consumed since they may also negatively interact with other NAD- consuming enzymes and increase disease risk. It has been documented that not all NAD+ precursors can equally boost NAD+ levels. Additionally, alternative approaches should be considered to ensure the stability and bioavailability of NAD+ precursors. NAD+ itself is not absorbed well by the human gut system, making its oral supplementation not clinically practical [77]. However, NR has been reported to be orally bioavailable. Moreover, other strategies of drug delivery such as transdermal, sub-buccal or intranasal delivery should be evaluated to maximize the health benefits of NAD+ precursors for individuals with disorders of the digestive system.
Summary of ongoing and finished clinical trials of NAD+ precursors related to Alzheimer’s disease
A recent review of the clinical trials register (clinicaltrials.gov), indicates that several human trials to elucidate the effect of NAD+ precursors for AD have commenced or currently recruiting participants (Fig. 1). These include a single-group, open-label study “Effects of Nicotinamide Riboside on Bioenergetics and Oxidative Stress in Mild Cognitive Impairment/Alzheimer’s Dementia” to investigate the neurobiological mechanisms and clinical effects of NR in patients with MCI and mild AD using in vivo novel neuroimaging techniques; and a double-blinded placebo controlled randomized trial “N-DOSE AD: A Dose Optimization Trial of Nicotinamide Riboside in Alzheimer’s Disease” to determine the optimal dose of NR, in individuals with AD. A few other studies have been completed although results are not yet available. These include a randomized placebo controlled study “Safety study of nicotinamide to treat Alzheimer’s disease” to determine whether NAM, or vitamin B3, is safe and effective in the treatment of AD; and a double-blind, randomized, placebo-controlled, investigator-initiated, multi-center trial “Metabolic Cofactor Supplementation in Alzheimer’s Disease (AD) and Parkinson’s Disease (PD) Patients” to establish metabolic improvements in AD and PD subjects by dietary supplementation with cofactors N- acetylcysteine, L-carnitine tartrate, nicotinamide riboside and serine. A single-arm non- interventional study using the Emerald device “Passive Sensor Identification of Digital Biomarkers to Assess Effects of Orally Administered Nicotinamide Riboside (Emerald- NRAD)” to monitor biomarkers that relate to bioenergetic changes in the brain due to NR supplementation in those with mild cognitive impairment and mild AD has been initiated but not yet recruiting. Further clinical trials are required to evaluate the pharmacokinetics, safety, and efficacy of NAD+ precursors in healthy and disease models to develop targeted novel formulations therapies that can slow down and/or delay the underlying neurodegenerative processes and prevent cognitive decline in AD and other related dementias.
AUTHOR CONTRIBUTIONS
Mohammed Alghamdi (Data curation; Formal analysis; Investigation; Methodology; Software; Validation; Writing – original draft; Writing – review & editing); Nady Braidy (Conceptualization; Investigation; Supervision; Writing – original draft; Writing – review & editing).
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This research was supported by a scholarship from Prince Sattam bin Abdulaziz University, Ministry of Education, Saudi Arabia.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article.