
Abstract
Animal models, particularly transgenic mice, are extensively used in Alzheimer’s disease (AD) research to emulate key disease hallmarks, such as amyloid plaques and neurofibrillary tangles formation. Although these models have contributed to our understanding of AD pathogenesis and can be helpful in testing potential therapeutic interventions, their reliability is dubious. While preclinical studies have shown promise, clinical trials often yield disappointing results, highlighting a notable gap and disparity between animal models and human AD pathology. Existing models frequently overlook early-stage human pathologies and other key AD characteristics, thereby limiting their application in identifying optimal therapeutic interventions. Enhancing model reliability necessitates rigorous study design, comprehensive behavioral evaluations, and biomarker utilization. Overall, a nuanced understanding of each model’s neuropathology, its fidelity to human AD, and its limitations is essential for accurate interpretation and successful translation of findings. This article analyzes the discrepancies between animal models and human AD pathology that complicate the translation of findings from preclinical studies to clinical applications. We also delve into AD pathogenesis and attributes to propose a new perspective on this pathology and deliberate over the primary limitations of key experimental models. Additionally, we discuss several fundamental problems that may explain the translational failures and suggest some possible directions for more effective preclinical studies.
INTRODUCTION
Alzheimer’s disease (AD) stands as the most prevalent form of dementia and a foremost neurodegenerative disorder, marked by a gradual deterioration in short-term memory and cognitive abilities, exerting a significant impact on daily functioning [1]. AD is a progressively debilitating condition that ultimately leads to complete functional dependence and culminates in the individual’s demise. The classical hallmarks of AD include brain atrophy, the accumulation of amyloid plaques, and neurofibrillary tangles (NFT), which have been thought for decades to play a crucial role in the progression of the disease [2]. Besides the presence of plaques and NFT, AD-related pathology is characterized by persistent neuroinflammation and metabolic malfunction [3]. The precise way these factors interact and how they lead to neurodegeneration and manifest in cognitive disability still remain partially elucidated [4].
Of importance, our current understanding of the complex AD pathology largely stems from examining pathological features in animal models. These models offer a platform for in-depth exploration of cellular and molecular aspects, enabling the study of AD intricacies. Additionally, they serve as invaluable arenas for testing innovative therapeutic approaches to treat the disease. The predominant experimental AD models are rodents. Most of them express human genes responsible for the formation of amyloid plaques (through human amyloid-β precursor protein (APP) expression alone or combined with human presenilin-1 (PS-1)) and NFT (via human microtubule-associated protein tau expression), closely resembling key aspects of AD seen in humans [5].
The first transgenic AD mouse model by Games et al. (1995) with substantial amyloid plaque buildup fueled interest and lead to an increase in the generation of various transgenic models [6]. Of importance, the Games et al. mice express high levels of human mutant APP, with valine at residue 717 substituted with phenylalanine, and develop amyloid-β deposits and plaques. Moreover, they demonstrated synaptic loss, astrocytosis and microgliosis. These animals served as a popular preclinical model for testing potential drugs (Fig. 1).
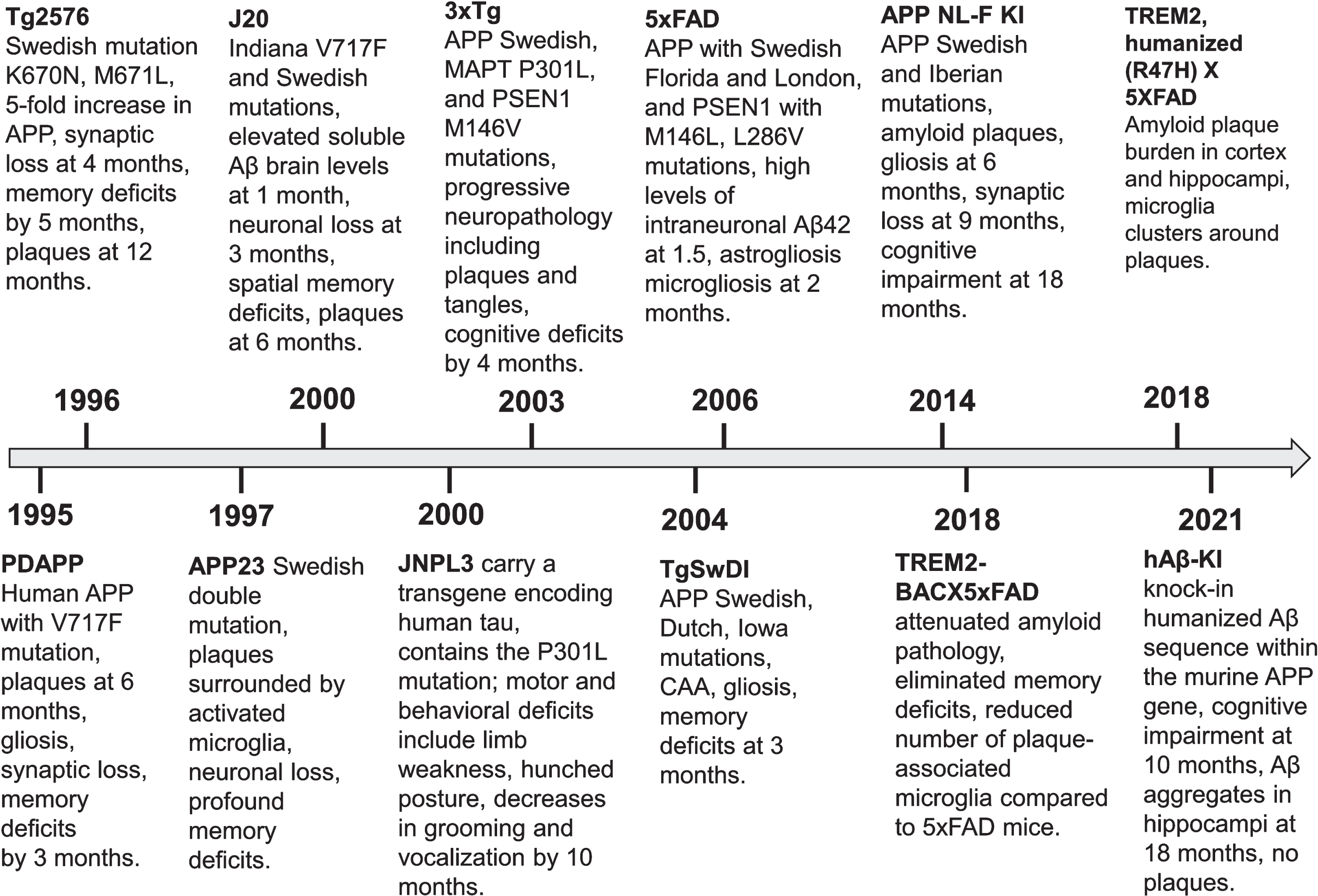
Timeline showcasing the progression of transgenic Alzheimer’s disease mouse models’ development and their phenotypes.
The emergence of more complex transgenic models holds substantial potential for enhancing our understanding of AD pathogenesis. These models have provided the means to explore questions beyond our reach in human studies [7]. Nonetheless, serious concerns have grown over the reliability of depending on these existing models, especially given the remarkably high rate of failure in clinical trials, despite promising successes in preclinical assessments [8].
These findings emphasize a frequently overlooked reality. In fact, mice and rats do not develop Alzheimer’s-like pathology in nature, and even artificially created animal models do not have AD [9]. This phenomenon is most likely attributed to the three amino acid variations in the amyloid-β sequence between humans and rodents [10]. Therefore, rodents merely replicate particular pathological characteristics of the disease. Moreover, these features are often recreated in a non-physiological manner to facilitate efficient experimentation. Most of these models primarily exhibit the accumulation of amyloid, a hallmark of AD. Amyloid accumulation is typically followed by specific cognitive impairments and memory deficits. Despite severe abnormalities in amyloid metabolism, these rodent models lack the broader presence of other pathological elements that define AD, particularly the development of NFT and neuronal loss [11]. The absence of these additional AD-associated features could, at least in part, contribute to the limited translation of results from preclinical studies to clinical trials.
The repertoire of rodent models for AD is expanding steadily. Currently, the commercially available compendium comprises 204 mouse models and 17 rat models. The early models incorporated mutations linked exclusively to familial AD, while subsequent research endeavors expanded to encompass mutations in tau, associated with frontotemporal dementia and parkinsonism. Novel cutting-edge models have been engineered to explore AD-like pathology in mice, integrating humanized amyloid-β to probe its interaction with triggering receptor expressed on myeloid cells-2 (TREM2) (Fig. 1).
Of importance, early studies in APP transgenic mice found no substantial brain atrophy in these animals in spite of significant memory deficiency [12]. In vivo high-resolution MRI studies prove that aged APP/PS1 mice show only a moderate global brain atrophy in the midbrain area and not at isocortical/hippocampal levels reported in AD patients [13]. Moreover, the severity of atrophy does not correlate with the amyloid load. Consequently, the relevance of these models for the study of AD-related neurodegeneration remains questionable.
There were attempts to create AD models using human wild-type APP [14]. Scientists used techniques like pronuclear injection or embryonic stem cell-based gene targeting to introduce the human wild-type APP gene into the mouse genome. As a result, these genetically engineering mice express the human APP gene in its natural, unmodified form. This transgene is typically under the promoter driving expression in neurons or specific brain regions affected by AD. These models aim to mimic the overproduction and accumulation of amyloid-β peptides without introducing mutations into the APP gene itself. Notably, these animals only develop mild neuropathology without deposition of typical plaques [15], which indicates that wildtype APP overexpression may not recapitulate efficiently the human disease in mice.
These mouse models are used in studies of potential therapeutic interventions, including drugs targeting amyloid-β production, aggregation, and clearance [16]. By using human wild-type APP in mouse models, researchers can better replicate the molecular and cellular processes underlying amyloid-β accumulation.
For example, the cysteine protease cathepsin B has been proposed as an alternative candidate β–secretase residing in the regulated secretory pathway of neurons [17]. In transgenic mice expressing human APP with the WT β-secretase site, inhibitors of cathepsin B were shown to reduce brain levels of amyloid-β derivatives, and brain levels of C-terminal β–secretase fragment derived from APP by β–secretase [18].
Mouse models using human wild-type APP replicate certain aspects of amyloid-β pathology. They can be beneficial in advancing Down syndrome research [19]. However, these models have limitations in replicating the complexity and progression of senile dementia in humans. As such, further research and validation are necessary to realize the potential of this approach.
Despite growing funding globally, the increasing number of potential AD-modifying agents successfully tested preclinically in mouse models has indeed led to disappointment when assessed in human trials. In the fiscal year 2021, the NIH alone allocated approximately $3.1 billion for AD and related dementias research [20]. Major pharmaceutical companies have incurred considerable financial losses in their efforts to discover an effective drug for AD. The pharmaceutical giant Pfizer dropped out of the dementia drug race and announced that it was pulling out of early-stage research into AD [21], which indicates that establishing a more solid knowledge foundation and enhancing the translation of successful results from animal studies is imperative. Thus, it becomes crucial to comprehend the precise neuropathology evident in each model, especially in terms of how accurately it mirrors the human pathology. This precision could enable an accurate interpretation of findings and improve meaningfully the prospects of translating outcomes to human-based studies.
This review endeavors to thoroughly examine crucial facets of AD modeling within laboratory settings. It seeks to meticulously analyze the constraints inherent in prominent experimental models employed for understanding AD pathology. Additionally, the review aspires to provide valuable insights into the current comprehension of AD pathology, elucidating the reliability and relevance of experimental models in advancing our understanding of this intricate neurodegenerative condition.
ALZHEIMER’S DISEASE ETIOLOGY AND PATHOGENESIS
Early- versus late-onset AD
AD is becoming more prevalent as the population ages. The increase in life expectancy, coupled with changes in lifestyle factors such as diet and physical activity, have contributed to the rise of AD cases across various populations. While advanced age is the primary risk factor for AD, its early onset still constitutes around 5% of all cases [22]. The inaugural case documented by Dr. Alois Alzheimer was about a woman who tragically succumbed to the disease at the age of 55. In fact, this instance should be categorized as an early-onset AD (EOAD) occurrence. In medical literature, EOAD refers to cases where clinical symptoms manifest before age 61 [23].
Typically, EOAD is inherited in a dominant Mendelian manner, although it represents a genetically diverse group. Epidemiological evidence indicates that autosomal dominant familial AD associated with mutations in genes like PSEN1, PSEN2, and APP accounts for approximately 0.5% of all AD cases [24, 25]. However, the proportion of familial cases within the EOAD subgroup increases significantly to 13% [23, 26]. On the other hand, late-onset AD (LOAD) displays a substantial hereditary component involving a broader array of genes in its development. Moreover, the progression of LOAD is believed to be influenced by a combination of genetic and environmental factors, which implies that its fundamental cause remains mainly unidentified [27, 28]. Although extensive research has been conducted on the mechanistic contribution of genes in AD pathogenesis, the specific biology involved in the disease progression still needs to be determined, which highlights the need for further investigation of the complex genetic and environmental factors contributing to AD’s development.
Notably, EOAD often displays a distinct cognitive profile compared to LOAD [22]. EOAD may lack evident amnesia and can instead manifest through language discrepancies, apraxia, and other atypical functional deficits. Additionally, LOAD patients experience more pronounced deficits in semantic memory than EOAD patients.
Recent objective data have proven significant differences between EOAD and LOAD. Cerebrospinal fluid total tau levels are notably higher in EOAD patients. Moreover, fludeoxyglucose F18 positron emission tomography reveals asymmetric hypometabolism in EOAD localized differently from those observed in LOAD [29]. Moreover, distinct patterns of amyloid-β oligomeric subtypes are observed in each AD form. Pentameric amyloid species within the insoluble fraction are more prevalent in EOAD compared to LOAD [30]. Also, elevated levels of inflammatory markers are distinguishing features of LOAD [31]. Together, these differences underscore substantial variations in the underlying mechanisms and progression between the two clinical presentations of the disease.
As we have noted, in contrast to EOAD, the LOAD etiology is notably more varied, involving a complex interplay of numerous genetic and environmental factors. Moreover, LOAD is frequently associated with coexisting conditions like hypertension and diabetes [32] and possesses metabolic signatures [33]. Accordingly, we believe that the shared features seen in both forms do not necessarily imply a common origin and development. Hence, attempts to link these two forms of AD to a single causal factor are likely unproductive.
Consequently, we suggest that EOAD and LOAD represent two distinctly separate pathological entities characterized by specific pathophysiological mechanisms attributed to their distinct genetic basis and uncommon clinical manifestations. Accordingly, we state that only EOAD truly embodies the classic characteristics of AD or presenile dementia of the Alzheimer’s type.
Of importance, mutations in the APP, PSEN1, and PSEN2 genes account for less than 1% of all AD cases; however, most transgenic murine AD models used in preclinical settings express mutated forms of APP and PSEN1 and mimic only certain aspects of AD pathology, particularly the brain accumulation of amyloid plaques. Therefore, we must be cautious when extrapolating findings from these models to human AD cases.
Furthermore, the exact role of amyloid-β plaques and NFT in disease progression and neural dysfunction is still a subject of active research and debate [34]. To date, it is not entirely clear whether these aggregates are triggers or just byproducts of complex molecular pathways involving protein misfolding and aggregation or even evolutionary conserved protective mechanisms of the brain [35–37]. It is noteworthy that some elderly individuals with extremely high levels of the typical brain lesions do not manifest clinical dementia [38, 39]. Moreover, even in demented patients the burden of Aβ plaques purely correlates with memory deficiency [34, 40]. This intriguing phenomenon challenges the straightforward connection between AD hallmarks and cognitive decline and emphasizes the intricate nature of brain responses to such challenges.
In this context, it is plausible to assume that hallmarks of the disease might not be directly responsible for neurodegeneration. Instead, they could be epiphenomena of underlying molecular events that cause the primary damage. In this view, the aggregates might accumulate due to the brain’s attempts to repair itself. Therefore, the mere existence of amyloid plaques should not automatically imply a specific cause or presume etiology.
Accordingly, an urgent need exists for reliable LOAD animal models that mirror the key aspects of a disease and share the same underlying disease mechanisms. These new models should enable researchers to gain insights into the disease development, provide effective and safe tools to assess potential treatments and therapies, aid in the discovery of biomarkers associated with the disease, and facilitate genetic and molecular investigations to unravel the genetic factors and molecular pathways involved in the disease, potentially leading to targeted therapeutic approaches.
Late-onset Alzheimer’s disease
Despite a thorough investigation spanning a century, no scientific consensus on the origins of AD achieved. The predominant viewpoint held by scientists revolves around the amyloid cascade hypothesis [41]. Nonetheless, increasing clinical and empirical data highlight an intricate systemic pathophysiology accompanying AD-associated cognitive decline. This complexity might even play a role in its development many years before noticeable clinical symptoms emerge. Beyond the recognizable hallmarks, the illness involves broader systemic irregularities and deviations in brain metabolism discernible at the molecular and biochemical tiers [42]. Consequently, the modern portrayal of AD-related pathology includes neuroinflammation, apoptosis, mitochondrial dysfunction, compromised metabolism, and persistent oxidative stress [43].
Oxidative damage is frequently regarded as the initial and central step in AD pathology [44]. Brain oxidative stress can arise from various sources, including reduced blood flow to the brain due to advanced atherosclerosis or impaired endothelial function, traumatic brain injury, infections, autoimmune disorders, insulin resistance, and other conditions that prompt neuroinflammation [3]. Recent evidence indicates gradual development of chronic cerebral hypoperfusion with aging, resulting from cerebral atherosclerosis and dysfunction in the endothelial cells lining the blood vessels in the brain [45], which, in turn, contribute to the brain energy insufficiency and, eventually, neurodegeneration. Of note, amyloid-β is a powerful antioxidant due to its ability to chelate transition metal ions, and copper in particular [46]. This view of LOAD pathogenesis with amyloid-β acting as a physiological antioxidant embraces general pathological mechanisms developing with aging, such as cerebral hypoperfusion and increased production of reactive oxygen species that are positioned upstream to the Aβ overproduction [47].
Original murine experiments have proven that focal ischemic insults and chronic cerebral hypoperfusion can lead to increased levels of APP translation, followed by the deposition of amyloid-β in the brain tissue [48]. In rodent models, prolonged vascular insufficiency causes the cleavage of APP into fragments similar in size to amyloid-β [49]. The chronic occlusion of blood vessels in these animals leads to a gradual buildup of amyloid-β peptide, with a shift from neuronal to extracellular deposition, resembling the characteristics of LOAD. This hypoxia-induced response is associated with increased activity of β- and γ-secretases, enzymes responsible for cleaving APP to form amyloid, while the activity of the non-amyloidogenic α-secretase decreases [50]. An objective study by Roher et al. convincingly demonstrated that atherosclerotic occlusion of the circle of Willis is significantly more extensive in the AD group than the non-demented control group [51], which points to intracranial atherosclerosis as a contributing factor to AD-associated dementia.
The association between atherosclerotic vascular disease and AD is a topic of ongoing research and high interest in the medical community [52, 53]. Converging data strongly suggest that atherosclerosis-related brain hypoperfusion contributes to the AD pathological and clinical manifestations [54, 55]. Remarkably, AD and cerebral arteriosclerosis share mutual risk factors, such as age, diabetes, high cholesterol, and high blood pressure, which points to reciprocal overlapping mechanisms. Accordingly, the idea that interventions maintaining vascular health may mitigate the progression of AD is an appealing hypothesis. Rigorous and well-designed studies are needed to understand the multifaceted mechanisms underlying atherosclerotic vascular disease and AD association and to determine the most effective strategies for preventing the AD development by addressing vascular health.
Understanding the role of tau protein in Alzheimer’s disease
Tau, a microtubule-associated protein, is expressed at high levels in neurons and is mainly localized in axons [56]. It plays an important role in the normal functioning of neurons and maintaining the structural integrity of the neuronal network. Any disruption in tau function can lead to a variety of neurodegenerative disorders, including AD. Tau protein hyperphosphorylation causes the development of NFT in neurons and contributes to neuronal dysfunction and eventual dementia. Notably, the incidence of NFT correlates with cognitive decline and neuronal death [57]. Genetic studies have linked tau protein coding gene mutations, especially P301 L/S mutations, to frontotemporal dementia and highlighted the significant role of tau in AD pathogenesis [58].
The JNPL3 mouse model expressing the P301 L mutation was the first single transgenic tauopathy model [59]. Transgenic rodent models have been instrumental in advancing our understanding of tau pathology in AD and other tauopathies. These models, which express either mutant forms of human tau gene (e.g., JNPL3, Line 2541) [60, 61] or overexpress wild-type tau [62], have led to the development of tau pathology mimicking certain aspects of AD and related conditions.
Of note, mouse models engineered to express mutant forms of human tau, such as the widely studied P301 L/S mutation, have been vital in replicating certain aspects of AD-related neurodegeneration. Significantly, the promoter region of these models determines the expression pattern of the mutant tau, allowing researchers to target expression to specific types of neurons or brain regions.
Numerous models have been created to study tau pathology, including regulatable transgenic models, such as rTg4510, which express P301 L mutant tau [63]. This model is particularly useful in studying the reversibility of tau pathology as the expression of tau can be turned on or off [64]. Another model, THY-Tau22, expresses human 4 R tau with G272 V and P301 S mutations under the Thy1.2 promoter and displays typical tau pathology and memory deficits [65]. Knock-in models that involve inserting mutations directly into the mouse tau gene to preserve the gene’s regulatory elements provide a more physiologically relevant expression pattern of the mutant protein [66].
However, despite the usefulness of these models, they have serious limitations. Specifically, some models rely on tau overexpression to induce pathology, leading to artifacts that do not represent the human disease process. Moreover, the insertion of transgenes in some mouse models can disrupt the coding sequence of endogenous genes, potentially impacting the pathogenesis of tauopathy and the overall model utility [67]. Additionally, most models focus on tau pathology in isolation, while AD involves multiple interrelated pathological processes, including amyloid-β accumulation and neuroinflammation. Thus, there is a need for models that incorporate various aspects of AD pathology for a more comprehensive understanding. In this context, it might be interesting to note an original model combining tau and amyloid-β pathologies. This model was created by crossing a mouse expressing two human MAPT mutations with 5xFAD mice expressing human mutated APP and PS1 [68]. These mice showed accelerated cognitive impairment at two months of age, increased amyloid-β depositions at four months, and neuritic plaques at six months.
Of note, current tau-based mouse models reflect some aspects of early-stage but not fully capture the complexity of tau pathology at the molecular level observed in late-stage, symptomatic human dementia, as recent studies have shown [69]. This discrepancy may be a potential reason why drugs that target tau and show promise in mouse models fail in clinical trials. Furthermore, while phosphorylation-driven tau accumulation is the primary focus of current models, chemical modifications of tau, such as acetylation and ubiquitination, play a crucial role in symptomatic and late-stage AD and are not adequately represented in these models [70, 71].
The development of next-generation models aims to address these limitations by creating more accurate AD representations. This includes the use of CRISPR/Cas9 technology for precise genetic editing, the creation of models expressing mutant tau at physiological levels, and the engineering of multi-transgenic models recapitulating both tau and amyloid pathologies, which can lead to the development of more advanced and adequate models mimicking the complexities of human disease. Additionally, using other rodent species, such as rats with larger brains and more complex behaviors, may offer new research opportunities.
The role of APOE
Apolipoprotein E gene allele ɛ4 (APOE4) represents a main risk factor for LOAD [72]. People with one APOE4 allele are at a 3–4 times higher risk of developing clinical dementia compared to those with the common APOE3 allele. If an individual possesses two copies of the APOE4 gene, the risk of AD upsurges by 8–15 times [73].
ApoE4 is a glycoprotein that serves as a lipid transporter facilitating the transport of cholesterol and phospholipids [74]. In peripheral tissues, ApoE4 is primarily synthesized by hepatocytes in the liver and macrophages [75]. ApoE4 cannot cross the blood-brain barrier; however, it is abundant within the brain where it is produced by various cell types, including astrocytes, activated microglia, vascular mural cells, choroid plexus cells, and, under conditions of stress, by neurons [76]. ApoE plays a crucial role in lipid binding and serves as the primary transporter of cholesterol in the brain.
A single amino acid substitution distinguishes ApoE4 from ApoE3 and ApoE3 from ApooE2, leading to significant changes in the functionality of these isoforms [77]. These substitutions result in isoform-specific structural variations that influence aspects such as lipid binding, receptor binding, propensity to form oligomers, and overall stability. Notably, ApoE isoforms exhibit distinct associations with peripheral lipoprotein particles. For example, ApoE4 is primarily found in triglyceride-rich particles like chylomicrons and very low-density lipoproteins, while ApoE2 and ApoE3 have a preference for high-density lipoproteins [78].
ApoE3 is considered the standard when comparing the functions of ApoE2 and ApoE4 alleles. ApoE2 exhibits reduced affinity to the receptor and lowers the risk of LOAD through mechanisms involving both amyloid-dependent and independent pathways [79]. In contrast, ApoE4 has an enhanced ability to bind to lipids, resulting in increased accumulation of cholesterol [80]. Recent research by Blanchard and colleagues demonstrated that abnormal cholesterol deposition in ApoE4 oligodendrocytes adversely affects cellular functions, including the process of myelination [81]. This finding aligns with the observed reduced myelination in the brains of individuals with APOE4. By pharmacologically facilitating cholesterol transport, Blanchard et al. observed improvements in axonal myelination and enhanced learning and memory in APOE4 mice. This research highlights potential therapeutic avenues for addressing AD by modulating cholesterol transport.
Numerous transgenic mouse lines expressing either APOE3 or APOE4 have been created, with the expression driven by the neuron-specific enolase promoter. These models ensure that human APOE3 or APOE4 is expressed at comparable levels within the neurons of transgenic mice that lack their endogenous mouse APOE counterparts [82, 83]. These transgenic animals play a pivotal role in AD research, as they allow scientists to investigate the specific effects of human APOE3 and APOE4 in isolation, without the confounding influence of mouse apoe. This level of control is essential for gaining insights into how these ApoE isoforms contribute to the development and progression of the disease. These models are invaluable tools to unravel the complexities of the disease with a hope to develop targeted treatments.
ApoE4 was shown to stimulate the accumulation of amyloid-β and hyperphosphorylated tau, which is followed by cognitive impairments in mice [84]. Accordingly, this model was suggested as the most suitable for unbiased studies of the mechanisms underlying the pathological effects of ApoE, and these transgenic mice were used in the preclinical setting to target ApoE4 [85]. However, despite a significant improvement shown in the animal study, no disease-modifying drug based on this manipulation was approved.
Several other approaches have shown promise in rodents expressing human APOE alleles. They include manipulating the ApoE levels, enhancing its lipidation, blocking interactions between ApoE and amyloid-β, and genetical conversion APOE4 to APOE3 or APOE2 isoforms [86]. Though, translating these findings into successful human clinical trials has proven to be a challenging endeavor.
APOE–/– mice, were instrumental in elucidating the role of ApoE in lipid transport, atherosclerosis and suggested to be a model of AD. These animals show severe hypercholesterolemia and develop atherosclerosis. Several studies utilized APOE–/– mice in AD research. Michaelson et al. have shown that ApoE-deficient mice develop several characteristic AD-related pathologies, such as memory deficits, cholinergic impairments [87], and tau hyperphosphorylation [88]. Methia et al. evidenced compromised blood-brain barrier in these animals [89]. In addition, ApoE deficiency leads to increased levels of protein oxidation [90], and age-dependent synaptic alterations [91], which supported the contention that ApoE play important role in maintaining the stability of the synapto-dendritic apparatus. Consequently, it was proposed that altered or deficient ApoE functioning may underlie the AD-related synaptic and cytoskeletal alterations.
However, a more recent study by Ophira Salomon group (2018) in elderly APOE–/– mice convincingly demonstrated that these animals do not develop AD-like pathologies, including plaques and neuroinflammation, despite advanced atherosclerotic lesions in the central and peripheral blood vessels [92]. The authors suggest that their results advocate a dichotomy between the brain and peripheral organs. Moreover, they state that APOE–/– mice cannot serve as an AD mouse model. Accordingly, this direction in AD research has become questionable and looks like another dead end. Similarly, other single-gene AD animal models could misrepresent the complex pathology found in AD.
Undoubtedly, ApoE-related pathogenesis extends beyond traditional mechanisms centered solely on amyloid-β peptides and tau neurofibrillary degeneration, but encompasses various glial responses and disruption of the blood-brain barrier. Since all these pathological processes have the potential to contribute to cognitive impairment, it is crucial to leverage this newfound knowledge for the development of efficient therapies.
RODENT MODELS OF ACCELERATED AGING
We have mentioned above that age is one of the most significant risk factors for AD. However, even very old mice do not show the characteristic AD hallmarks. Nevertheless, some murine models of accelerated aging demonstrate the typical features of the disease. Accordingly, some groups use these animals to investigate the aging process, age-related diseases, and test potential interventions.
Progeroid mice are engineered to exhibit accelerated aging phenotypes, resembling the features of human progeria syndromes, such as Hutchinson-Gilford Progeria Syndrome [93]. These mice often have mutations in genes like Lamin A, which play a role in maintaining the structural integrity of the cell nucleus. Progeria background, such as the Ercc1 mutant mouse, provides an aging component with age-related co-morbidities [94]. These mice develop conditions common in elderly such as osteoporosis, sarcopenia, cardiovascular disease, peripheral neuropathy, cognitive decline, etc. [95]. They show gradual impairment in learning and memory [96], but do not exhibit forthright AD pathology and the complexity of the human disease. In fact, progeroid mice represent a predisposed environment, where pathogenic elements can be introduced, either by crossing with well-established AD transgenic mouse lines, or transcranial delivery [97]. Through a six-fold acceleration of aging, these animals expedite therapeutic testing, streamlining experimental processes.
Senescence-accelerated mouse prone (SAMP) mice are a series of inbred strains that display accelerated aging characteristics. These models are particularly useful for studying various aspects of aging, including cognitive decline, immune system dysfunction, and the development of age-related diseases, including AD. The SAMP8 is a commonly used model to study AD and age-related cognitive decline [98]. These mice exhibit progressive age-related learning and memory deficits [99]. Mounting evidence collectively suggest that SAMP8 mice can serve as a good model for studying AD given their behavioral, neurochemical, and neuropathological similarities to the human disease. Morley et al. (2004) highlighted the role of amyloid-β overproduction in cognitive defects in SAMP8 mice [100]. Akiguchi et al. (2017) noted age-related histological changes in SAMP8 mice similar to those seen in aging human brains, including amyloid-β deposition in the hippocampus [101]. In addition, SAMP8 mice display severe oxidative and endoplasmic reticulum stress, abnormal autophagy, neuroinflammation, and tau hyperphosphorylation. Hence, there is hope that these animals can offer valuable insights into age-related declines in learning and memory, ultimately identifying new therapeutic targets for AD.
There are several other much less popular in Alzheimer’s research mouse models that show accelerated aging. Among them Klotho-deficient mice, telomerase-deficient mice, and DNA repair-deficient mice. These animals display premature aging, skin atrophy, and cognitive decline [102]. Researchers use these models to test potential interventions, drugs, gene therapies, or lifestyle modifications, to slow down or mitigate the effects of aging and improve overall health in both rodents and potentially in humans.
OCTODON DEGUS AS A RODENT MODEL OF LOAD
Accruing evidence robustly substantiates a moderate-sized, diurnal, and precocial rodent species, the common degu (Octodon degus), as a valuable naturalistic model for investigating the early neurodegenerative processes associated with sporadic AD. It closely mirrors certain aspects of human physiology and behavior [103]. This convergence of traits positions the degus as a helpful resource for delving into the nuanced interplay of genetic, vascular, and psychosocial factors contributing to the onset and progression of LOAD [104].
Of importance, degus, exhibits a distinctive expression pattern of neuronal APP, showcasing both intracellular and extracellular deposits of amyloid-β peptide. Moreover, intracellular accumulations of tau-protein and ubiquitin, a robust astrocytic response are evident in the aged animals. The remarkable amino acid homology, accounting for 97.5% similarity, between degus and human amyloid-β sequences emerges as a pivotal factor likely contributing to the manifestation of AD markers in this aging rodent [105].
These findings firmly establish the aged degu as an important wild-type rodent model for investigating neurodegenerative processes associated with normal aging and AD. The observed spectrum of AD-related pathology and markers in this naturally aging rodent highlights the species’ potential as an invaluable tool for studying AD and testing potential therapies [106].
OTHER MAMMALIAN ANIMAL MODELS OF AD
In the realm of experimental model organisms for a diverse array of human diseases, rodents, notably the rat and the mouse, stand out as the most extensively employed. The prolific nature of these small mammals, characterized by a brief reproductive cycle, coupled with their elevated birth rate and compact size, renders them notably facile to sustain and oversee in laboratory settings. While rodents have been extensively utilized in AD research, recent advancements underscore the advantages offered by alternative animal models, highlighting their distinct benefits over traditional rodent models. These emerging models not only contribute to a more comprehensive understanding of AD but also offer unique insights that complement and enhance our knowledge, fostering a more nuanced and translational approach to investigating this complex neurodegenerative disorder.
Guinea pigs
The guinea pig (Cavia porcellus), a distinct wild-type rodent employed in AD research, exhibits an amyloid-β peptide sequence mirroring the human counterpart, offering a physiologically relevant model for in vivo exploration of the enduring impacts stemming from experimental manipulations on APP processing. Notably, the fidelity of APP processing in guinea pig primary neuronal cultures aligns demonstrably with analogous cultures of human origin [107]. Despite these advantages, the utility of guinea pigs is constrained due to their lack of pathological features characteristic of AD, such as senile plaques and neurofibrillary tangles. Moreover, the absence of well-established behavioral tests further limits the comprehensive assessment of this species in AD research.
Rabbits
Rabbits (Oryctolagus cuniculus) constitute another rodent model in AD research, boasting an amyloid-β peptide sequence identical to humans but lacking spontaneous presentation of AD pathology. Intriguingly, when copper is introduced into the diet of cholesterol-fed rabbits, they manifest cortical amyloid deposits and up to twelve other pathological markers akin to those observed in AD, accompanied by a discernible impairment in the acquisition of complex learning tasks [108]. This observation posits copper, in contrast to other heavy metals like aluminum or zinc, as a potential modulator of AD progression through its impact on the diminished clearance of amyloid-β from the brain. The significance of this discovery extends to underscore the utility of the rabbit model, with its validity now expanding to encompass diverse treatment modalities in the realm of AD research [109, 110].
Dogs
Dog (Canis Lupus spp.) have also emerged as a particularly apt model for delving into the intricacies of human brain aging and neurodegenerative diseases. Amyloid-β deposits do not manifest universally in aged dogs, challenging the notion that they are merely a consequence of normal aging. Instead, akin to humans, the susceptibility of individual dogs to developing amyloid pathology at a given age appears to be influenced by a combination of exogenous and/or genetic factors [111].
Within the expansive spectrum of domestic dog breeds, unparalleled diversity in body size exists, surpassing that observed in any other terrestrial vertebrate. Geneticists leverage this diversity to probe the genetic underpinnings of size and its correlation with lifespan [112]. Evolving in close phylogenetic proximity to humans, dogs help to interpret human social and communicative cues that are impaired in AD [113].
Dogs, frequently enlisted in preclinical drug development, serve as a robust testing ground for a diverse array of pharmaceuticals, ranging from cholinergic agonists to antioxidant and mitochondrial enzymatic cofactors, in the context of AD treatment strategies [114]. The growing interest in this model is underscored by the dog’s propensity to naturally develop age-related cognitive dysfunction, mirroring several facets of AD pathology [115]. This cognitive decline is found to be correlated with the extent of amyloid-β deposits in the brain, as observed in rigorous examinations [116, 117]. These findings highlight the utility of canine models in replicating age-related cognitive dysfunction, offering valuable insights into the progression of cognitive impairments akin to those observed in AD.
Dolphins
Cetaceans, particularly dolphins, represent an intriguing group of animals that may exhibit brain pathology reminiscent of AD. Examination of stranded bottlenose dolphins (Tursiops truncates) has revealed widespread amyloid-β deposits throughout the brain [118]. Notably, the observed amino acid homology between the APP, beta-site amyloid precursor protein cleaving enzyme, presenilin-1, and presenilin-2 in various dolphin species and humans suggests a striking similarity.
Another recent study by Vacher et al. (2023) examined brains from 22 stranded odontocetes of five species, namely Risso’s dolphin (Grampus griseus), long-finned pilot whale (Globicephala melas), white-beaked dolphin (Lagenorhynchus albirostris), harbor porpoise (Phocoena phocoena), and bottlenose dolphin (Tursiops truncates), using immunohistochemistry and revealed AD-like neuropathology [119]. All aged animals showed characteristic amyloid plaques. In three individuals, there was co-occurrence of amyloid-β plaques, NFT, and neuropil threads. Another animal displayed tau pathology and neuritic plaques without amyloid plaques. Microglia and astrocytes were present in expected patterns, with variations in morphology and numbers among individuals. The concurrent presence of amyloid-β plaques and hyperphosphorylated tau suggests spontaneous development of AD-like neuropathology in these odontocete species.
Dolphins and other cetaceans, with their prolonged lifespan and unique brain characteristics, show potential as a natural model for AD [120]. Their longevity, coupled with AD-like brain lesions, offers a distinctive avenue for studying neurodegenerative processes. Investigating the molecular and pathological aspects of AD in dolphins may reveal insights that broaden our understanding of this complex disorder. Delving into dolphin neurobiology, particularly in the context of AD, offers a novel perspective to enhance our comprehension of underlying mechanisms and etiological factors. Exploring dolphins as a natural AD model not only overcomes traditional model limitations but also unveils new avenues for therapeutic interventions and preventive strategies.
Of note, studying the intricate behavior of marine mammals poses unique challenges for researchers, given the size, mobility, and intermittent visibility of these animals. Moreover, conducting intricate behavioral experiments in cetaceans, particularly to assess spatial memory acquisition and recall, is still in its early stages, indicating a gap in preclinical studies for AD therapies.
Primates
Primates, owing to their remarkable phylogenetic proximity to humans, stand as ideal experimental models, particularly in cutting-edge studies on neurodegeneration. The fundamental similarities in the physiology and behavior to humans make them invaluable tools for deciphering complex pathophysiological processes [121]. However, the widespread adoption of primate models poses serious challenges, including high costs, limited availability, maintenance demands, and ethical concerns. These factors collectively pose significant barriers, effectively curbing the accessibility of primate models to only a select few laboratories with the resources and infrastructure to overcome these challenges.
Similar to dogs, primates exhibit age-dependent brain pathologies reminiscent of AD [122, 123]. The utilization of primate models in AD research has unveiled intriguing parallels, particularly in terms of tau pathology. While not all primate models showcase AD-like features, those that do significantly enrich the comprehensiveness of the disease model. One such example is the mouse lemur (Microcebus murinus), which, despite its relatively short lifespan, becomes particularly relevant in the study of aging-related neurodegeneration [124]. Remarkably, individuals aged over 5 years are considered elderly, and some of them show significant brain atrophy [125]. This atrophy is accompanied by the presence of abundant amyloid plaques, cytoskeletal tau pathology, and the loss of cholinergic neurons, providing a valuable and intricate representation of multifaceted aspects of AD pathology in a primate model with a relatively accelerated aging process.
A groundbreaking discovery unveiled the first documented instance of tauopathy with paired helical filaments in an aged chimpanzee (Pan troglodytes). Structurally identical to those observed in AD, pathologic tau forms were identified in neuronal compartments, including somata, neuropil threads, and plaque-like neurite clusters in the neocortex [126]. This finding highlights the profound similarity between tau pathology in the chimpanzee and AD, emphasizing the potential of non-human primates as valuable models for studying neurodegenerative disorders.
More recent investigation examined a substantial cohort of aged chimpanzee brains, revealing evidence of amyloid-β and tau lesions in regions affected by AD in humans [127]. Amyloid-β was detected in plaques and blood vessels, while tau lesions manifested as NFT and tau-immunoreactive neuritic clusters. Higher amyloid deposition in vessels compared to plaques correlated with increased tau lesions, implying that amyloid accumulation in microvasculature precedes plaque formation in chimpanzees. Tangle pathology was observed in individuals with plaques and cerebral amyloid angiopathy, indicating a link between amyloid and tau pathology in aged chimpanzees, highlighting the non-specificity of these AD lesions to the human brain, and pointing to primates as an excellent model for preclinical tests.
However, despite their close phylogenetic proximity to humans, primates face inherent limitations as experimental models, including high costs, limited availability, demanding maintenance, and ethical concerns.
THERAPEUTIC WINDOW
AD is a progressive pathology that gradually develops over the years and ultimately leads to clinical dementia. Dementia itself refers to the very final stage of this detrimental disease marked by severe brain atrophy and other irreversible changes in the central nervous system. By the time symptoms become noticeable, significant brain damage is already in place. The damage to brain cells and neural networks in AD is often irreversible. Once these cells are lost, they cannot be easily regenerated, limiting the potential for recovery.
The primary issue is that AD is often diagnosed only at this late stage, resulting in delayed and inadequately tailored care. This situation disappoints patients, their relatives, and health care providers. Moreover, it imposes significant financial costs and affects overall quality of life for a great part of the society. Accordingly, to reduce the prevalence of dementia and alleviate the burden upon individuals and society we must discover and implement effective strategies delaying the onset of dementia and slowing down the progression of the disease [1]. So, it is important to provide therapy and support to individuals in a timely and adequate manner.
The treatment of AD presents substantial challenges, primarily due to the undefined and presumably narrow therapeutic window. This poses a critical problem, as the disease progresses insidiously. Accordingly, effective intervention during the early stages is crucial. However, the current lack of clear biomarkers or easily identifiable symptoms in the initial phases makes it extremally challenging to detect and treat individuals before any irreversible damage occurs. Then, in our view, the AD research should focus upon developing strategies to extend and optimize the therapeutic window. This includes early detection methods, novel treatment approaches, and a deeper understanding of the disease’s underlying mechanisms. The complexity of AD requires a comprehensive and interdisciplinary approach to address the challenges and improve the chances of successful interventions.
In this context, we propose that testing various therapeutic strategies in animals should adhere to schedules that overlap with the period preceding the onset of any learning and memory deficits. The primary objective of a successful preclinical study is to demonstrate the agent’s capacity to prevent the development of cognitive decline rather than attempting to reverse existing deficits.
VALIDITY ISSUES
Undoubtedly, animal models are instrumental in advancing our knowledge of human biology. Moreover, they are valuable tools to study diseases, test potential treatments, and, eventually, bridge the gap between basic research and clinical applications. However, assessing the value of an animal model involves several key considerations. These include how closely the model resembles human biology, the specific research objectives it addresses, ethical concerns related to animal welfare, its ability to predict human outcomes, and its place in relation to alternative testing methods. The complexity of the disease being studied, transparency in research, regulatory requirements, and ongoing technological advancements also determine an animal model’s value in modern medical research.
In the preclinical setting, an animal model validity evaluation can be difficult due to the issue of species differences. Even though mammals share a significant amount of genetic material, the differences still dramatically affect research outcomes. Moreover, the animals’ environment and behavior in a laboratory are drastically different from their natural habitats, which introduce biases. Lastly, ethical considerations seriously limit the types and spectrum of experimentation that can be done, which reduces the research comprehensiveness. Subsequently, in AD research animal models may not accurately represent the genetic and environmental factors that contribute to AD in humans. The genetic makeup of animals perplexes the investigation of the impact of specific genetic variations on disease development and progression. Moreover, animal models do not replicate the influence of environmental factors that play a role in the onset and progression of AD in humans.
McKinney and Bunney (1969) pioneered the field of animal model research by introducing criteria for assessing the external validity of these models, with a primary focus on affective disorders [128]. In 1984, Wilner proposed a simplified framework consisting of three external validations criteria: predictive validity, face validity, and construct validity [129]. These are currently widely accepted model validation criteria that benchmark accuracy and reliability.
According to the framework, predictive validity indicates a model’s ability to predict future outcomes. Therefore, high predictive validity of a drug tested in an animal model successfully predicts its efficacy and safety in humans. Face validity assesses whether the model accurately represents the human condition or disease. A model with high face validity precisely represents the symptoms, pathology, or behavior characteristic to a human disease. Construct validity assesses the degree to which a model represents the underlying theoretical constructs and mimics the biological, physiological, or pathological processes of the human condition. Accordingly, a model with high construct validity exactly replicates the molecular mechanisms of a human disease [130].
In fact, evaluating the validity rate provides researchers with a tool to establish the relevance and assess reliability of a specific animal model in research of human conditions. However, the scientific community lacks consensus regarding AD etiology and pathogenesis, which poses significant challenges. Consequently, achieving high construct validity in creating a model becomes a complex task. The inherent ambiguity surrounding AD’s underlying mechanisms and causative factors poses objective obstacles, underscoring the intricacies of aligning animal models with the diverse aspects of this multifaceted human condition.
Significant strides have been made in developing numerous animal models with high face validity for AD, as evidenced by their successful utilization in preclinical studies. These models have demonstrated promising results, showcasing their efficacy in reversing cognitive decline and reducing the rate of amyloidosis. The achievement of high face validity in these animal models reflects a closer resemblance to the observable features of AD in humans, enhancing their translational relevance and contributing to the advancement of therapeutic strategies.
Nevertheless, the current absence of an effective disease-modifying therapy accentuates a potential challenge with the predictive validity of these models. Serious translational gap between promising outcomes and the development of clinically impactful therapies suggests a need for further scrutiny. Addressing the limitations in predictive validity becomes imperative to ensure that insights gained from preclinical studies can robustly inform the discovery and development of transformative treatments that transcend the confines of the laboratory and effectively target the complex pathology of AD in human patients (Fig. 2).
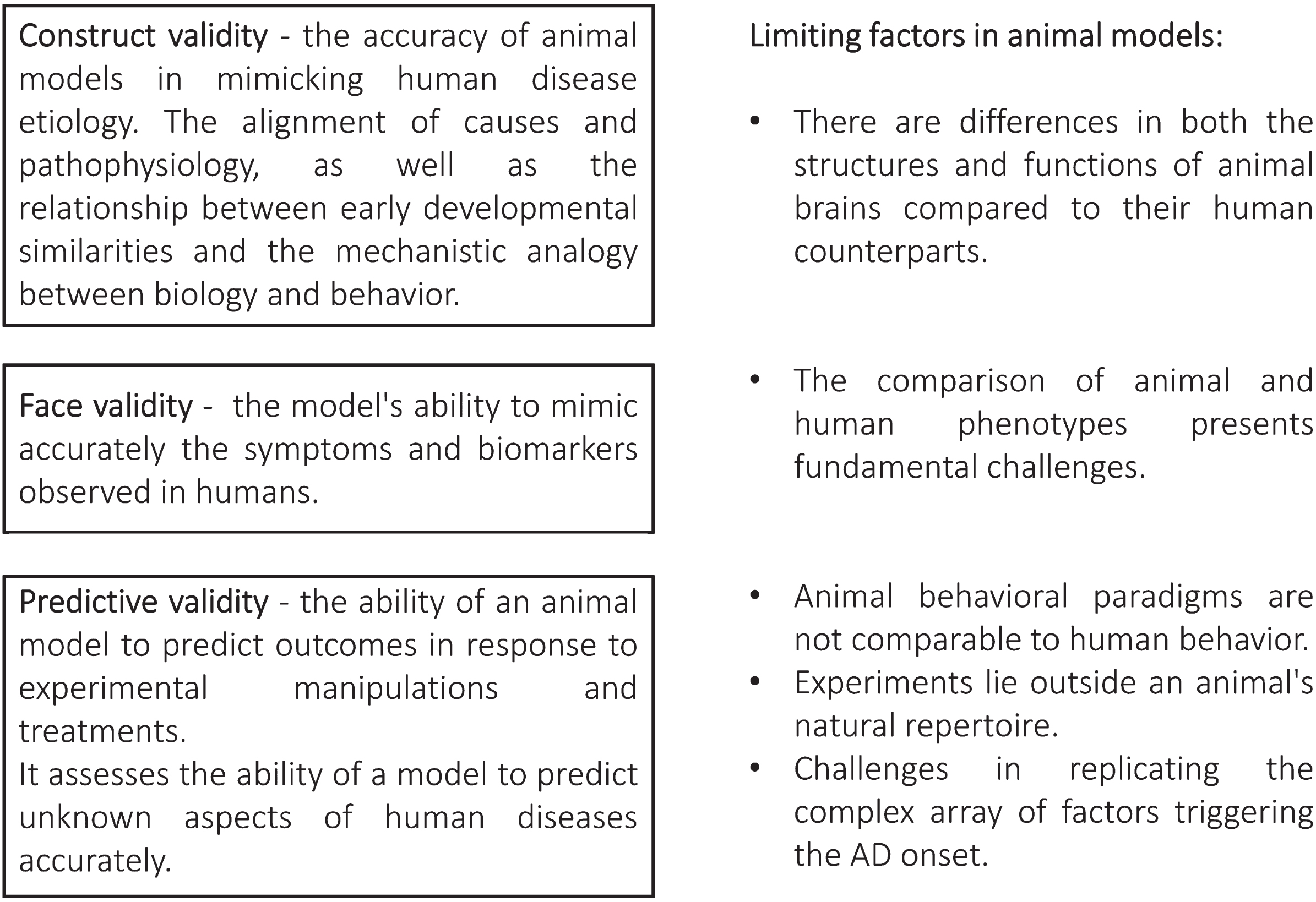
Schematic overview outlining the criteria used to validate animal models of Alzheimer’s disease, along with the challenges involved in their evaluation.
We have to keep in mind that no animal model is perfect. Inherent limitations and differences between animal and human biology curb the successful application of animal models in medical research. Therefore, the accurate extrapolation of the animal studies’ results to human populations requires caution and further validation in clinical setting. In addition, continued refinement and validation of existing and creation new animal models remains crucial for ensuring the robustness and reliability of preclinical research. Navigating these challenges is paramount for enhancing the translational potential.
Although a single model cannot completely replicate the clinical setting, it is plausible to come close by using a combination of models demonstrating validity across broad spectrums. Thus, a multifactorial approach can be applied more efficiently to study and treat AD with selection of a suitable combination of models. Still, the inherent complexity of AD continues to impose notable constraints on accessing specific realms of validity.
NON-ANIMAL MODELS
Several emerging approaches and novel laboratory techniques promise improvement in our grasp of the AD pathogenesis and may bridge the existing gap between animal models and human disease. One such approach is the utilization of human brain organoids, which are three-dimensional models mimicking the structure and function of the human brain [131]. Organoids represent more accurately human brain complexity than traditional in vitro models and allow researchers to study the disease progression in a highly controlled laboratory setting.
Another promising direction is the application of advanced computational models. These models incorporate data from human studies and simulate the complex interactions between genetic, environmental, and biological factors involved in AD [132]. In fact, these models already provide researchers with valuable insights into disease mechanisms and indicate potential therapeutic targets by utilizing artificial intelligence and machine learning tools [133].
These alternative approaches offer novel possibilities for improving our understanding of AD and addressing the discrepancies between animal models and human pathology. Hence, we hope to develop more accurate and effective AD treatments via combining the emerging techniques with existing methodologies. In this context, non-animal models offer advantages in reflecting inter-individual differences observed in patients and have the potential to provide a more accurate representation of the disease [134].
Another promising in vitro approach involves the use of cortical neurons that are derived from induced pluripotent stem cells of individuals who have early symptomatic AD. The main objective of this method is to match the disease characteristics of the patient with analogous features in the cells. This personalized model allows for the study of AD in a more personalized way and could provide valuable insights into the clinical vulnerability of patients [134].
While non-animal models demonstrate great potential for advancing AD research and preclinical screening of drugs, it is important to recognize the fundamental limitations in replicating the clinical complexity of AD within a single model. The intricate AD nature poses serious challenges as certain aspects of the disease remain beyond the reach of any single animal, in vitro, or in silico model. We state that a multilayered approach combining various models is essential to comprehensively understand and effectively treat AD. This holistic approach should capture the diverse aspects of the disease and contribute to resolution of the problem.
DISCUSSION
Mice have served as ubiquitous models in the exploration of diverse facets of AD; nonetheless, it is imperative to acknowledge that they do not spontaneously exhibit AD pathology akin to humans. Several compelling rationales underpin this disparity. Primarily, disparities in brain size, structural complexity, metabolic processes, and anatomical intricacies between mouse and human cerebral architectures pointedly influence the manifestation of AD-like pathology in murine models. Likewise, the dissimilarities in genetic backgrounds between mice and humans are paramount, as the genetic mutations implicated in human AD pathogenesis are not inherently recapitulated in murine counterparts. Mice also exhibit considerably truncated lifespans relative to their human counterparts, and as AD predominantly afflicts the aging population, mice often fail to attain the requisite longevity to faithfully replicate the spectrum of age-related changes observed in human AD patients.
As we elucidated, numerous murine AD models are meticulously crafted through genetic engineering techniques, resulting in the overexpression of mutant forms of APP or tau. While these models can recapitulate select facets of AD, they may fall short of encapsulating the full spectrum of intricacies characterizing the human condition. Accordingly, we have to judiciously acknowledge the inherent constraints of mouse models and exercise circumspection in the interpretation of research findings. These models undeniably serve as invaluable instruments for the dissection of discrete elements of AD pathophysiology and for the assessment of prospective therapeutic interventions. However, it is important to note that they cannot perfectly replicate the complex nature of human disease.
Brain-resident immune cells activation, peripheral cells migration across the blood-brain barrier, and release of inflammatory factors contribute to neuroinflammatory responses and progression of neurodegeneration in AD [135, 136]. The immune system functions as a nuanced orchestra, where subtle variations in its intricate composition can significantly influence treatment outcomes across species. Given the substantial differences between human and rodent immune systems, translational research based on murine models presents a formidable challenge. Therefore, effectively navigating these differences requires a profound understanding of species-specific immunological intricacies and an appreciation for the dynamic nature of the immune system.
The absence of consensus on AD etiology and pathogenesis hinders successful preclinical research [137]. We conceptualize AD as a spectrum of disorders. Within this continuum, a common downstream pathway and an identifiable pattern of aberrant biological responses converge, ultimately culminating in the onset of clinical dementia. We interpret the process of brain amyloidogenesis as an evolutionarily conserved response to oxidative and metabolic stresses. Various factors have the potential to incite this intricate biological reaction. This perspective harmonizes with the intriguing hypothesis that amyloid-β serves as an antioxidant agent within the aging brain and amid the context of AD [46, 138]. A compelling proposition posits that the accrual of amyloid-β within neuronal structures may, in fact, embody a cellular defense mechanism directed towards mitigating the deleterious effects of oxidative damage.
The process of amyloid aggregation leading to the formation of extracellular amyloid plaques, characterized by insoluble amyloid forms, can be seen as an adaptive mechanism employed by the brain. This biological process decreases the soluble toxic oligomeric and fibrillar amyloid-β species levels, which detrimentally affect synaptic function and trigger an inflammatory reaction. As a result, there is a gradual decrease in amyloid-β concentration within the cerebrospinal fluid of individuals with AD [139].
In this context, it becomes evident that rodent models of AD, characterized by the overexpression of APP or mutant variants of β- and γ-secretases, fall short of faithfully recapitulating the pathogenesis of LOAD. Consequently, these models are deemed inadequate for preclinical assessments of potential disease-modifying interventions. Moreover, focusing solely on eradicating amyloid-β plaques seems to lack foresight.
Furthermore, recent research indicates that the relationship between protein aggregates and neural dysfunction is extremely complex and might vary depending on the stage of AD progression, the specific forms of aggregates, sex, and individual differences [140, 141]. The toxicity of amyloid-β aggregates likely involves a combination of factors, including size and conformation, their interaction with cellular processes, inflammation, and genetic factors [142, 143]. While human amyloid plaques exhibit an apparent structural similarity to those found in mice, a striking divergence emerges when examining the resilience of their core constituents [144]. Human amyloid plaques contain cores that are remarkably resistant to both chemical and physical disruption. In stark contrast, the amyloid cores produced in mice readily dissolve in buffers containing sodium dodecyl sulfate. Therefore, amyloid-β aggregates in animal models poorly reflect human pathology. Accordingly, the evaluation of therapeutic agents or protocols in these animal models necessitates a discerning assessment, recognizing the absence of complete equivalence between the transgenic mouse plaques and the pathological lesions characteristic of human disease.
CONCLUSIONS
In order to address translational challenges, it is imperative that preclinical investigations encompass a diverse array of models, spanning transgenic mice, non-human primates, and models characterized by distinct genetic backgrounds. Engaging in extended experiments and longitudinal AD research can provide a more comprehensive understanding of the temporal aspects of its pathology. The accuracy of translating findings from animal models to human patients may be enhanced by aligning preclinical study methodologies with clinical research practices and incorporating more pertinent outcome measures.
Exploring innovative methodologies, including the development and utilization of models featuring humanized characteristics, such as the integration of human neurons or glial cells, and the use of human-derived neurons and 3D brain organoids, represents a high-priority strategy. These approaches aim to advance our comprehension and replication of the human-specific aspects of AD, ultimately contributing to the validation of findings in human tissues.
Besides, funding and educational institutions should actively promote robust international collaboration and data sharing among researchers to cultivate a more extensive and integrated knowledge base. Such initiatives are pivotal in advancing the collective pursuit of effective AD treatments, mitigating duplication of efforts, and expediting the pace of research.
AUTHOR CONTRIBUTIONS
Baruh Polis (Conceptualization; Formal analysis; Methodology; Writing – original draft; Writing – review & editing); Abraham O. Samson (Conceptualization; Methodology; Supervision; Writing – review & editing).
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.